
Implication of Adult Hippocampal Neurogenesis in Alzheimer’s Disease and Potential Therapeutic Approaches




Abstract
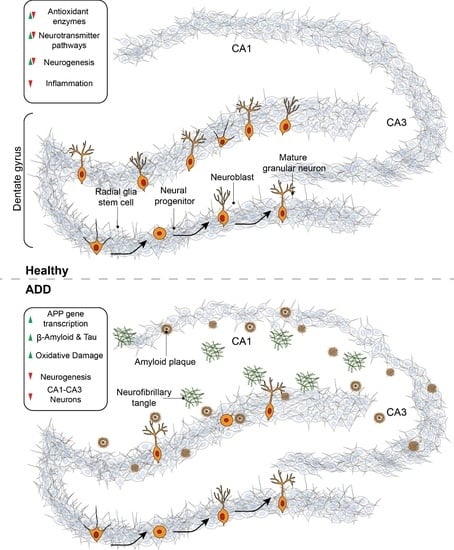
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Alzheimer’s Disease Pathogenesis
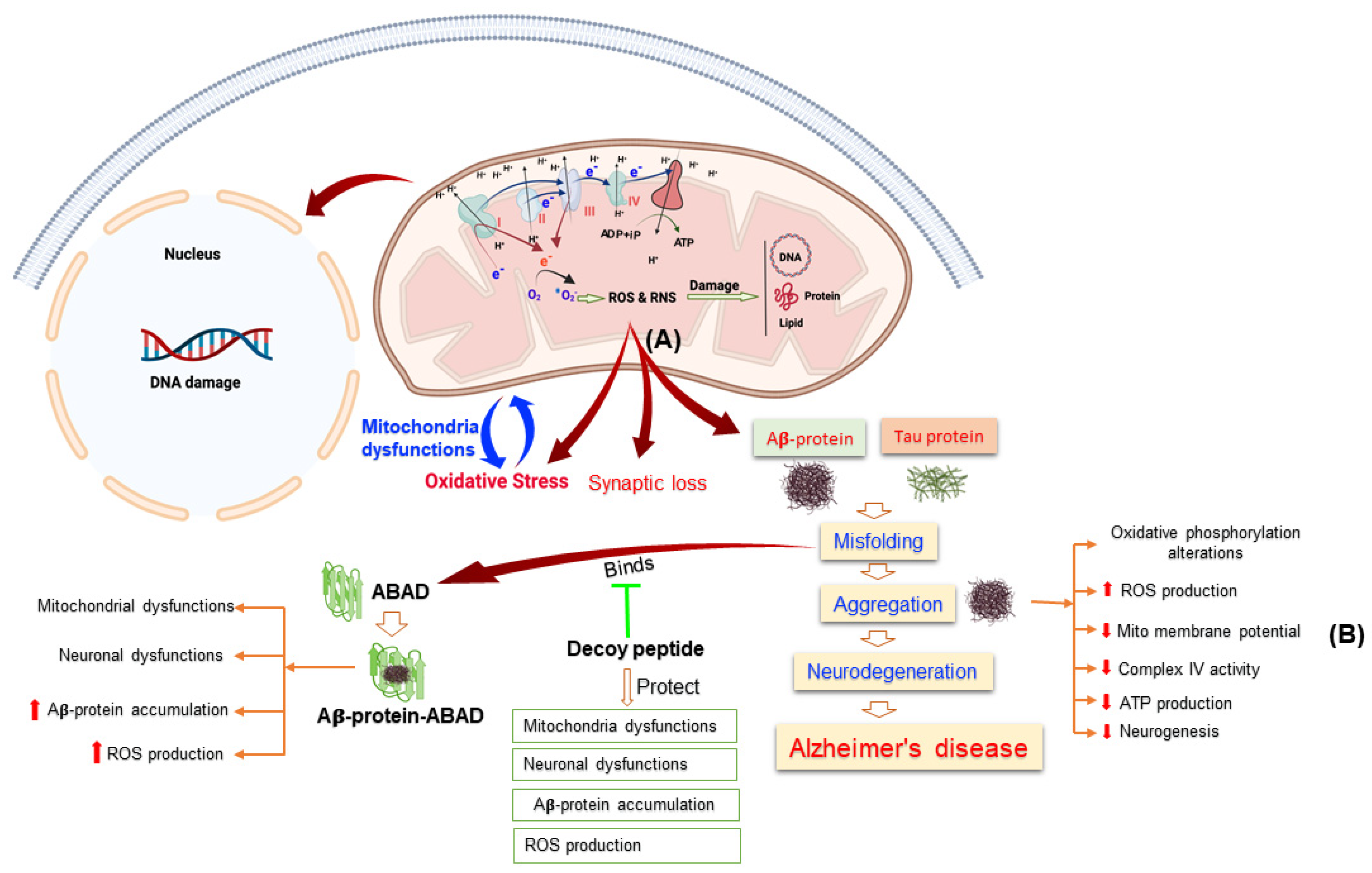
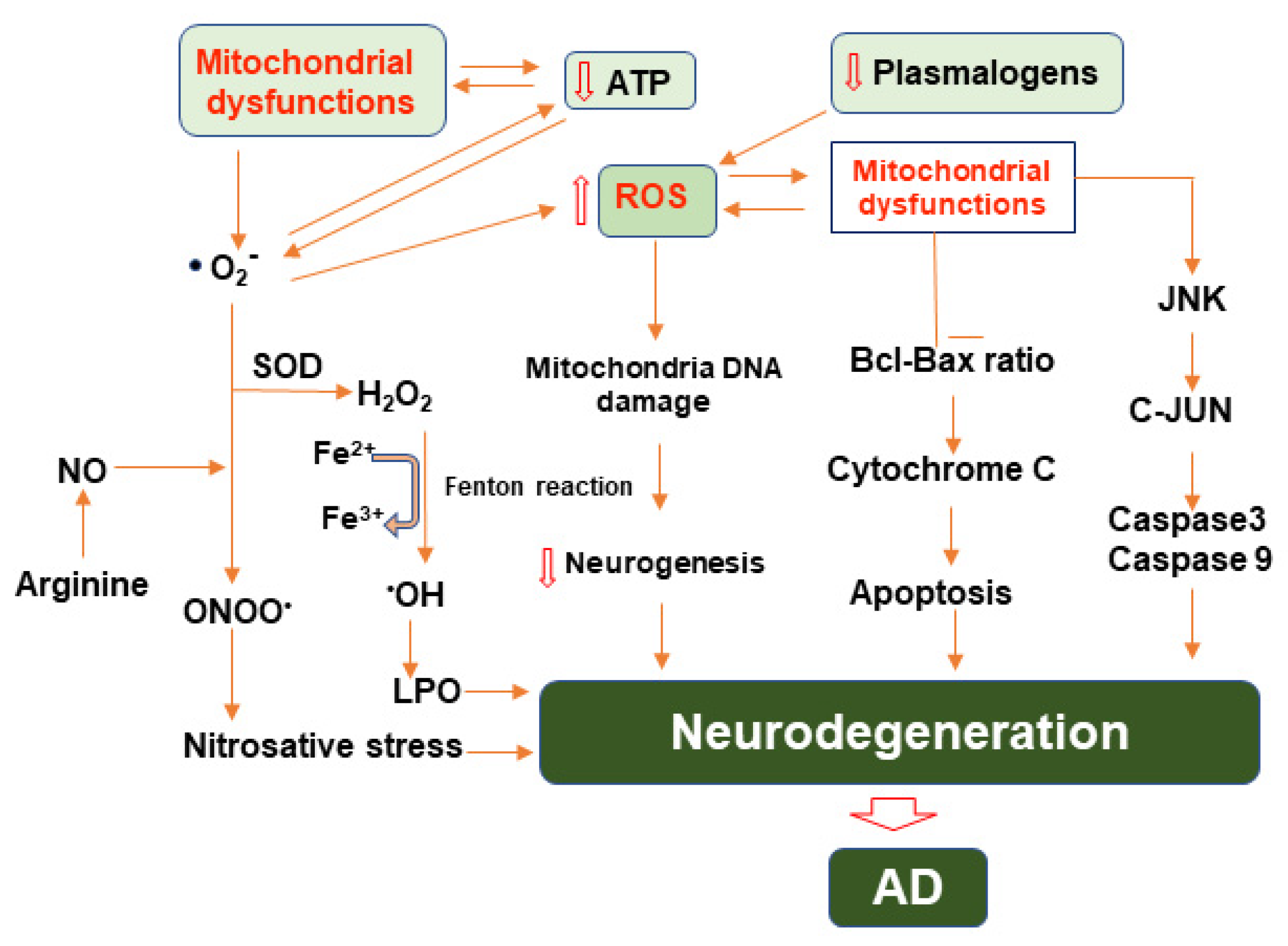
3. Mitochondrial Dysfunction and Oxidative Stress in AD
3.1. Mitochondrial Dysfunction in Alzheimer’s Disease
3.2. Alteration in Metabolism of Energy Associates with Mitochondrial Dysfunction in AD
3.3. Reactive Oxygen Species and Their Role in Alzheimer’s Disease
3.4. Alteration in Homeostasis of Mitochondrial Genomic in AD
3.5. Interrupted Bioenergetics of Mitochondrial in AD
3.6. Mitophagy in Alzheimer’s Disease
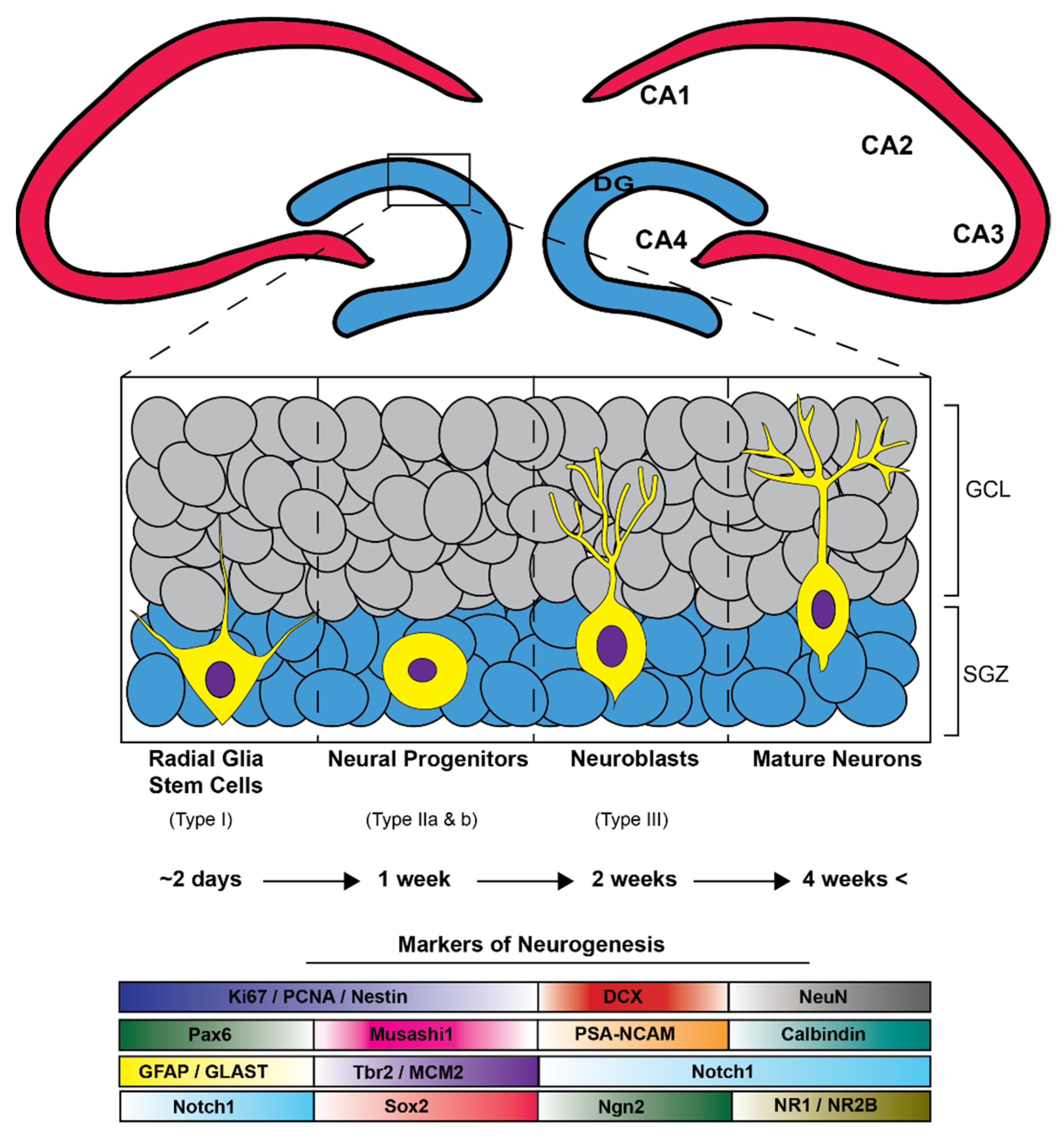
4. Neurogenesis
5. Neurogenesis in Alzheimer’s Disease
6. Neurogenesis as a Therapeutic Target in Alzheimer’s Disease
7. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- John Wiley & Sons. Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef]
- Hurd, M.D.; Martorell, P.; Delavande, A.; Mullen, K.J.; Langa, K. Monetary Costs of Dementia in the United States. N. Engl. J. Med. 2013, 368, 1326–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devanand, D.P.; Jacobs, D.; Tang, M.-X.; Del Castillo-Castaneda, C.; Sano, M.; Marder, K.; Bell, K.; Bylsma, F.W.; Brandt, J.; Albert, M.; et al. The Course of Psychopathologic Features in Mild to Moderate Alzheimer Disease. Arch. Gen. Psychiatry 1997, 54, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Rodriguez, G.; Herman, M.; Emrani, S.; Nahmani, E.; Barrett, G.; Figueroa, H.Y.; Goldberg, E.; Hussaini, S.A.; Duff, K.E. Tau Pathology Induces Excitatory Neuron Loss, Grid Cell Dysfunction, and Spatial Memory Deficits Reminiscent of Early Alzheimer’s Disease. Neuron 2017, 93, 533–541.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, R.J.; Wong, P.C. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Babcock, K.R.; Page, J.S.; Fallon, J.R.; Webb, A.E. Adult Hippocampal Neurogenesis in Aging and Alzheimer’s Disease. Stem Cell Rep. 2021, 16, 681–693. [Google Scholar] [CrossRef]
- Altman, J.; Das, G.D.; Altman, G.D.D.J. Postnatal Neurogenesis in the Guinea-pig. Nature 1967, 214, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, P.S.; Perfilieva, E.; Bjork-Eriksson, T.; Alborn, A.-M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, J.T.; Berger, E.P.; Lansbury, P.T. The carboxy terminus of the beta. amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry 1993, 32, 4693–4697. [Google Scholar] [CrossRef]
- Kojro, E.; Fahrenholz, F. The Non-Amyloidogenic Pathway: Structure and Function of α-Secretases. Alzheimer’s Dis. 2005, 38, 105–127. [Google Scholar] [CrossRef]
- Bayer, T.A.; Wirths, O.; Majtényi, K.; Hartmann, T.; Multhaup, G.; Beyreuther, K.; Czech, C. Key factors in Alzheimer’s disease: Beta-amyloid precursor protein processing, metabolism and intraneuronal transport. Brain Pathol. 2006, 11, 1–11. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broussard, G.; Mytar, J.; Li, R.-C.; Klapstein, G.J. The role of inflammatory processes in Alzheimer’s disease. Inflammopharmacology 2012, 20, 109–126. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2017, 217, 459–472. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 Variants in Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Kleinberger, G.; Yamanishi, Y.; Suárez-Calvet, M.; Czirr, E.; Lohmann, E.; Cuyvers, E.; Struyfs, H.; Pettkus, N.; Wenninger-Weinzierl, A.; Mazaheri, F.; et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 2014, 6, 243ra86. [Google Scholar] [CrossRef]
- Mazaheri, F.; Snaidero, N.; Kleinberger, G.; Madore, C.; Daria, A.; Werner, G.; Krasemann, S.; Capell, A.; Trümbach, D.; Wurst, W.; et al. TREM 2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep. 2017, 18, 1186–1198. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devine, M.J.; Kittler, J.T. Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Magrassi, L.; Leto, K.; Rossi, F. Lifespan of neurons is uncoupled from organismal lifespan. Proc. Natl. Acad. Sci. USA 2013, 110, 4374–4379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Vicente, M. Neuronal Mitophagy in Neurodegenerative Diseases. Front. Mol. Neurosci. 2017, 10, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, V.K.; Singh, T.G.; Mehta, V. Stressed mitochondria: A target to intrude alzheimer’s disease. Mitochondrion 2021, 59, 48–57. [Google Scholar] [CrossRef]
- John, A.; Reddy, P.H. Synaptic basis of Alzheimer’s disease: Focus on synaptic amyloid beta, P-tau and mitochondria. Ageing Res. Rev. 2020, 65, 101208. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, J.M.P.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. J. Cereb. Blood Flow Metab. 2019, 176, 3489–3507. [Google Scholar] [CrossRef]
- Carvalho, C.; Correia, S.C.; Cardoso, S.; Plácido, A.I.; Candeias, E.; Duarte, A.I.; Moreira, P. The role of mitochondrial disturbances in Alzheimer, Parkinson and Huntington diseases. Expert Rev. Neurother. 2015, 15, 867–884. [Google Scholar] [CrossRef]
- Correia, S.C.; Santos, R.X.; Cardoso, S.; Carvalho, C.; Candeias, E.; Duarte, A.I.; Plácido, A.I.; Santos, M.S.; Moreira, P.I. Alzheimer disease as a vascular disorder: Where do mitochondria fit? Exp. Gerontol. 2012, 47, 878–886. [Google Scholar] [CrossRef]
- Uddin, S.; Al Mamun, A.; Rahman, A.; Behl, T.; Perveen, A.; Hafeez, A.; Bin-Jumah, M.N.; Abdel-Daim, M.M.; Ashraf, G.M. Emerging Proof of Protein Misfolding and Interactions in Multifactorial Alzheimer’s Disease. Curr. Top. Med. Chem. 2020, 20, 2380–2390. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Knight, G.E. Cellular Distribution and Functions of P2 Receptor Subtypes in Different Systems. Int. Rev. Cytol. 2004, 240, 31–304. [Google Scholar] [CrossRef] [PubMed]
- Trushina, E.; Nemutlu, E.; Zhang, S.; Christensen, T.; Camp, J.; Mesa, J.; Siddiqui, A.; Tamura, Y.; Sesaki, H.; Wengenack, T.M.; et al. Defects in Mitochondrial Dynamics and Metabolomic Signatures of Evolving Energetic Stress in Mouse Models of Familial Alzheimer’s Disease. PLoS ONE 2012, 7, e32737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [Green Version]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD Directly Links Aβ to Mitochondrial Toxicity in Alzheimer’s Disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [Green Version]
- Morsy, A.; Trippier, P.C. Amyloid-Binding Alcohol Dehydrogenase (ABAD) Inhibitors for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2018, 62, 4252–4264. [Google Scholar] [CrossRef]
- Cameron, B.; Landreth, G.E. Inflammation, microglia, and alzheimer’s disease. Neurobiol. Dis. 2010, 37, 503–509. [Google Scholar] [CrossRef] [Green Version]
- Ji, K.; Akgul, G.; Wollmuth, L.P.; Tsirka, S.E. Microglia Actively Regulate the Number of Functional Synapses. PLoS ONE 2013, 8, e56293. [Google Scholar] [CrossRef] [Green Version]
- Heppner, F.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate immunity in Alzheimer’s disease. Nat. Immunol. 2015, 16, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Siegel, G.J.; Agranoff, B.W.; Albers, R.W.; Molinoff, P. Basic Neurochemistry: Molecular, Cellular, and Medical Aspects; Raven Press: New York, NY, USA, 1999. [Google Scholar]
- Pedrós, I.; Petrov, D.; Allgaier, M.; Sureda, F.X.; Barroso, E.; Beas-Zarate, C.; Auladell, C.; Pallàs, M.; Vázquez-Carrera, M.; Casadesus, G.; et al. Early alterations in energy metabolism in the hippocampus of APPswe/PS1dE9 mouse model of Alzheimer’s disease. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2014, 1842, 1556–1566. [Google Scholar] [CrossRef] [Green Version]
- Szablewski, L. Glucose Transporters in Brain: In Health and in Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 55, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Croteau, E.; Castellano, C.-A.; Fortier, M.; Bocti, C.; Fulop, T.; Paquet, N.; Cunnane, S. A cross-sectional comparison of brain glucose and ketone metabolism in cognitively healthy older adults, mild cognitive impairment and early Alzheimer’s disease. Exp. Gerontol. 2018, 107, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Kapogiannis, D.; Mattson, M.P. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. Lancet Neurol. 2011, 10, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Gordon, B.A.; Blazey, T.M.; Su, Y.; Hari-Raj, A.; Dincer, A.; Flores, S.; Christensen, J.; McDade, E.; Wang, G.; Xiong, C.; et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: A longitudinal study. Lancet Neurol. 2018, 17, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Hakim, J. Reactive oxygen species and inflammation. C. R. Seances Soc. Biol. Fil. 1993, 187, 286–295. (In French) [Google Scholar]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Jones, D.P.; Sies, H. The Redox Code. Antioxidants Redox Signal. 2015, 23, 734–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, X.; Cueto, R.; Effi, C.; Zhang, Y.; Tan, H.; Qin, X.; Ji, Y.; Yang, X.; Wang, H. Biochemical basis and metabolic interplay of redox regulation. Redox Biol. 2019, 26, 101284. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Kim, H.; Min, S.H.; Rhee, S.G.; Jeong, W. Dynein Light Chain LC8 Negatively Regulates NF-κB through the Redox-dependent Interaction with IκBα. J. Biol. Chem. 2008, 283, 23863–23871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.-F.; Kuo, H.-P.; Liu, M.; Chou, C.-K.; Xia, W.; Du, Y.; Shen, J.; Chen, C.-T.; Huo, L.; Hsu, M.-C.; et al. KEAP1 E3 Ligase-Mediated Downregulation of NF-κB Signaling by Targeting IKKβ. Mol. Cell 2009, 36, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [Green Version]
- Woo, J.R.; Kim, S.J.; Jeong, W.; Cho, Y.H.; Lee, S.C.; Chung, Y.J.; Rhee, S.G.; Ryu, S.E. Structural Basis of Cellular Redox Regulation by Human TRP14. J. Biol. Chem. 2004, 279, 48120–48125. [Google Scholar] [CrossRef] [Green Version]
- Schreck, R.; Rieber, P.; Baeuerle, P. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991, 10, 2247–2258. [Google Scholar] [CrossRef]
- Aslan, M.; Ozben, T. Reactive Oxygen and Nitrogen Species in Alzheimers Disease. Curr. Alzheimer Res. 2004, 1, 111–119. [Google Scholar] [CrossRef]
- Tu, W.; Wang, H.; Li, S.; Liu, Q.; Sha, H. The Anti-Inflammatory and Anti-Oxidant Mechanisms of the Keap1/Nrf2/ARE Signaling Pathway in Chronic Diseases. Aging Dis. 2019, 10, 637–651. [Google Scholar] [CrossRef] [Green Version]
- Branca, C.; Ferreira, E.; Nguyen, T.-V.; Doyle, K.; Caccamo, A.; Oddo, S. Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 4823–4835. [Google Scholar] [CrossRef]
- Rahman, M.M.; Sykiotis, G.; Nishimura, M.; Bodmer, R.; Bohmann, D. Declining signal dependence of N rf2- M af S -regulated gene expression correlates with aging phenotypes. Aging Cell 2013, 12, 554–562. [Google Scholar] [CrossRef] [Green Version]
- Suh, J.H.; Shenvi, S.V.; Dixon, B.M.; Liu, H.; Jaiswal, A.K.; Liu, R.-M.; Hagen, T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA 2004, 101, 3381–3386. [Google Scholar] [CrossRef] [Green Version]
- Krauss, S.; Zhang, C.-Y.; Lowell, B.B. The mitochondrial uncoupling-protein homologues. Nat. Rev. Mol. Cell Biol. 2005, 6, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Abdul, H.M.; Joshi, G.; Opii, W.O.; Butterfield, D.A. Protective effect of quercetin in primary neurons against Aβ(1–42): Relevance to Alzheimer’s disease. J. Nutr. Biochem. 2009, 20, 269–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.-G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Castellani, R.J.; Moreira, P.; Aliev, G.; Shenk, J.C.; Siedlak, S.L.; Harris, P.L.; Fujioka, H.; Sayre, L.M.; Szweda, P.A.; et al. Hydroxynonenal-generated crosslinking fluorophore accumulation in Alzheimer disease reveals a dichotomy of protein turnover. Free. Radic. Biol. Med. 2012, 52, 699–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leßig, J. Plasmalogens in Biological Systems: Their Role in Oxidative Processes in Biological Membranes, their Contribution to Pathological Processes and Aging and Plasmalogen Analysis. Curr. Med. Chem. 2009, 16, 2021–2041. [Google Scholar] [CrossRef] [PubMed]
- Su, X.Q.; Wang, J.; Sinclair, A.J. Plasmalogens and Alzheimer’s disease: A review. Lipids Heal. Dis. 2019, 18, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Jain, J.; Hasan, W.; Jat, D. Role of Mitochondria in Cancer. Int. J. Adv. Res. Rev. 2020, 5, 70–93. [Google Scholar]
- D’Souza, A.R.; Minczuk, M. Mitochondrial transcription and translation: Overview. Essays Biochem. 2018, 62, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Boczonadi, V.; Ricci, G.; Horvath, R. Mitochondrial DNA transcription and translation: Clinical syndromes. Essays Biochem. 2018, 62, 321–340. [Google Scholar] [CrossRef] [PubMed]
- Inczedy-Farkas, G.; Trampush, J.W.; Perczel Forintos, D.; Beech, D.; Andrejkovics, M.; Varga, Z.; Remenyi, V.; Bereznai, B.; Gal, A.; Molnar, M.J. Mitochondrial DNA Mutations and Cognition: A Case-Series Report. Arch. Clin. Neuropsychol. 2014, 29, 315–321. [Google Scholar] [CrossRef]
- Keogh, M.J.; Chinnery, P.F. Mitochondrial DNA mutations in neurodegeneration. Biochim. Biophys. Acta 2015, 1847, 1401–1411. [Google Scholar] [CrossRef] [Green Version]
- Phillips, N.R.; Simpkins, J.W.; Roby, R.K. Mitochondrial DNA deletions in Alzheimer’s brains: A review. Alzheimer’s Dement. 2013, 10, 393–400. [Google Scholar] [CrossRef] [Green Version]
- Corral-Debrinski, M.; Horton, T.; Lott, M.; Shoffner, J.M.; McKee, A.C.; Beal, M.; Graham, B.; Wallace, D.C. Marked Changes in Mitochondrial DNA Deletion Levels in Alzheimer Brains. Genomics 1994, 23, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Damas, J.; Samuels, D.C.; Carneiro, J.; Amorim, A.; Pereira, F. Mitochondrial DNA Rearrangements in Health and Disease-A Comprehensive Study. Hum. Mutat. 2014, 35, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.S.; Reiman, E.M.; Valla, J.; Dunckley, T.; Beach, T.G.; Grover, A.; Niedzielko, T.L.; Schneider, L.E.; Mastroeni, D.; Caselli, R.; et al. Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 4441–4446. [Google Scholar] [CrossRef] [Green Version]
- Brooks, W.M.; Lynch, P.J.; Ingle, C.C.; Hatton, A.; Emson, P.C.; Faull, R.; Starkey, M.P. Gene expression profiles of metabolic enzyme transcripts in Alzheimer’s disease. Brain Res. 2007, 1127, 127–135. [Google Scholar] [CrossRef]
- Mastroeni, D.; Khdour, O.M.; Delvaux, E.; Nolz, J.; Olsen, G.; Berchtold, N.; Cotman, C.; Hecht, S.M.; Coleman, P.D. Nuclear but not mitochondrial-encoded oxidative phosphorylation genes are altered in aging, mild cognitive impairment, and Alzheimer’s disease. Alzheimer’s Dement. 2016, 13, 510–519. [Google Scholar] [CrossRef] [Green Version]
- Hroudová, J.; Singh, N.; Fišar, Z. Mitochondrial Dysfunctions in Neurodegenerative Diseases: Relevance to Alzheimer’s Disease. BioMed Res. Int. 2014, 2014, 175062. [Google Scholar] [CrossRef]
- Zhang, L.; Guo, X.; Chu, J.; Zhang, X.; Yan, Z.; Li, Y. Potential hippocampal genes and pathways involved in Alzheimer’s disease: A bioinformatic analysis. Genet. Mol. Res. 2015, 14, 7218–7232. [Google Scholar] [CrossRef] [PubMed]
- Minjarez, B.; Calderón-González, K.G.; Rustarazo, M.L.V.; Herrera-Aguirre, M.E.; Labra-Barrios, M.L.; Rincon-Limas, D.E.; del Pino, M.M.S.; Mena, R.; Luna-Arias, J.P. Identification of proteins that are differentially expressed in brains with Alzheimer’s disease using iTRAQ labeling and tandem mass spectrometry. J. Proteom. 2016, 139, 103–121. [Google Scholar] [CrossRef] [PubMed]
- Adav, S.S.; Park, J.E.; Sze, S.K. Quantitative profiling brain proteomes revealed mitochondrial dysfunction in Alzheimer’s disease. Mol. Brain 2019, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Nie, L.; Zhou, Q.; Rahman, S.U.; Liu, J.; Yang, X.; Li, S. Proteomic Profiles of the Early Mitochondrial Changes in APP/PS1 and ApoE4 Transgenic Mice Models of Alzheimer’s Disease. J. Proteome Res. 2019, 18, 2632–2642. [Google Scholar] [CrossRef]
- Hasan, W.; Kori, R.K.; Thakre, K.; Yadav, R.S.; Jat, D. Synthesis, characterization and efficacy of mitochondrial targeted delivery of TPP-curcumin in rotenone-induced toxicity. DARU J. Pharm. Sci. 2019, 27, 557–570. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-Dependent And Independent Signals In Selective Autophagy. Trends Cell Biol. 2015, 26, 6–16. [Google Scholar] [CrossRef]
- Gouspillou, G.; Godin, R.; Piquereau, J.; Picard, M.; Mofarrahi, M.; Mathew, J.; Purves-Smith, F.M.; Sgarioto, N.; Hepple, R.T.; Burelle, Y.; et al. Protective role of Parkin in skeletal muscle contractile and mitochondrial function. J. Physiol. 2018, 596, 2565–2579. [Google Scholar] [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Meissner, C.; Lorenz, H.; Weihofen, A.; Selkoe, D.J.; Lemberg, M.K. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 2011, 117, 856–867. [Google Scholar] [CrossRef]
- Rasool, S.; Soya, N.; Truong, L.; Croteau, N.; Lukacs, G.L.; Trempe, J. PINK 1 autophosphorylation is required for ubiquitin recognition. EMBO Rep. 2018, 19, e44981. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.-M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W.; Heo, J.-M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Sharma, C.; Kaur, A.; Thind, S.S.; Singh, B.; Raina, S. Advanced glycation End-products (AGEs): An emerging concern for processed food industries. J. Food Sci. Technol. 2015, 52, 7561–7576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bongarzone, S.; Savickas, V.; Luzi, F.; Gee, A.D. Targeting the Receptor for Advanced Glycation Endproducts (RAGE): A Medicinal Chemistry Perspective. J. Med. Chem. 2017, 60, 7213–7232. [Google Scholar] [CrossRef] [Green Version]
- Pinkas, A.; Aschner, M. Advanced Glycation End-Products and Their Receptors: Related Pathologies, Recent Therapeutic Strategies, and a Potential Model for Future Neurodegeneration Studies. Chem. Res. Toxicol. 2016, 29, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Tobin, M.K.; Musaraca, K.; Disouky, A.; Shetti, A.; Bheri, A.; Honer, W.G.; Kim, N.; Dawe, R.J.; Bennett, D.A.; Arfanakis, K.; et al. Human Hippocampal Neurogenesis Persists in Aged Adults and Alzheimer’s Disease Patients. Cell Stem Cell 2019, 24, 974–982.e3. [Google Scholar] [CrossRef]
- Demars, M.P.; Hollands, C.; Zhao, K.D.; Lazarov, O. Soluble amyloid precursor protein-α rescues age-linked decline in neural progenitor cell proliferation. Neurobiol. Aging 2013, 34, 2431–2440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, H.; Wang, D.; Zhang, X.; Zhou, D.; Zhang, H.; Qian, Q.; He, X.; Liu, Z.; Liu, Y.; Zheng, T.; et al. Amyloid β Is Not the Major Factor Accounting for Impaired Adult Hippocampal Neurogenesis in Mice Overexpressing Amyloid Precursor Protein. Stem Cell Rep. 2016, 7, 707–718. [Google Scholar] [CrossRef] [Green Version]
- Briley, D.; Ghirardi, V.; Woltjer, R.; Renck, A.; Zolochevska, O.; Taglialatela, G.; Micci, M.-A. Preserved neurogenesis in non-demented individuals with AD neuropathology. Sci. Rep. 2016, 6, 27812. [Google Scholar] [CrossRef] [Green Version]
- Pereira-Caixeta, A.R.; Guarnieri, L.O.; Medeiros, D.C.; Mendes, E.M.; Ladeira, L.C.; Pereira, M.T.; Moraes, M.F.; Pereira, G.S. Inhibiting constitutive neurogenesis compromises long-term social recognition memory. Neurobiol. Learn. Mem. 2018, 155, 92–103. [Google Scholar] [CrossRef]
- West, M.J.; Kawas, C.H.; Martin, L.J.; Troncoso, J.C. The CA1 Region of the Human Hippocampus Is a Hot Spot in Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 2006, 908, 255–259. [Google Scholar] [CrossRef]
- Lovell, M.A.; Geiger, H.; Van Zant, G.E.; Lynn, B.C.; Markesbery, W.R. Isolation of neural precursor cells from Alzheimer’s disease and aged control postmortem brain. Neurobiol. Aging 2006, 27, 909–917. [Google Scholar] [CrossRef]
- Velasco, M.X.; Kosti, A.; Guardia, G.D.; Santos, M.C.; Tegge, A.; Qiao, M.; Correa, B.R.S.; Hernández, G.; Kokovay, E.; Galante, P.A.; et al. Antagonism between the RNA-binding protein Musashi1 and miR-137 and its potential impact on neurogenesis and glioblastoma development. RNA 2019, 25, 768–782. [Google Scholar] [CrossRef]
- Montalbano, M.; McAllen, S.; Puangmalai, N.; Sengupta, U.; Bhatt, N.; Johnson, O.D.; Kharas, M.G.; Kayed, R. RNA-binding proteins Musashi and tau soluble aggregates initiate nuclear dysfunction. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Fukutani, Y.; Cairns, N.J.; Shiozawa, M.; Sasaki, K.; Sudo, S.; Isaki, K.; Lantos, P.L. Neuronal loss and neurofibrillary degeneration in the hippocampal cortex in late-onset sporadic Alzheimer’s disease. Psychiatry Clin. Neurosci. 2000, 54, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Wirths, O. Altered neurogenesis in mouse models of Alzheimer disease. Neurogenesis 2017, 4, e1327002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrous, D.N.; Koehl, M.; Le Moal, M. Adult Neurogenesis: From Precursors to Network and Physiology. Physiol. Rev. 2005, 85, 523–569. [Google Scholar] [CrossRef] [Green Version]
- Haughey, N.J.; Nath, A.; Chan, S.L.; Borchard, A.C.; Rao, M.S.; Mattson, M.P. Disruption of neurogenesis by amyloid β-peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer’s disease. J. Neurochem. 2002, 83, 1509–1524. [Google Scholar] [CrossRef]
- Rodríguez, J.J.; Jones, V.; Tabuchi, M.; Allan, S.; Knight, E.; LaFerla, F.M.; Oddo, S.; Verkhratsky, A. Impaired Adult Neurogenesis in the Dentate Gyrus of a Triple Transgenic Mouse Model of Alzheimer’s Disease. PLoS ONE 2008, 3, e2935. [Google Scholar] [CrossRef] [Green Version]
- Yetman, M.J.; Jankowsky, J.L. Wild-Type Neural Progenitors Divide and Differentiate Normally in an Amyloid-Rich Environment. J. Neurosci. 2013, 33, 17335–17341. [Google Scholar] [CrossRef] [Green Version]
- Mathews, P.; Levy, E. Cystatin C in aging and in Alzheimer’s disease. Ageing Res. Rev. 2016, 32, 38–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, E.; Sastre, M.; Kumar, A.; Gallo, G.; Piccardo, P.; Ghetti, B.; Tagliavini, F. Codeposition of Cystatin C with Amyloid-β Protein in the Brain of Alzheimer Disease Patients. J. Neuropathol. Exp. Neurol. 2001, 60, 94–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, Y.; Yamada, M.; Hayakawa, M.; Otomo, E.; Miyatake, T. Cerebral amyloid angiopathy: A significant cause of cerebellar as well as lobar cerebral hemorrhage in the elderly. J. Neurol. Sci. 1993, 116, 135–141. [Google Scholar] [CrossRef]
- Khacho, M.; Clark, A.; Svoboda, D.S.; Maclaurin, J.G.; Lagace, D.C.; Park, D.; Slack, R.S. Mitochondrial dysfunction underlies cognitive defects as a result of neural stem cell depletion and impaired neurogenesis. Hum. Mol. Genet. 2017, 26, 3327–3341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.; Ure, K.; Ables, J.L.; Lagace, D.C.; Nave, K.-A.; Goebbels, S.; Eisch, A.; Hsieh, J. Neurod1 is essential for the survival and maturation of adult-born neurons. Nat. Neurosci. 2009, 12, 1090–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richetin, K.; Moulis, M.; Millet, A.; Arràzola, M.S.; Andraini, T.; Hua, J.; Davezac, N.; Roybon, L.; Belenguer, P.; Miquel, M.-C.; et al. Amplifying mitochondrial function rescues adult neurogenesis in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2017, 102, 113–124. [Google Scholar] [CrossRef]
- Rego, A.C.; Oliveira, C. Mitochondrial Dysfunction and Reactive Oxygen Species in Excitotoxicity and Apoptosis: Implications for the Pathogenesis of Neurodegenerative Diseases. Neurochem. Res. 2003, 28, 1563–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, F.Q.; Buettner, G. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009, 16, 1040–1052. [Google Scholar] [CrossRef] [Green Version]
- Gray, B.; Carmichael, A.J. Kinetics of superoxide scavenging by dismutase enzymes and manganese mimics determined by electron spin resonance. Biochem. J. 1992, 281, 795–802. [Google Scholar] [CrossRef]
- Rola, R.; Zou, Y.; Huang, T.-T.; Fishman, K.; Baure, J.; Rosi, S.; Milliken, H.; Limoli, C.L.; Fike, J.R. Lack of extracellular superoxide dismutase (EC-SOD) in the microenvironment impacts radiation-induced changes in neurogenesis. Free. Radic. Biol. Med. 2007, 42, 1133–1145. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.-T.; Zou, Y.; Corniola, R. Oxidative stress and adult neurogenesis—Effects of radiation and superoxide dismutase deficiency. Semin. Cell Dev. Biol. 2012, 23, 738–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martino, G.; Pluchino, S. The therapeutic potential of neural stem cells. Nat. Rev. Neurosci. 2006, 7, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Yagita, Y.; Kitagawa, K.; Ohtsuki, T.; Takasawa, K.-I.; Miyata, T.; Okano, H.; Hori, M.; Matsumoto, M. Neurogenesis by Progenitor Cells in the Ischemic Adult Rat Hippocampus. Stroke 2001, 32, 1890–1896. [Google Scholar] [CrossRef] [Green Version]
- Levenstein, M.E.; Ludwig, T.E.; Xu, R.-H.; Llanas, R.A.; VanDenHeuvel-Kramer, K.; Manning, D.; Thomson, J.A. Basic Fibroblast Growth Factor Support of Human Embryonic Stem Cell Self-Renewal. Stem Cells 2005, 24, 568–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAvoy, K.M.; Sahay, A. Targeting Adult Neurogenesis to Optimize Hippocampal Circuits in Aging. Neurotherapeutics 2017, 14, 630–645. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, A.; Sandler, V.M.; Toni, N.; Zhao, C.; Gage, F.H. NMDA-receptor-mediated, cell-specific integration of new neurons in adult dentate gyrus. Nature 2006, 442, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Pieper, A.A.; Xie, S.; Capota, E.; Estill, S.J.; Zhong, J.; Long, J.M.; Becker, G.L.; Huntington, P.; Goldman, S.; Shen, C.-H.; et al. Discovery of a Proneurogenic, Neuroprotective Chemical. Cell 2010, 142, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadhwa, M.; Prabhakar, A.; Ray, K.; Roy, K.; Kumari, P.; Jha, P.K.; Kishore, K.; Kumar, S.; Panjwani, U. Inhibiting the microglia activation improves the spatial memory and adult neurogenesis in rat hippocampus during 48 h of sleep deprivation. J. Neuroinflammation 2017, 14, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Lue, L. Modeling microglial activation in Alzheimer’s disease with human postmortem microglial cultures. Neurobiol. Aging 2001, 22, 945–956. [Google Scholar] [CrossRef]
- Chakrabarti, M.; McDonald, A.J.; Reed, J.W.; Moss, M.A.; Das, B.C.; Ray, S.K. Molecular Signaling Mechanisms of Natural and Synthetic Retinoids for Inhibition of Pathogenesis in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 50, 335–352. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Qiao, A.; Wang, Z.; Goodwin, J.S.; Lee, E.-S.; Block, M.L.; Allsbrook, M.; McDonald, M.P.; Fan, G.-H. Retinoic Acid Attenuates -Amyloid Deposition and Rescues Memory Deficits in an Alzheimer’s Disease Transgenic Mouse Model. J. Neurosci. 2008, 28, 11622–11634. [Google Scholar] [CrossRef]
- van Praag, H.; Kempermann, G.; Gage, F.H. Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat. Neurosci. 1999, 2, 266–270. [Google Scholar] [CrossRef] [PubMed]
- van Praag, H.; Christie, B.; Sejnowski, T.J.; Gage, F.H. Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc. Natl. Acad. Sci. USA 1999, 96, 13427–13431. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.Z.; Nusslock, R. Exercise-Mediated Neurogenesis in the Hippocampus via BDNF. Front. Neurosci. 2018, 12, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bath, K.G.; Akins, M.; Lee, F.S. BDNF control of adult SVZ neurogenesis. Dev. Psychobiol. 2011, 54, 578–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montiel, T.; Quiroz-Baez, R.; Massieu, L.; Arias, C. Role of oxidative stress on β-amyloid neurotoxicity elicited during impairment of energy metabolism in the hippocampus: Protection by antioxidants. Exp. Neurol. 2006, 200, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G.; Atkinson, J. Vitamin E, antioxidant and nothing more. Free Radic. Biol. Med. 2007, 43, 4–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devore, E.; Grodstein, F.; Van Rooij, F.; Hofman, A.; Stampfer, M.; Witteman, J.; Breteler, M. Dietary Antioxidants and Long-term Risk of Dementia. Arch. Neurol. 2010, 67, 819–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padayatty, S.J.; Katz, A.; Wang, Y.; Eck, P.; Kwon, O.; Lee, J.-H.; Chen, S.; Corpe, C.P.; Dutta, A.; Dutta, S.K.; et al. Vitamin C as an Antioxidant: Evaluation of Its Role in Disease Prevention. J. Am. Coll. Nutr. 2003, 22, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Murata, N.; Ozawa, Y.; Kinoshita, N.; Irie, K.; Shirasawa, T.; Shimizu, T. Vitamin C Restores Behavioral Deficits and Amyloid-β Oligomerization without Affecting Plaque Formation in a Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2011, 26, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Ak, T.; Gülçin, I. Antioxidant and radical scavenging properties of curcumin. Chem. Interact. 2008, 174, 27–37. [Google Scholar] [CrossRef]
- Kim, G.-Y.; Kim, K.-H.; Lee, S.-H.; Yoon, M.-S.; Lee, H.-J.; Moon, D.-O.; Lee, C.-M.; Ahn, S.-C.; Park, Y.C.; Park, Y.-M. Curcumin Inhibits Immunostimulatory Function of Dendritic Cells: MAPKs and Translocation of NF-κB as Potential Targets. J. Immunol. 2005, 174, 8116–8124. [Google Scholar] [CrossRef] [Green Version]
- Lim, G.P.; Chu, T.; Yang, F.; Beech, W.; Frautschy, S.A.; Cole, G.M. The Curry Spice Curcumin Reduces Oxidative Damage and Amyloid Pathology in an Alzheimer Transgenic Mouse. J. Neurosci. 2001, 21, 8370–8377. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Essa, H.; Peyton, L.; Hasan, W.; León, B.E.; Choi, D.-S. Implication of Adult Hippocampal Neurogenesis in Alzheimer’s Disease and Potential Therapeutic Approaches. Cells 2022, 11, 286. https://doi.org/10.3390/cells11020286
Essa H, Peyton L, Hasan W, León BE, Choi D-S. Implication of Adult Hippocampal Neurogenesis in Alzheimer’s Disease and Potential Therapeutic Approaches. Cells. 2022; 11(2):286. https://doi.org/10.3390/cells11020286
Chicago/Turabian StyleEssa, Hesham, Lee Peyton, Whidul Hasan, Brandon Emanuel León, and Doo-Sup Choi. 2022. "Implication of Adult Hippocampal Neurogenesis in Alzheimer’s Disease and Potential Therapeutic Approaches" Cells 11, no. 2: 286. https://doi.org/10.3390/cells11020286
APA StyleEssa, H., Peyton, L., Hasan, W., León, B. E., & Choi, D. -S. (2022). Implication of Adult Hippocampal Neurogenesis in Alzheimer’s Disease and Potential Therapeutic Approaches. Cells, 11(2), 286. https://doi.org/10.3390/cells11020286