
Multifaceted Microcephaly-Related Gene MCPH1


Abstract
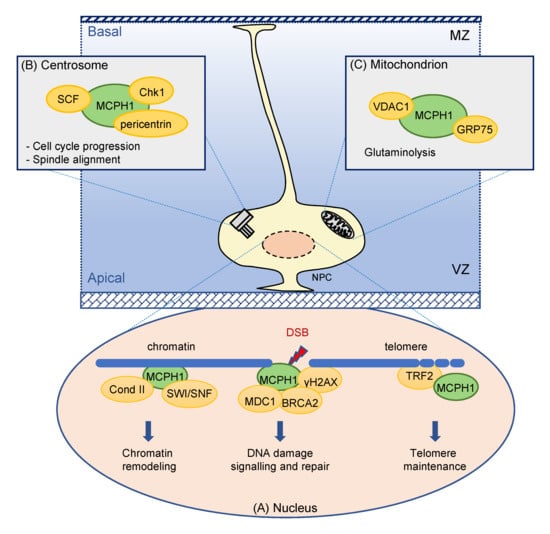
:
1. Neurogenesis and Brain Size Determination
2. Genetics and Etiology of Microcephaly (MCPH)
3. Primary Microcephaly Type 1—MCPH1
4. MCPH1 Protein Structure
5. Cellular Function of MCPH1
5.1. MCPH1 Subcellular Localization
5.2. Cellular Toxicity
5.3. MCPH1 and DNA Repair
5.4. MCPH1 and Cell Cycle Control
5.5. Emerging Role of MCPH1 in Metabolism
6. Animal Models of MCPH1
6.1. Mcph1−/− Mice
6.2. Mcph1gt/gt Mice
6.3. Mcph1-del Mice
6.4. Mcph1tm1a/tm1a Mice
6.5. Mcph1lox/lox; Emx1kiCre/+
6.6. Mcph1-ΔBR1 Mice
7. Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kaindl, A.M.; Passemard, S.; Kumar, P.; Kraemer, N.; Issa, L.; Zwirner, A.; Gerard, B.; Verloes, A.; Mani, S.; Gressens, P. Many roads lead to primary autosomal recessive microcephaly. Prog. Neurobiol. 2010, 90, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.R.; Hong, C.J.; Kim, J.Y.; Kim, E.K.; Sun, W.; Yu, S.W. Control of adult neurogenesis by programmed cell death in the mammalian brain. Mol. Brain 2016, 9, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotz, M.; Huttner, W.B. The cell biology of neurogenesis. Nat. Rev. Mol. Cell Biol. 2005, 6, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Hakanen, J.; Ruiz-Reig, N.; Tissir, F. Linking Cell Polarity to Cortical Development and Malformations. Front. Cell Neurosci. 2019, 13, 244. [Google Scholar] [CrossRef]
- Paridaen, J.T.; Huttner, W.B. Neurogenesis during development of the vertebrate central nervous system. EMBO Rep. 2014, 15, 351–364. [Google Scholar] [CrossRef] [Green Version]
- Bonnefont, J.; Vanderhaeghen, P. Neuronal fate acquisition and specification: Time for a change. Curr. Opin. Neurobiol. 2021, 66, 195–204. [Google Scholar] [CrossRef]
- Chenn, A.; Zhang, Y.A.; Chang, B.T.; McConnell, S.K. Intrinsic polarity of mammalian neuroepithelial cells. Mol. Cell Neurosci. 1998, 11, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Sauer, F.C. Mitosis in the neural tube and the cellular structure of the neural tube. Philadelphia 1935, 377–405, 313–323. [Google Scholar]
- Kosodo, Y. Interkinetic nuclear migration: Beyond a hallmark of neurogenesis. Cell Mol. Life Sci. 2012, 69, 2727–2738. [Google Scholar] [CrossRef]
- Molina, A.; Pituello, F. Playing with the cell cycle to build the spinal cord. Dev. Biol. 2017, 432, 14–23. [Google Scholar] [CrossRef]
- Cheffer, A.; Tarnok, A.; Ulrich, H. Cell cycle regulation during neurogenesis in the embryonic and adult brain. Stem Cell Rev. Rep. 2013, 9, 794–805. [Google Scholar] [CrossRef]
- Lange, C.; Huttner, W.B.; Calegari, F. Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell Stem Cell 2009, 5, 320–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilaz, L.J.; Patti, D.; Marcy, G.; Ollier, E.; Pfister, S.; Douglas, R.J.; Betizeau, M.; Gautier, E.; Cortay, V.; Doerflinger, N.; et al. Forced G1-phase reduction alters mode of division, neuron number, and laminar phenotype in the cerebral cortex. Proc. Natl. Acad. Sci. USA 2009, 106, 21924–21929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas-Hurtado, D.; Brault, J.B.; Piolot, T.; Leconte, L.; Da Silva, N.; Pennetier, C.; Baffet, A.; Marthiens, V.; Basto, R. Differences in Mitotic Spindle Architecture in Mammalian Neural Stem Cells Influence Mitotic Accuracy during Brain Development. Curr. Biol. 2019, 29, 2993–3005. [Google Scholar] [CrossRef]
- di Pietro, F.; Echard, A.; Morin, X. Regulation of mitotic spindle orientation: An integrated view. EMBO Rep. 2016, 17, 1106–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzaki, F.; Shitamukai, A. Cell Division Modes and Cleavage Planes of Neural Progenitors during Mammalian Cortical Development. Cold Spring Harb. Perspect. Biol. 2015, 7, a015719. [Google Scholar] [CrossRef]
- Woods, C.G.; Bond, J.; Enard, W. Autosomal recessive primary microcephaly (MCPH): A review of clinical, molecular, and evolutionary findings. Am. J. Hum. Genet. 2005, 76, 717–728. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Markandaya, M.; Girimaji, S.C. Primary microcephaly: Microcephalin and ASPM determine the size of the human brain. J. Biosci. 2002, 27, 629–632. [Google Scholar] [CrossRef]
- O’Driscoll, M.; Jackson, A.P.; Jeggo, P.A. Microcephalin: A causal link between impaired damage response signalling and microcephaly. Cell Cycle 2006, 5, 2339–2344. [Google Scholar] [CrossRef] [PubMed]
- Siskos, N.; Stylianopoulou, E.; Skavdis, G.; Grigoriou, M.E. Molecular Genetics of Microcephaly Primary Hereditary: An Overview. Brain Sci. 2021, 11, 581. [Google Scholar] [CrossRef]
- Jean, F.; Stuart, A.; Tarailo-Graovac, M. Dissecting the Genetic and Etiological Causes of Primary Microcephaly. Front. Neurol. 2020, 11, 570830. [Google Scholar] [CrossRef] [PubMed]
- Bond, J.; Woods, C.G. Cytoskeletal genes regulating brain size. Curr. Opin. Cell Biol. 2006, 18, 95–101. [Google Scholar] [CrossRef]
- Thomas, S.; Boutaud, L.; Reilly, M.L.; Benmerah, A. Cilia in hereditary cerebral anomalies. Biol. Cell 2019, 111, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Joukov, V.; De Nicolo, A. The Centrosome and the Primary Cilium: The Yin and Yang of a Hybrid Organelle. Cells 2019, 8, 701. [Google Scholar] [CrossRef] [Green Version]
- Andreu-Cervera, A.; Catala, M.; Schneider-Maunoury, S. Cilia, ciliopathies and hedgehog-related forebrain developmental disorders. Neurobiol. Dis. 2021, 150, 105236. [Google Scholar] [CrossRef] [PubMed]
- Bornens, M. Cell polarity: Having and making sense of direction-on the evolutionary significance of the primary cilium/centrosome organ in Metazoa. Open Biol. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Nulty, J.; Alsaffar, M.; Barry, D. Radial glial cells organize the central nervous system via microtubule dependant processes. Brain Res. 2015, 1625, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, D.; Bae, B.I.; Walsh, C.A. The Genetics of Primary Microcephaly. Annu. Rev. Genom. Hum. Genet. 2018, 19, 177–200. [Google Scholar] [CrossRef] [Green Version]
- Jackson, A.P.; Eastwood, H.; Bell, S.M.; Adu, J.; Toomes, C.; Carr, I.M.; Roberts, E.; Hampshire, D.J.; Crow, Y.J.; Mighell, A.J.; et al. Identification of microcephalin, a protein implicated in determining the size of the human brain. Am. J. Hum. Genet. 2002, 71, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Trimborn, M.; Bell, S.M.; Felix, C.; Rashid, Y.; Jafri, H.; Griffiths, P.D.; Neumann, L.M.; Krebs, A.; Reis, A.; Sperling, K.; et al. Mutations in microcephalin cause aberrant regulation of chromosome condensation. Am. J. Hum. Genet. 2004, 75, 261–266. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.Y.; Elledge, S.J. Multiple tumor suppressor pathways negatively regulate telomerase. Cell 2003, 113, 881–889. [Google Scholar] [CrossRef] [Green Version]
- Jeffers, L.J.; Coull, B.J.; Stack, S.J.; Morrison, C.G. Distinct BRCT domains in Mcph1/Brit1 mediate ionizing radiation-induced focus formation and centrosomal localization. Oncogene 2008, 27, 139–144. [Google Scholar] [CrossRef] [Green Version]
- Journiac, N.; Gilabert-Juan, J.; Cipriani, S.; Benit, P.; Liu, X.; Jacquier, S.; Faivre, V.; Delahaye-Duriez, A.; Csaba, Z.; Hourcade, T.; et al. Cell Metabolic Alterations due to Mcph1 Mutation in Microcephaly. Cell Rep. 2020, 31, 107506. [Google Scholar] [CrossRef]
- Bilguvar, K.; Ozturk, A.K.; Louvi, A.; Kwan, K.Y.; Choi, M.; Tatli, B.; Yalnizoglu, D.; Tuysuz, B.; Caglayan, A.O.; Gokben, S.; et al. Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature 2010, 467, 207–210. [Google Scholar] [CrossRef]
- Jayaraman, D.; Kodani, A.; Gonzalez, D.M.; Mancias, J.D.; Mochida, G.H.; Vagnoni, C.; Johnson, J.; Krogan, N.; Harper, J.W.; Reiter, J.F.; et al. Microcephaly Proteins Wdr62 and Aspm Define a Mother Centriole Complex Regulating Centriole Biogenesis, Apical Complex, and Cell Fate. Neuron 2016, 92, 813–828. [Google Scholar] [CrossRef] [Green Version]
- Nicholas, A.K.; Khurshid, M.; Desir, J.; Carvalho, O.P.; Cox, J.J.; Thornton, G.; Kausar, R.; Ansar, M.; Ahmad, W.; Verloes, A.; et al. WDR62 is associated with the spindle pole and is mutated in human microcephaly. Nat. Genet. 2010, 42, 1010–1014. [Google Scholar] [CrossRef]
- Yu, T.W.; Mochida, G.H.; Tischfield, D.J.; Sgaier, S.K.; Flores-Sarnat, L.; Sergi, C.M.; Topcu, M.; McDonald, M.T.; Barry, B.J.; Felie, J.M.; et al. Mutations in WDR62, encoding a centrosome-associated protein, cause microcephaly with simplified gyri and abnormal cortical architecture. Nat. Genet. 2010, 42, 1015–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bond, J.; Roberts, E.; Springell, K.; Lizarraga, S.B.; Scott, S.; Higgins, J.; Hampshire, D.J.; Morrison, E.E.; Leal, G.F.; Silva, E.O.; et al. A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat. Genet. 2005, 37, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Kodani, A.; Yu, T.W.; Johnson, J.R.; Jayaraman, D.; Johnson, T.L.; Al-Gazali, L.; Sztriha, L.; Partlow, J.N.; Kim, H.; Krup, A.L.; et al. Centriolar satellites assemble centrosomal microcephaly proteins to recruit CDK2 and promote centriole duplication. Elife 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Genin, A.; Desir, J.; Lambert, N.; Biervliet, M.; Van Der Aa, N.; Pierquin, G.; Killian, A.; Tosi, M.; Urbina, M.; Lefort, A.; et al. Kinetochore KMN network gene CASC5 mutated in primary microcephaly. Hum. Mol. Genet. 2012, 21, 5306–5317. [Google Scholar] [CrossRef] [Green Version]
- Bond, J.; Roberts, E.; Mochida, G.H.; Hampshire, D.J.; Scott, S.; Askham, J.M.; Springell, K.; Mahadevan, M.; Crow, Y.J.; Markham, A.F.; et al. ASPM is a major determinant of cerebral cortical size. Nat. Genet. 2002, 32, 316–320. [Google Scholar] [CrossRef]
- Kumar, A.; Girimaji, S.C.; Duvvari, M.R.; Blanton, S.H. Mutations in STIL, encoding a pericentriolar and centrosomal protein, cause primary microcephaly. Am. J. Hum. Genet. 2009, 84, 286–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.J.; Lin, S.Y.; Hsu, W.B.; Lin, Y.N.; Wu, C.T.; Lin, Y.C.; Chang, C.W.; Wu, K.S.; Tang, T.K. The human microcephaly protein STIL interacts with CPAP and is required for procentriole formation. EMBO J. 2011, 30, 4790–4804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, M.S.; Baig, S.M.; Neumann, S.; Nurnberg, G.; Farooq, M.; Ahmad, I.; Alef, T.; Hennies, H.C.; Technau, M.; Altmuller, J.; et al. A truncating mutation of CEP135 causes primary microcephaly and disturbed centrosomal function. Am. J. Hum. Genet. 2012, 90, 871–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.C.; Chang, C.W.; Hsu, W.B.; Tang, C.J.; Lin, Y.N.; Chou, E.J.; Wu, C.T.; Tang, T.K. Human microcephaly protein CEP135 binds to hSAS-6 and CPAP, and is required for centriole assembly. EMBO J. 2013, 32, 1141–1154. [Google Scholar] [CrossRef] [Green Version]
- Guernsey, D.L.; Jiang, H.; Hussin, J.; Arnold, M.; Bouyakdan, K.; Perry, S.; Babineau-Sturk, T.; Beis, J.; Dumas, N.; Evans, S.C.; et al. Mutations in centrosomal protein CEP152 in primary microcephaly families linked to MCPH4. Am. J. Hum. Genet. 2010, 87, 40–51. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.J.; Baltus, A.E.; Mathew, R.S.; Murphy, E.A.; Evrony, G.D.; Gonzalez, D.M.; Wang, E.P.; Marshall-Walker, C.A.; Barry, B.J.; Murn, J.; et al. Microcephaly gene links trithorax and REST/NRSF to control neural stem cell proliferation and differentiation. Cell 2012, 151, 1097–1112. [Google Scholar] [CrossRef] [Green Version]
- Awad, S.; Al-Dosari, M.S.; Al-Yacoub, N.; Colak, D.; Salih, M.A.; Alkuraya, F.S.; Poizat, C. Mutation in PHC1 implicates chromatin remodeling in primary microcephaly pathogenesis. Hum. Mol. Genet. 2013, 22, 2200–2213. [Google Scholar] [CrossRef] [Green Version]
- Hussain, M.S.; Baig, S.M.; Neumann, S.; Peche, V.S.; Szczepanski, S.; Nurnberg, G.; Tariq, M.; Jameel, M.; Khan, T.N.; Fatima, A.; et al. CDK6 associates with the centrosome during mitosis and is mutated in a large Pakistani family with primary microcephaly. Hum. Mol. Genet. 2013, 22, 5199–5214. [Google Scholar] [CrossRef]
- Mirzaa, G.M.; Vitre, B.; Carpenter, G.; Abramowicz, I.; Gleeson, J.G.; Paciorkowski, A.R.; Cleveland, D.W.; Dobyns, W.B.; O’Driscoll, M. Mutations in CENPE define a novel kinetochore-centromeric mechanism for microcephalic primordial dwarfism. Hum. Genet. 2014, 133, 1023–1039. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.; Rupp, V.M.; Orpinell, M.; Hussain, M.S.; Altmuller, J.; Steinmetz, M.O.; Enzinger, C.; Thiele, H.; Hohne, W.; Nurnberg, G.; et al. A missense mutation in the PISA domain of HsSAS-6 causes autosomal recessive primary microcephaly in a large consanguineous Pakistani family. Hum. Mol. Genet. 2014, 23, 5940–5949. [Google Scholar] [CrossRef]
- Alakbarzade, V.; Hameed, A.; Quek, D.Q.; Chioza, B.A.; Baple, E.L.; Cazenave-Gassiot, A.; Nguyen, L.N.; Wenk, M.R.; Ahmad, A.Q.; Sreekantan-Nair, A.; et al. A partially inactivating mutation in the sodium-dependent lysophosphatidylcholine transporter MFSD2A causes a non-lethal microcephaly syndrome. Nat. Genet. 2015, 47, 814–817. [Google Scholar] [CrossRef]
- Guemez-Gamboa, A.; Nguyen, L.N.; Yang, H.; Zaki, M.S.; Kara, M.; Ben-Omran, T.; Akizu, N.; Rosti, R.O.; Rosti, B.; Scott, E.; et al. Inactivating mutations in MFSD2A, required for omega-3 fatty acid transport in brain, cause a lethal microcephaly syndrome. Nat. Genet. 2015, 47, 809–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, S.; Jaiswal, M.; Charng, W.L.; Gambin, T.; Karaca, E.; Mirzaa, G.; Wiszniewski, W.; Sandoval, H.; Haelterman, N.A.; Xiong, B.; et al. A drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases. Cell 2014, 159, 200–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, B.N.; Moccia, A.; Drunat, S.; Soukarieh, O.; Tubeuf, H.; Chitty, L.S.; Verloes, A.; Gressens, P.; El Ghouzzi, V.; Joriot, S.; et al. Mutations in Citron Kinase Cause Recessive Microlissencephaly with Multinucleated Neurons. Am. J. Hum. Genet. 2016, 99, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Bielas, S.L.; Zaki, M.S.; Ismail, S.; Farfara, D.; Um, K.; Rosti, R.O.; Scott, E.C.; Tu, S.; Chi, N.C.; et al. Biallelic Mutations in Citron Kinase Link Mitotic Cytokinesis to Human Primary Microcephaly. Am. J. Hum. Genet. 2016, 99, 501–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadir, R.; Harel, T.; Markus, B.; Perez, Y.; Bakhrat, A.; Cohen, I.; Volodarsky, M.; Feintsein-Linial, M.; Chervinski, E.; Zlotogora, J.; et al. ALFY-Controlled DVL3 Autophagy Regulates Wnt Signaling, Determining Human Brain Size. PLoS Genet. 2016, 12, e1005919. [Google Scholar] [CrossRef] [Green Version]
- DiStasio, A.; Driver, A.; Sund, K.; Donlin, M.; Muraleedharan, R.M.; Pooya, S.; Kline-Fath, B.; Kaufman, K.M.; Prows, C.A.; Schorry, E.; et al. Copb2 is essential for embryogenesis and hypomorphic mutations cause human microcephaly. Hum. Mol. Genet. 2017, 26, 4836–4848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carleton, M.; Mao, M.; Biery, M.; Warrener, P.; Kim, S.; Buser, C.; Marshall, C.G.; Fernandes, C.; Annis, J.; Linsley, P.S. RNA interference-mediated silencing of mitotic kinesin KIF14 disrupts cell cycle progression and induces cytokinesis failure. Mol. Cell Biol. 2006, 26, 3853–3863. [Google Scholar] [CrossRef] [Green Version]
- Moawia, A.; Shaheen, R.; Rasool, S.; Waseem, S.S.; Ewida, N.; Budde, B.; Kawalia, A.; Motameny, S.; Khan, K.; Fatima, A.; et al. Mutations of KIF14 cause primary microcephaly by impairing cytokinesis. Ann. Neurol. 2017, 82, 562–577. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.A.; Murray, J.E.; Carroll, P.; Leitch, A.; Mackenzie, K.J.; Halachev, M.; Fetit, A.E.; Keith, C.; Bicknell, L.S.; Fluteau, A.; et al. Mutations in genes encoding condensin complex proteins cause microcephaly through decatenation failure at mitosis. Genes Dev. 2016, 30, 2158–2172. [Google Scholar] [CrossRef] [Green Version]
- Braun, D.A.; Lovric, S.; Schapiro, D.; Schneider, R.; Marquez, J.; Asif, M.; Hussain, M.S.; Daga, A.; Widmeier, E.; Rao, J.; et al. Mutations in multiple components of the nuclear pore complex cause nephrotic syndrome. J. Clin. Investig. 2018, 128, 4313–4328. [Google Scholar] [CrossRef] [Green Version]
- Perez, Y.; Bar-Yaacov, R.; Kadir, R.; Wormser, O.; Shelef, I.; Birk, O.S.; Flusser, H.; Birnbaum, R.Y. Mutations in the microtubule-associated protein MAP11 (C7orf43) cause microcephaly in humans and zebrafish. Brain 2019, 142, 574–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parry, D.A.; Martin, C.A.; Greene, P.; Marsh, J.A.; Genomics England Research, C.; Blyth, M.; Cox, H.; Donnelly, D.; Greenhalgh, L.; Greville-Heygate, S.; et al. Heterozygous lamin B1 and lamin B2 variants cause primary microcephaly and define a novel laminopathy. Genet. Med. 2021, 23, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.; Lindbaek, L.; Krogh, N.; Doganli, C.; Keller, C.; Monnich, M.; Goncalves, A.B.; Sakthivel, S.; Mang, Y.; Fatima, A.; et al. RRP7A links primary microcephaly to dysfunction of ribosome biogenesis, resorption of primary cilia, and neurogenesis. Nat. Commun. 2020, 11, 5816. [Google Scholar] [CrossRef] [PubMed]
- Waseem, S.S.; Moawia, A.; Budde, B.; Tariq, M.; Khan, A.; Ali, Z.; Khan, S.; Iqbal, M.; Malik, N.A.; Haque, S.U.; et al. A Homozygous AKNA Frameshift Variant Is Associated with Microcephaly in a Pakistani Family. Genes 2021, 12, 1494. [Google Scholar] [CrossRef]
- Neitzel, H.; Neumann, L.M.; Schindler, D.; Wirges, A.; Tonnies, H.; Trimborn, M.; Krebsova, A.; Richter, R.; Sperling, K. Premature chromosome condensation in humans associated with microcephaly and mental retardation: A novel autosomal recessive condition. Am. J. Hum. Genet. 2002, 70, 1015–1022. [Google Scholar] [CrossRef] [Green Version]
- Tommerup, N.; Mortensen, E.; Nielsen, M.H.; Wegner, R.D.; Schindler, D.; Mikkelsen, M. Chromosomal breakage, endomitosis, endoreduplication, and hypersensitivity toward radiomimetric and alkylating agents: A possible new autosomal recessive mutation in a girl with craniosynostosis and microcephaly. Hum. Genet. 1993, 92, 339–346. [Google Scholar] [CrossRef]
- Farooq, M.; Baig, S.; Tommerup, N.; Kjaer, K.W. Craniosynostosis-microcephaly with chromosomal breakage and other abnormalities is caused by a truncating MCPH1 mutation and is allelic to premature chromosomal condensation syndrome and primary autosomal recessive microcephaly type 1. Am. J. Med. Genet. A 2010, 152A, 495–497. [Google Scholar] [CrossRef]
- Phillips, E.R.; McKinnon, P.J. DNA double-strand break repair and development. Oncogene 2007, 26, 7799–7808. [Google Scholar] [CrossRef] [Green Version]
- Alderton, G.K.; Galbiati, L.; Griffith, E.; Surinya, K.H.; Neitzel, H.; Jackson, A.P.; Jeggo, P.A.; O’Driscoll, M. Regulation of mitotic entry by microcephalin and its overlap with ATR signalling. Nat. Cell Biol. 2006, 8, 725–733. [Google Scholar] [CrossRef]
- Alcantara, D.; O’Driscoll, M. Congenital microcephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2014, 166C, 124–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, A.P.; McHale, D.P.; Campbell, D.A.; Jafri, H.; Rashid, Y.; Mannan, J.; Karbani, G.; Corry, P.; Levene, M.I.; Mueller, R.F.; et al. Primary autosomal recessive microcephaly (MCPH1) maps to chromosome 8p22-pter. Am. J. Hum. Genet. 1998, 63, 541–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darvish, H.; Esmaeeli-Nieh, S.; Monajemi, G.B.; Mohseni, M.; Ghasemi-Firouzabadi, S.; Abedini, S.S.; Bahman, I.; Jamali, P.; Azimi, S.; Mojahedi, F.; et al. A clinical and molecular genetic study of 112 Iranian families with primary microcephaly. J. Med. Genet. 2010, 47, 823–828. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.Y.; Rai, R.; Li, K.; Xu, Z.X.; Elledge, S.J. BRIT1/MCPH1 is a DNA damage responsive protein that regulates the Brca1-Chk1 pathway, implicating checkpoint dysfunction in microcephaly. Proc. Natl. Acad. Sci. USA 2005, 102, 15105–15109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulvers, J.N.; Journiac, N.; Arai, Y.; Nardelli, J. MCPH1: A window into brain development and evolution. Front. Cell Neurosci. 2015, 9, 92. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhou, Z.W.; Wang, Z.Q. The DNA damage response molecule MCPH1 in brain development and beyond. Acta Biochim. Biophys. Sin. (Shanghai) 2016, 48, 678–685. [Google Scholar] [CrossRef] [Green Version]
- Trimborn, M.; Richter, R.; Sternberg, N.; Gavvovidis, I.; Schindler, D.; Jackson, A.P.; Prott, E.C.; Sperling, K.; Gillessen-Kaesbach, G.; Neitzel, H. The first missense alteration in the MCPH1 gene causes autosomal recessive microcephaly with an extremely mild cellular and clinical phenotype. Hum. Mutat. 2005, 26, 496. [Google Scholar] [CrossRef]
- Ghani-Kakhki, M.; Robinson, P.N.; Morlot, S.; Mitter, D.; Trimborn, M.; Albrecht, B.; Varon, R.; Sperling, K.; Neitzel, H. Two Missense Mutations in the Primary Autosomal Recessive Microcephaly Gene MCPH1 Disrupt the Function of the Highly Conserved N-Terminal BRCT Domain of Microcephalin. Mol. Syndromol. 2012, 3, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.Y.; Liang, Y.; Li, K. Multiple roles of BRIT1/MCPH1 in DNA damage response, DNA repair, and cancer suppression. Yonsei Med. J. 2010, 51, 295–301. [Google Scholar] [CrossRef] [Green Version]
- Gruber, R.; Zhou, Z.; Sukchev, M.; Joerss, T.; Frappart, P.O.; Wang, Z.Q. MCPH1 regulates the neuroprogenitor division mode by coupling the centrosomal cycle with mitotic entry through the Chk1-Cdc25 pathway. Nat. Cell Biol. 2011, 13, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Gavvovidis, I.; Rost, I.; Trimborn, M.; Kaiser, F.J.; Purps, J.; Wiek, C.; Hanenberg, H.; Neitzel, H.; Schindler, D. A novel MCPH1 isoform complements the defective chromosome condensation of human MCPH1-deficient cells. PLoS ONE 2012, 7, e40387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glover, J.N.; Williams, R.S.; Lee, M.S. Interactions between BRCT repeats and phosphoproteins: Tangled up in two. Trends Biochem. Sci. 2004, 29, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Huyton, T.; Bates, P.A.; Zhang, X.; Sternberg, M.J.; Freemont, P.S. The BRCA1 C-terminal domain: Structure and function. Mutat. Res. 2000, 460, 319–332. [Google Scholar] [CrossRef]
- Derbyshire, D.J.; Basu, B.P.; Serpell, L.C.; Joo, W.S.; Date, T.; Iwabuchi, K.; Doherty, A.J. Crystal structure of human 53BP1 BRCT domains bound to p53 tumour suppressor. EMBO J. 2002, 21, 3863–3872. [Google Scholar] [CrossRef] [Green Version]
- Mesquita, R.D.; Woods, N.T.; Seabra-Junior, E.S.; Monteiro, A.N. Tandem BRCT Domains: DNA’s Praetorian Guard. Genes Cancer 2010, 1, 1140–1146. [Google Scholar] [CrossRef]
- Williams, R.S.; Bernstein, N.; Lee, M.S.; Rakovszky, M.L.; Cui, D.; Green, R.; Weinfeld, M.; Glover, J.N. Structural basis for phosphorylation-dependent signaling in the DNA-damage response. Biochem. Cell Biol. 2005, 83, 721–727. [Google Scholar] [CrossRef]
- Woods, N.T.; Mesquita, R.D.; Sweet, M.; Carvalho, M.A.; Li, X.; Liu, Y.; Nguyen, H.; Thomas, C.E.; Iversen, E.S., Jr.; Marsillac, S.; et al. Charting the landscape of tandem BRCT domain-mediated protein interactions. Sci. Signal. 2012, 5, rs6. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Lu, L.Y.; Yang, C.Y.; Wang, S.; Yu, X. The FHA and BRCT domains recognize ADP-ribosylation during DNA damage response. Genes Dev. 2013, 27, 1752–1768. [Google Scholar] [CrossRef] [Green Version]
- Wood, J.L.; Liang, Y.; Li, K.; Chen, J. Microcephalin/MCPH1 associates with the Condensin II complex to function in homologous recombination repair. J. Biol. Chem. 2008, 283, 29586–29592. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Schneble-Lohnert, N.; Kristofova, M.; Qing, X.; Labisch, J.; Hofmann, S.; Ehrenberg, S.; Sannai, M.; Jorss, T.; Ori, A.; et al. The N-terminal BRCT domain determines MCPH1 function in brain development and fertility. Cell Death Dis. 2021, 12, 143. [Google Scholar] [CrossRef]
- Kristofova, M.; Wang, Z.Q. MCPH1, beyond its role deciding the brain size. Aging (Albany NY) 2021, 13, 23437–23439. [Google Scholar] [CrossRef]
- Meyer, S.K.; Dunn, M.; Vidler, D.S.; Porter, A.; Blain, P.G.; Jowsey, P.A. Phosphorylation of MCPH1 isoforms during mitosis followed by isoform-specific degradation by APC/C-CDH1. FASEB J. 2019, 33, 2796–2808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.; Pfeifer, G.P.; Xu, X. Microcephalin encodes a centrosomal protein. Cell Cycle 2006, 5, 457–458. [Google Scholar] [CrossRef]
- Tibelius, A.; Marhold, J.; Zentgraf, H.; Heilig, C.E.; Neitzel, H.; Ducommun, B.; Rauch, A.; Ho, A.D.; Bartek, J.; Kramer, A. Microcephalin and pericentrin regulate mitotic entry via centrosome-associated Chk1. J. Cell Biol. 2009, 185, 1149–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hainline, S.G.; Rickmyre, J.L.; Neitzel, L.R.; Lee, L.A.; Lee, E. The Drosophila MCPH1-B isoform is a substrate of the APCCdh1 E3 ubiquitin ligase complex. Biol. Open 2014, 3, 669–676. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.A.; Bourke, E.; Liptrot, C.; Dockery, P.; Morrison, C.G. MCPH1/BRIT1 limits ionizing radiation-induced centrosome amplification. Oncogene 2010, 29, 5537–5544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denu, R.A.; Burkard, M.E. Analysis of the “centrosome-ome” identifies MCPH1 deletion as a cause of centrosome amplification in human cancer. Sci. Rep. 2020, 10, 11921. [Google Scholar] [CrossRef]
- Mai, L.; Yi, F.; Gou, X.; Zhang, J.; Wang, C.; Liu, G.; Bu, Y.; Yuan, C.; Deng, L.; Song, F. The overexpression of MCPH1 inhibits cell growth through regulating cell cycle-related proteins and activating cytochrome c-caspase 3 signaling in cervical cancer. Mol. Cell Biochem. 2014, 392, 95–107. [Google Scholar] [CrossRef]
- Zhou, L.; Bai, Y.; Li, Y.; Liu, X.; Tan, T.; Meng, S.; He, W.; Wu, X.; Dong, Z. Overexpression of MCPH1 inhibits uncontrolled cell growth by promoting cell apoptosis and arresting the cell cycle in S and G2/M phase in lung cancer cells. Oncol. Lett. 2016, 11, 365–372. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Lee, J.; Stern, D.F. Microcephalin is a DNA damage response protein involved in regulation of CHK1 and BRCA1. J. Biol. Chem. 2004, 279, 34091–34094. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, D.; Shintomi, K.; Ono, T.; Gavvovidis, I.; Schindler, D.; Neitzel, H.; Trimborn, M.; Hirano, T. MCPH1 regulates chromosome condensation and shaping as a composite modulator of condensin II. J. Cell Biol. 2011, 194, 841–854. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Mondal, G.; Wang, X.; Wu, J.; Yang, L.; Pankratz, V.S.; Rowley, M.; Couch, F.J. Microcephalin regulates BRCA2 and Rad51-associated DNA double-strand break repair. Cancer Res. 2009, 69, 5531–5536. [Google Scholar] [CrossRef] [Green Version]
- Wood, J.L.; Singh, N.; Mer, G.; Chen, J. MCPH1 functions in an H2AX-dependent but MDC1-independent pathway in response to DNA damage. J. Biol. Chem. 2007, 282, 35416–35423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Gao, H.; Lin, S.Y.; Peng, G.; Huang, X.; Zhang, P.; Goss, J.A.; Brunicardi, F.C.; Multani, A.S.; Chang, S.; et al. BRIT1/MCPH1 is essential for mitotic and meiotic recombination DNA repair and maintaining genomic stability in mice. PLoS Genet. 2010, 6, e1000826. [Google Scholar] [CrossRef] [Green Version]
- Peng, G.; Yim, E.K.; Dai, H.; Jackson, A.P.; Burgt, I.; Pan, M.R.; Hu, R.; Li, K.; Lin, S.Y. BRIT1/MCPH1 links chromatin remodelling to DNA damage response. Nat. Cell Biol. 2009, 11, 865–872. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Lee, O.H.; Xin, H.; Chen, L.Y.; Qin, J.; Chae, H.K.; Lin, S.Y.; Safari, A.; Liu, D.; Songyang, Z. TRF2 functions as a protein hub and regulates telomere maintenance by recognizing specific peptide motifs. Nat. Struct Mol. Biol. 2009, 16, 372–379. [Google Scholar] [CrossRef]
- Cicconi, A.; Rai, R.; Xiong, X.; Broton, C.; Al-Hiyasat, A.; Hu, C.; Dong, S.; Sun, W.; Garbarino, J.; Bindra, R.S.; et al. Microcephalin 1/BRIT1-TRF2 interaction promotes telomere replication and repair, linking telomere dysfunction to primary microcephaly. Nat. Commun. 2020, 11, 5861. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Li, M.; Su, B. MCPH1/BRIT1 represses transcription of the human telomerase reverse transcriptase gene. Gene 2012, 495, 1–9. [Google Scholar] [CrossRef]
- Yang, S.Z.; Lin, F.T.; Lin, W.C. MCPH1/BRIT1 cooperates with E2F1 in the activation of checkpoint, DNA repair and apoptosis. EMBO Rep. 2008, 9, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Trimborn, M.; Ghani, M.; Walther, D.J.; Dopatka, M.; Dutrannoy, V.; Busche, A.; Meyer, F.; Nowak, S.; Nowak, J.; Zabel, C.; et al. Establishment of a mouse model with misregulated chromosome condensation due to defective Mcph1 function. PLoS ONE 2010, 5, e9242. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zong, W.; Li, T.; Wang, Y.; Xu, X.; Zhou, Z.W.; Wang, Z.Q. The E3 ubiquitin ligase APC/C(C)(dh1) degrades MCPH1 after MCPH1-betaTrCP2-Cdc25A-mediated mitotic entry to ensure neurogenesis. EMBO J. 2017, 36, 3666–3681. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Wiltshire, T.D.; Thompson, J.R.; Mer, G.; Couch, F.J. Molecular basis for the association of microcephalin (MCPH1) protein with the cell division cycle protein 27 (Cdc27) subunit of the anaphase-promoting complex. J. Biol. Chem. 2012, 287, 2854–2862. [Google Scholar] [CrossRef] [Green Version]
- Houlard, M.; Cutts, E.E.; Shamim, M.S.; Godwin, J.; Weisz, D.; Presser Aiden, A.; Lieberman-Aiden, E.; Schermelleh, L.; Nasmyth, K.; Vannini, A. MCPH1 inhibits condensin II during interphase by regulating its SMC2-kleisin interface. Elife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Green, L.C.; Kalitsis, P.; Chang, T.M.; Cipetic, M.; Kim, J.H.; Marshall, O.; Turnbull, L.; Whitchurch, C.B.; Vagnarelli, P.; Samejima, K.; et al. Contrasting roles of condensin I and condensin II in mitotic chromosome formation. J. Cell Sci. 2012, 125, 1591–1604. [Google Scholar] [CrossRef] [Green Version]
- Vincent, E.E.; Sergushichev, A.; Griss, T.; Gingras, M.C.; Samborska, B.; Ntimbane, T.; Coelho, P.P.; Blagih, J.; Raissi, T.C.; Choiniere, L.; et al. Mitochondrial Phosphoenolpyruvate Carboxykinase Regulates Metabolic Adaptation and Enables Glucose-Independent Tumor Growth. Mol. Cell 2015, 60, 195–207. [Google Scholar] [CrossRef] [Green Version]
- Lange, C.; Turrero Garcia, M.; Decimo, I.; Bifari, F.; Eelen, G.; Quaegebeur, A.; Boon, R.; Zhao, H.; Boeckx, B.; Chang, J.; et al. Relief of hypoxia by angiogenesis promotes neural stem cell differentiation by targeting glycolysis. EMBO J. 2016, 35, 924–941. [Google Scholar] [CrossRef]
- Asimakopoulou, A.; Fulop, A.; Borkham-Kamphorst, E.; de Leur, E.V.; Gassler, N.; Berger, T.; Beine, B.; Meyer, H.E.; Mak, T.W.; Hopf, C.; et al. Altered mitochondrial and peroxisomal integrity in lipocalin-2-deficient mice with hepatic steatosis. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2093–2110. [Google Scholar] [CrossRef]
- Hans, F.; Dimitrov, S. Histone H3 phosphorylation and cell division. Oncogene 2001, 20, 3021–3027. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.W.; Tapias, A.; Bruhn, C.; Gruber, R.; Sukchev, M.; Wang, Z.Q. DNA damage response in microcephaly development of MCPH1 mouse model. DNA Repair. (Amst) 2013, 12, 645–655. [Google Scholar] [CrossRef]
- Gavvovidis, I.; Pohlmann, C.; Marchal, J.A.; Stumm, M.; Yamashita, D.; Hirano, T.; Schindler, D.; Neitzel, H.; Trimborn, M. MCPH1 patient cells exhibit delayed release from DNA damage-induced G2/M checkpoint arrest. Cell Cycle 2010, 9, 4893–4899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Ingham, N.; Clare, S.; Raisen, C.; Vancollie, V.E.; Ismail, O.; McIntyre, R.E.; Tsang, S.H.; Mahajan, V.B.; Dougan, G.; et al. Mcph1-deficient mice reveal a role for MCPH1 in otitis media. PLoS ONE 2013, 8, e58156. [Google Scholar] [CrossRef] [Green Version]
- Woods, R.P.; Freimer, N.B.; De Young, J.A.; Fears, S.C.; Sicotte, N.L.; Service, S.K.; Valentino, D.J.; Toga, A.W.; Mazziotta, J.C. Normal variants of Microcephalin and ASPM do not account for brain size variability. Hum. Mol. Genet. 2006, 15, 2025–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobson-Stone, C.; Gatt, J.M.; Kuan, S.A.; Grieve, S.M.; Gordon, E.; Williams, L.M.; Schofield, P.R. Investigation of MCPH1 G37995C and ASPM A44871G polymorphisms and brain size in a healthy cohort. Neuroimage 2007, 37, 394–400. [Google Scholar] [CrossRef]
- Rai, R.; Dai, H.; Multani, A.S.; Li, K.; Chin, K.; Gray, J.; Lahad, J.P.; Liang, J.; Mills, G.B.; Meric-Bernstam, F.; et al. BRIT1 regulates early DNA damage response, chromosomal integrity, and cancer. Cancer Cell 2006, 10, 145–157. [Google Scholar] [CrossRef] [Green Version]
- Mantere, T.; Winqvist, R.; Kauppila, S.; Grip, M.; Jukkola-Vuorinen, A.; Tervasmaki, A.; Rapakko, K.; Pylkas, K. Targeted Next-Generation Sequencing Identifies a Recurrent Mutation in MCPH1 Associating with Hereditary Breast Cancer Susceptibility. PLoS Genet. 2016, 12, e1005816. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Gao, H.; Lin, S.Y.; Goss, J.A.; Du, C.; Li, K. Mcph1/Brit1 deficiency promotes genomic instability and tumor formation in a mouse model. Oncogene 2015, 34, 4368–4378. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Luo, X.; Jiang, J.; Chen, Y.; Liu, C.; Hu, T.; Li, M.; Lin, Q.; Li, Y.; Huang, J.; et al. Transgenic rhesus monkeys carrying the human MCPH1 gene copies show human-like neoteny of brain development. Natl. Sci. Rev. 2019, 6, 480–493. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Disease ID | Chromosome Location | Inheritance | Gene | Protein | Subcellular Location | Cellular Function | Reference |
|---|---|---|---|---|---|---|---|
| MCPH1 | 8p23.1 | AR | MCPH1 | Microcephalin 1/ BRIT1 | Nucleus/Centrosome/ Mitochondria | DNA damage response, chromatin condensation, cell cycle control | [29,30,31,32,33] |
| MCPH2 | 19q13.12 | AR | WDR62 | WD Repeat-containing protein 62 | Centrosome/Spindle poles | Centriole biogenesis, spindle assembly | [34,35,36,37] |
| MCPH3 | 9q33.2 | AR | CDK5RAP2 | CDK5 regulatory subunit-associated protein 2 | Nucleus/ Centrosome | Centriole biogenesis, spindle checkpoint, cytokinesis | [38,39] |
| MCPH4 | 15q15.1 | AR | CASC5 (KNL1) | Cancer susceptibility candidate 5/Kinetochore scaffold 1 | Kinetochore | Kinetochore attachment Spindle checkpoint | [40] |
| MCPH5 | 1q31.3 | AR | ASPM | Abnormal spindle microtubule assembly | Nucleus/ Centrosome/ Midbody | Centriole biogenesis, spindle assembly, cytokinesis | [35,41] |
| MCPH6 | 13q12.12-q12.13 | AR | CENPJ (SAS-4, CPAP) | Centromere protein J, | Centrosome | Centriole biogenesis | [38] |
| MCPH7 | 1p33 | AR | STIL | SCL/TAL1 interrupting locus | Centrosome | Centriole biogenesis, spindle assembly | [42,43] |
| MCPH8 | 4q12 | AR | CEP135 | Centrosomal protein 135 | Centrosome | Centriole biogenesis | [44,45] |
| MCPH9 | 15q21.1 | AR | CEP152 | Centrosomal protein 152 | Centrosome | Centriole biogenesis | [39,46] |
| MCPH10 | 20q13.12 | AR | ZNF335 | Zinc finger protein 335 | Nucleus | Transcription, chromatin remodeling | [47] |
| MCPH11 | 12p13.31 | AR | PHC1 | Polyhomeotic-like 1 | Nucleus | Transcription, chromatin remodeling | [48] |
| MCPH12 | 7q21.2 | AR | CDK6 | Cyclin-dependent kinase 6 | Cytosol/ Nucleus/ Centrosome/ Spindle poles | Cell cycle | [49] |
| MCPH13 | 4q24 | AR | CENPE | Centromere protein E | Kinetochore | Kinetochore attachment, spindle checkpoint | [50] |
| MCPH14 | 1p21.2 | AR | SASS6 (SAS6) | Spindle assembly abnormal protein 6 homolog | Centrosome | Centriole biogenesis | [45,51] |
| MCPH15 | 1p34.2 | AR | MFSD2A | Major facilitator superfamily domain-containing 2A | Plasma membrane | Metabolism, cell cycle | [52,53] |
| MCPH16 | 12q24.33 | AR | ANKLE2 (LEM4) | Ankyrin repeat and LEM domain-containing protein 2 | Endoplasmic reticulum/ Nucleus | Nuclear envelope assembly | [54] |
| MCPH17 | 12q24.23 | AR | CIT | Citron rho-interacting serine/threonine kinase | Spindle/ Midbody | Spindle assembly, cytokinesis | [55,56] |
| MCPH18 | 4q21.23 | AD | ALFY (WDFY3) | Autophagy-linked FYVE protein | Cytoplasm/ Nucleus | Wnt signaling | [57] |
| MCPH19 | 3q23 | AR | COPB2 | Coatomer protein complex subunit Beta 2 | Non-clathrin vesicles | Vesicle trafficking, apoptosis | [58] |
| MCPH20 | 1q32.1 | AR | KIF14 | Kinesin family member 14 | Spindle poles/ Midbody | Spindle assembly, cytokinesis | [59,60] |
| MCPH21 | 12p13.31 | AR | NCAPD2 (CNAP1) | Non-SMC condensin I complex subunit D2/Centrosomal Nek2-associated protein 1 | Nucleus | Chromatin condensation | [61] |
| MCPH22 | 11q25 | AR | NCAPD3 | Non-SMC condensin II complex subunit D3 | Nucleus | Chromatin condensation | [61] |
| MCPH23 | 2q11.2 | AR | NCAPH | Non-SMC condensin I complex subunit H | Nucleus | Chromatin condensation | [61] |
| MCPH24 | 12q23.2 | AR | NUP37 | Nucleoporin 37 | Nucleus/ Kinetochore | Nuclear pore assembly, spindle assembly | [62] |
| MCPH25 | 7q22.1 | AR | MAP11 (TRAPPC14) | Microtubule-associated protein 11 | Spindle/ Midbody/ Golgi | Spindle assembly, cytokinesis, Golgi trafficking | [63] |
| MCPH26 | 5q23.2 | AD | LMNB1 | Lamin B1 | Nucleus/ Spindle | Nuclear envelope assembly, spindle assembly | [64] |
| MCPH27 | 19p13.3 | AD | LMNB2 | Lamin B2 | Nucleus/ Spindle | Nuclear envelope assembly, spindle assembly | [64] |
| MCPH28 | 22q13 | AR | RRP7A | Ribosomal RNA processing 7 homolog A | Nucleolus | Ribosome biogenesis, primary cilia resorption | [65] |
| MCPH29 | 9q32 | AR | AKNA | AT-Hook Transcription Factor | Centrosome | Microtubule organization | [66] |
| Mouse Model | Mutation | Phenotypes | Reference | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Microcephaly | Smaller Body | Gonad Atrophy | Infertility | Tumors | DDR Defects | PCC | |||
| Mcph1−/− | Exon 2 deletion | No | Yes | Yes | Yes | No | Yes | NR | [105] |
| Mcph1gt/gt | BRCT3 deletion | No | No | No | No | No | No | Yes | [111] |
| Mcph1-del | Exon 4-5 deletion | Yes | Yes | Yes | Yes | Yes | Yes | Yes | [81,120] |
| Mcph1tm1a/tm1a | Exon 4 deletion | Yes | No | NR | Yes | NR | Yes | NR | [122] |
| Mcph1-ΔBR1 | BRCT1 deletion | Yes | Yes | Yes | Yes | Yes | Yes | Yes | [91] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kristofova, M.; Ori, A.; Wang, Z.-Q. Multifaceted Microcephaly-Related Gene MCPH1. Cells 2022, 11, 275. https://doi.org/10.3390/cells11020275
Kristofova M, Ori A, Wang Z-Q. Multifaceted Microcephaly-Related Gene MCPH1. Cells. 2022; 11(2):275. https://doi.org/10.3390/cells11020275
Chicago/Turabian StyleKristofova, Martina, Alessandro Ori, and Zhao-Qi Wang. 2022. "Multifaceted Microcephaly-Related Gene MCPH1" Cells 11, no. 2: 275. https://doi.org/10.3390/cells11020275
APA StyleKristofova, M., Ori, A., & Wang, Z. -Q. (2022). Multifaceted Microcephaly-Related Gene MCPH1. Cells, 11(2), 275. https://doi.org/10.3390/cells11020275