

From Differential DNA Methylation in COPD to Mitochondria: Regulation of AHRR Expression Affects Airway Epithelial Response to Cigarette Smoke

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Characteristics of the Subjects Included in the Study
2.1.1. Subject Characteristics of the Lifelines Cohort Study
2.1.2. Subject Characteristics of the Donors of the Primary Airway Epithelial Cells
2.2. DNA Methylation Measurement in the Lifelines Cohort
2.3. Cell Culture
2.3.1. Isolation and Culture of Primary AECs from COPD Patients and Non-COPD Controls
2.3.2. Culture of 16HBE Cells
2.4. Generation, Selection and Culture of Wild-Type and AHRR Knockout 16HBE Cells
2.5. Preparation of Cigarette Smoke Extract (CSE)
2.6. In Vitro Cell Treatment
2.6.1. Treatment of AECs
2.6.2. Treatment of 16HBE Cells
2.7. RNA and DNA Isolation
2.8. DNA Methylation Analysis
2.9. qPCR
2.10. Cell Proliferation Assay
2.11. Mitochondrial Membrane Potential (Δψm) Assay
2.12. Live/Dead Cell Analysis
2.13. Cytochrome C ELISA Assay
2.14. dsDNA Measurement Assay
2.15. Statistics
2.15.1. DNA Methylation Data Analysis in Lifelines
2.15.2. In Vitro Experiments
3. Results
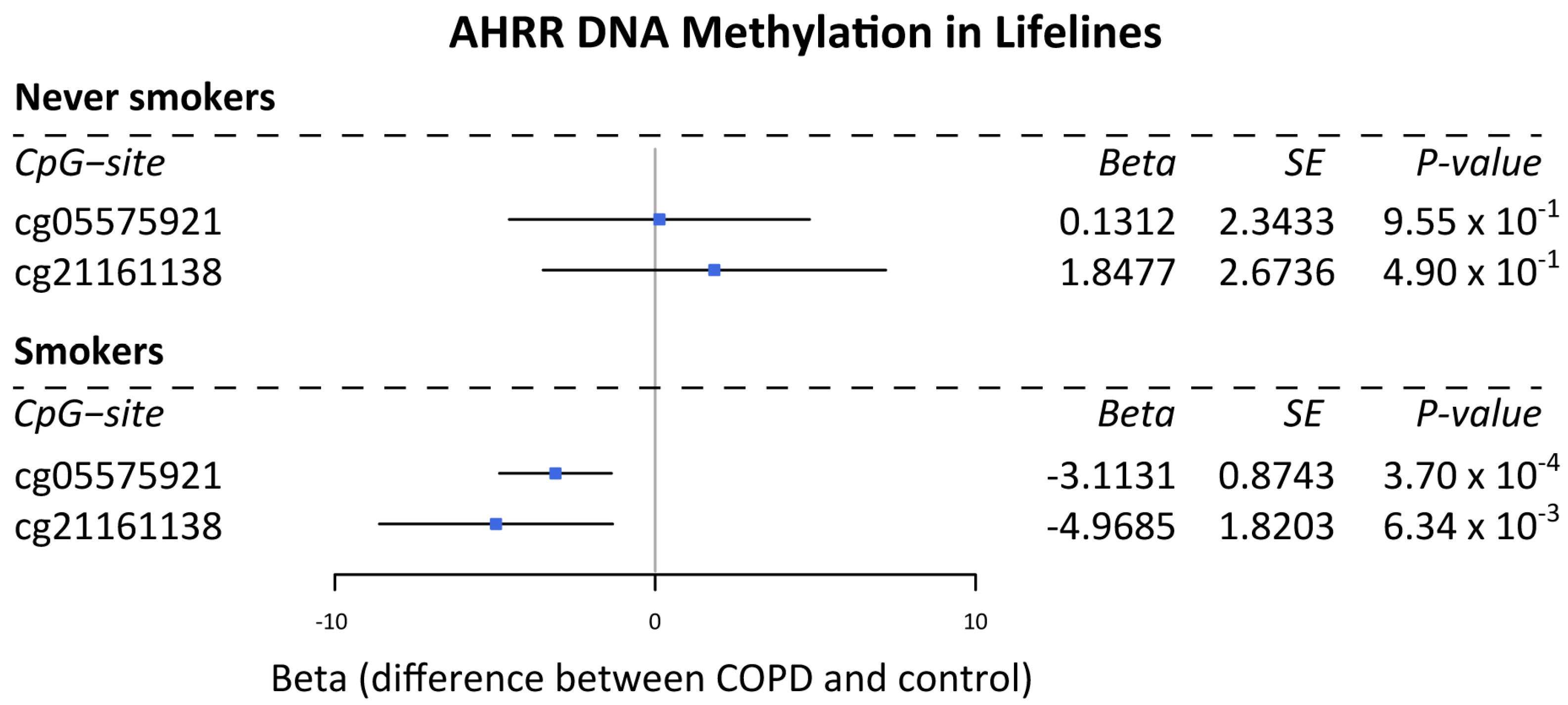
3.1. DNA Methylation Levels of AHRR Are Lower in Subjects with COPD Compared to Non-COPD Controls in Current Smokers
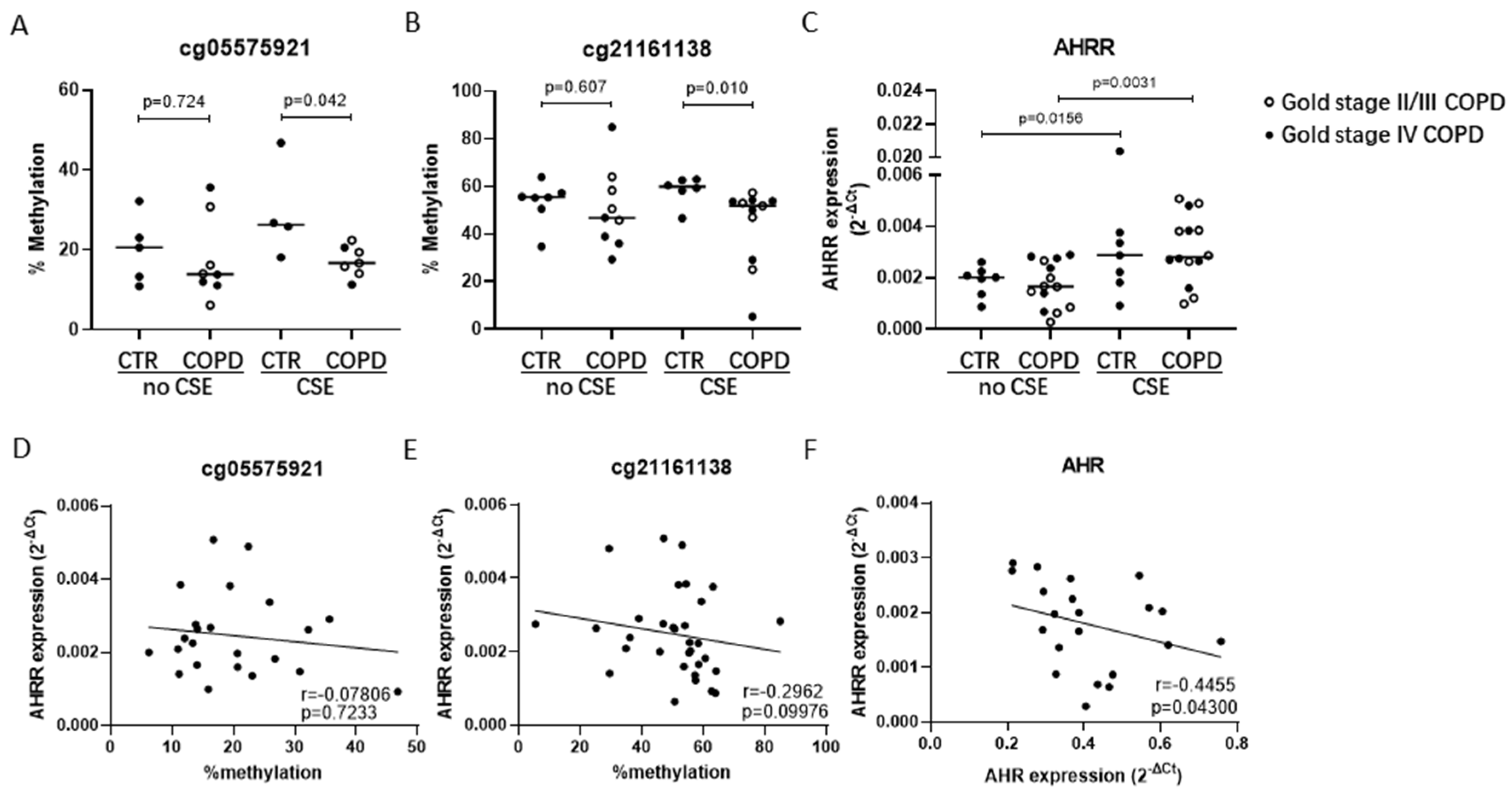
3.2. Lower AHRR DNA Methylation in COPD- vs. Non-COPD-Derived AECs upon CSE Exposure
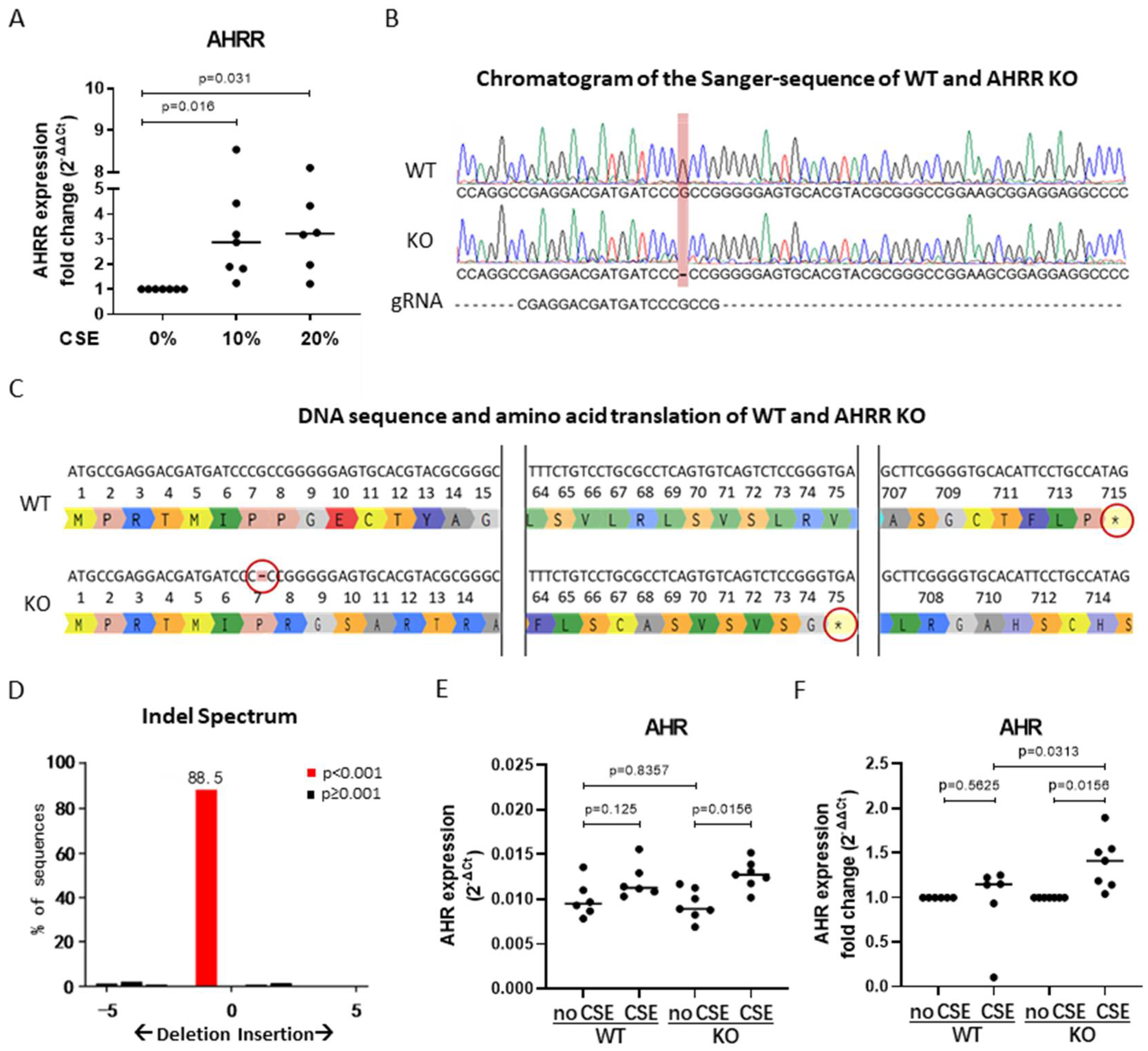
3.3. AHRR Knockout in 16HBE Cells
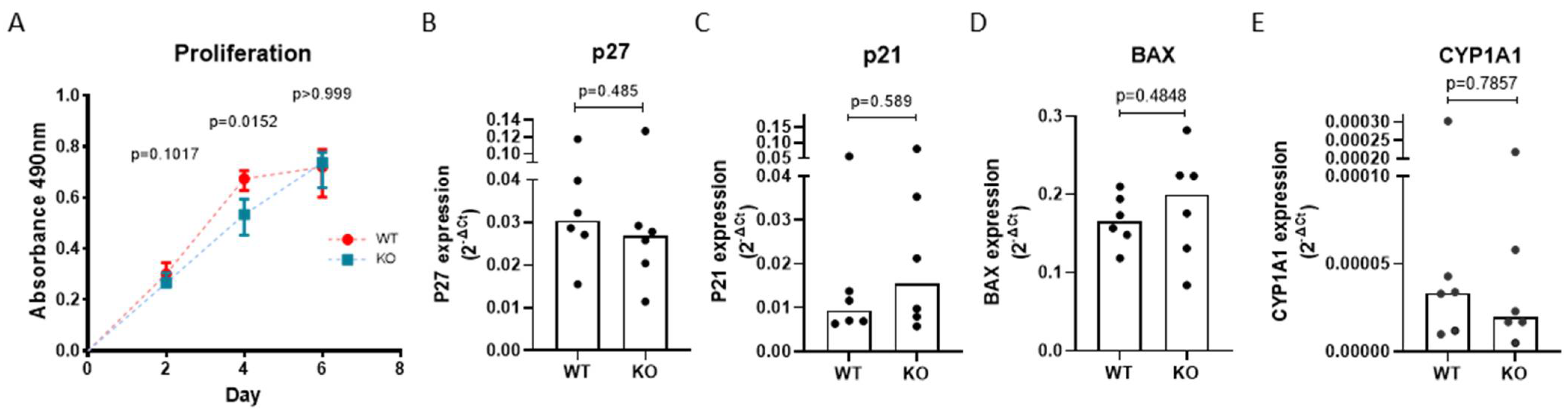
3.4. AHRR Knockout Attenuates the Proliferation of 16HBE Cells
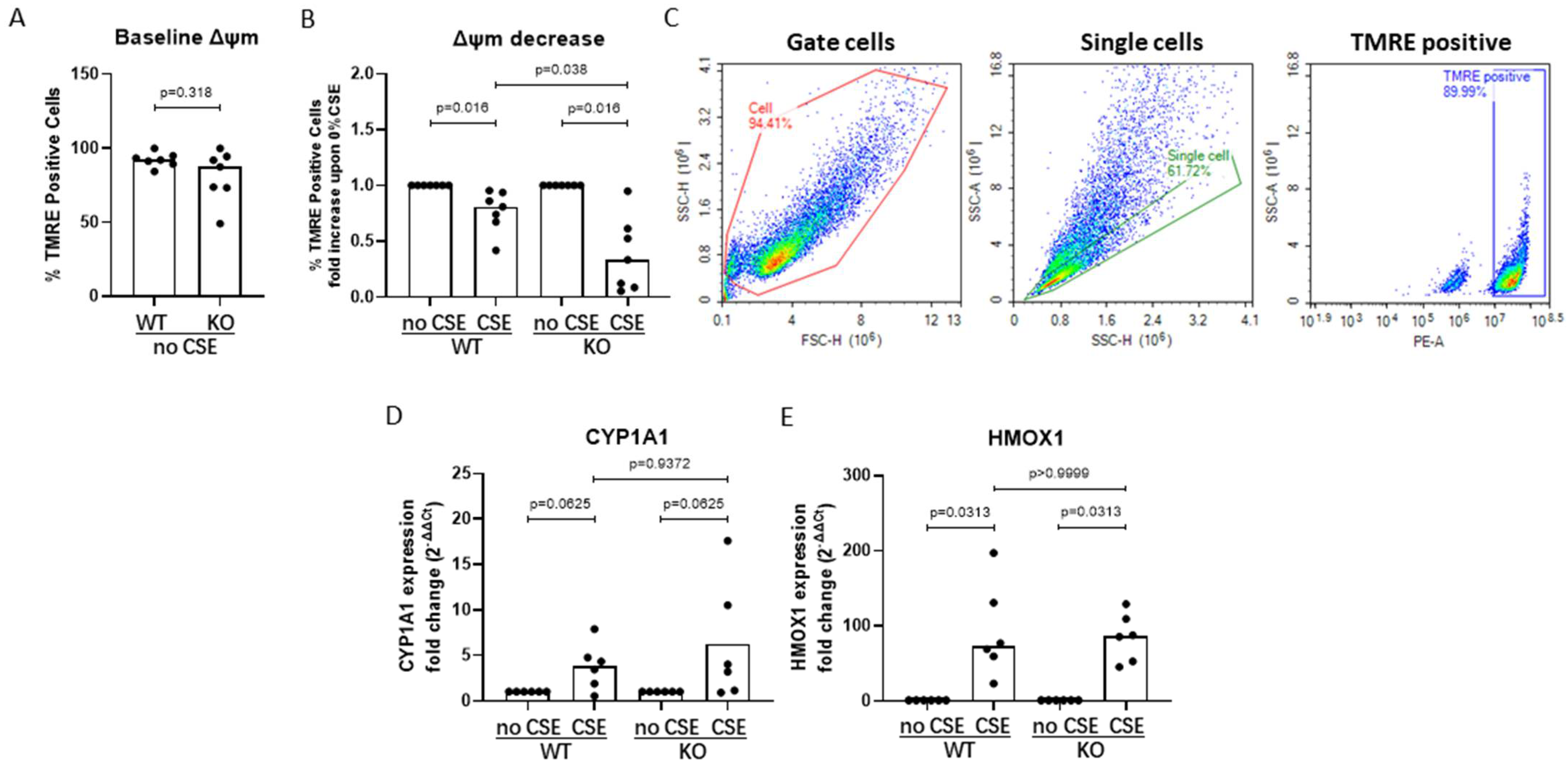
3.5. AHRR Knockout Decreases Mitochondrial Membrane Potential upon CSE Exposure
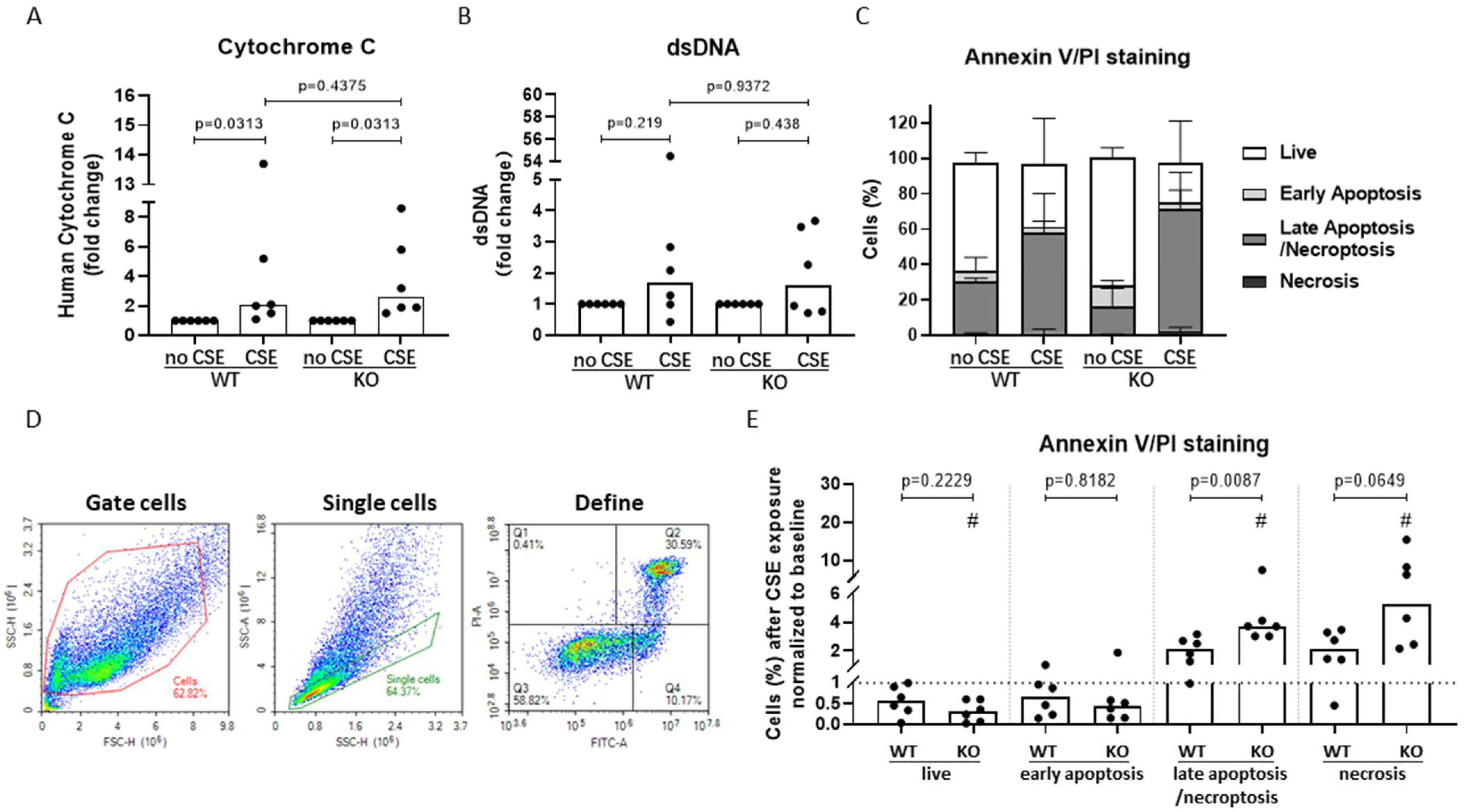
3.6. AHRR Knockout Decreases Bronchial Epithelial Cell Viability and Increases Apoptotic Cell Death upon CSE Exposure
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Global Strategy for the Diangosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease 2021 Report; © 2020, Global Initiative for Chronic Obstructive Lung Disease: Fontana, WI, USA, 2021.
- Barnes, P.J.; Burney, P.G.J.; Silverman, E.K.; Celli, B.R.; Vestbo, J.; Wedzicha, J.A.; Wouters, E.F.M. Chronic obstructive pulmonary disease. Nat. Rev. Dis. Prim. 2015, 1, 15076. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; de Vries, M.; Nwozor, K.O.; Noordhoek, J.A.; Brandsma, C.-A.; Boezen, H.M.; Heijink, I.H. A Protective Role of FAM13A in Human Airway Epithelial Cells Upon Exposure to Cigarette Smoke Extract. Front. Physiol. 2021, 12, 690936. [Google Scholar] [CrossRef] [PubMed]
- Hiromichi, H.; Jun, A.; Saburo, I.; Kenji, K.; Naoki, T.; Yutaka, Y.; Hiroshi, W.; Jun, K.; Kenichiro, S.; Takanori, N.; et al. Mitochondrial fragmentation in cigarette smoke-induced bronchial epithelial cell senescence. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L737–L746. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, R.F.; Zarrintan, S.; Brandenburg, S.M.; Kol, A.; de Bruin, H.G.; Jafari, S.; Dijk, F.; Kalicharan, D.; Kelders, M.; Gosker, H.R.; et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir. Res. 2013, 14, 97. [Google Scholar] [CrossRef] [Green Version]
- Aghapour, M.; Raee, P.; Moghaddam, S.J.; Hiemstra, P.S.; Heijink, I.H. Airway epithelial barrier dysfunction in chronic obstructive pulmonary disease: Role of cigarette smoke exposure. Am. J. Respir. Cell Mol. Biol. 2018, 58, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Van Der Toorn, M.; Slebos, D.-J.; De Bruin, H.G.; Leuvenink, H.G.; Bakker, S.J.L.; Gans, R.O.B.; Koëter, G.H.; Van Oosterhout, A.J.M.; Kauffman, H.F. Cigarette smoke-induced blockade of the mitochondrial respiratory chain switches lung epithelial cell apoptosis into necrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, 1211–1218. [Google Scholar] [CrossRef] [Green Version]
- Pouwels, S.D.; Zijlstra, G.J.; van der Toorn, M.; Hesse, L.; Gras, R.; Ten Hacken, N.H.T.; Krysko, D.V.; Vandenabeele, P.; De Vries, M.; Van Oosterhout, A.J.M.M.; et al. Cigarette smoke-induced necroptosis and DAMP release trigger neutrophilic airway inflammation in mice. Am. J. Physiol. Cell Physiol. Lung Cell. Mol. Physiol. 2016, 310, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Pouwels, S.D.; Heijink, I.H.; Ten Hacken, N.H.T.; Vandenabeele, P.; Krysko, D.V.; Nawijn, M.C.; Van Oosterhout, A.J.M. DAMPs activating innate and adaptive immune responses in COPD. Mucosal Immunol. 2014, 7, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Janciauskiene, S. The beneficial effects of antioxidants in health and diseases. Chronic Obstr. Pulm. Dis. 2020, 7, 182–202. [Google Scholar] [CrossRef]
- Nyunoya, T.; Mebratu, Y.; Contreras, A.; Delgado, M.; Chand, H.S.; Tesfaigzi, Y. Molecular processes that drive cigarette smoke-induced epithelial cell fate of the lung. Am. J. Respir. Cell Mol. Biol. 2014, 50, 471–482. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, M.; Van Der Plaat, D.A.; Nedeljkovic, I.; Verkaik-Schakel, R.N.; Kooistra, W.; Amin, N.; Van Duijn, C.M.; Brandsma, C.A.; Van Diemen, C.C.; Vonk, J.M.; et al. From blood to lung tissue: Effect of cigarette smoke on DNA methylation and lung function. Respir. Res. 2018, 19, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojesen, S.E.; Timpson, N.; Relton, C.; Davey Smith, G.; Nordestgaard, B.G. AHRR (cg05575921) hypomethylation marks smoking behaviour, morbidity and mortality. Thorax 2017, 72, 646–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodal, J.B.; Kobylecki, C.J.; Vedel-Krogh, S.; Nordestgaard, B.G.G.; Bojesen, S.E. AHRR hypomethylation, lung function, lung function decline and respiratory symptoms. Eur. Respir. J. 2018, 51, 1701512. [Google Scholar] [CrossRef] [Green Version]
- Mimura, J.; Fujii-Kuriyama, Y. Functional role of AhR in the expression of toxic effects by TCDD. Biochim. Biophys. Acta 2003, 1619, 263–268. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Hankinson, O. An aryl hydrocarbon receptor agonist suppresses the growth of human umbilical vein endothelial cells in vitro: Potent effect with polyunsaturated fatty acids. Int. J. Exp. Pathol. 2020, 101, 248–263. [Google Scholar] [CrossRef]
- Formosa, R.; Borg, J.; Vassallo, J. Aryl hydrocarbon receptor (AHR) is a potential tumour suppressor in pituitary adenomas. Endocr. Relat. Cancer 2017, 24, 445–457. [Google Scholar] [CrossRef] [Green Version]
- Guerrina, N.; Traboulsi, H.; Eidelman, D.H.; Baglole, C.J. The Aryl Hydrocarbon Receptor and the Maintenance of Lung Health. Int. J. Mol. Sci. 2018, 19, 3882. [Google Scholar] [CrossRef] [Green Version]
- Nothdurft, S.; Thumser-Henner, C.; Breitenbücher, F.; Okimoto, R.A.; Dorsch, M.; Opitz, C.A.; Sadik, A.; Esser, C.; Hölzel, M.; Asthana, S.; et al. Functional screening identifies aryl hydrocarbon receptor as suppressor of lung cancer metastasis. Oncogenesis 2020, 9, 102. [Google Scholar] [CrossRef]
- Haarmann-Stemmann, T.; Abel, J. The arylhydrocarbon receptor repressor (AhRR): Structure, expression, and function. Biol. Chem. 2006, 387, 1195–1199. [Google Scholar] [CrossRef]
- Sijtsma, A.; Rienks, J.; van der Harst, P.; Navis, G.; Rosmalen, J.G.M.; Dotinga, A. Cohort Profile Update: Lifelines, a three-generation cohort study and biobank. Int. J. Epidemiol. 2022, 51, e295–e302. [Google Scholar] [CrossRef] [PubMed]
- De Vries, M.; Van Der Plaat, D.A.; Vonk, J.M.; Boezen, H.M. No association between DNA methylation and COPD in never and current smokers. BMJ Open Respir. Res. 2018, 5, e000282. [Google Scholar] [CrossRef] [PubMed]
- Heijink, I.H.; De Bruin, H.G.; Dennebos, R.; Jonker, M.R.; Noordhoek, J.A.; Brandsma, C.A.; Van Den Berge, M.; Postma, D.S. Cigarette smoke-induced epithelial expression of WNT-5B: Implications for COPD. Eur. Respir. J. 2016, 48, 504–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broome, S.T.; Fisher, T.; Faiz, A.; Keay, K.A.; Musumeci, G.; Al-Badri, G.; Castorina, A. Assessing the anti-inflammatory activity of the anxiolytic drug buspirone using crispr-cas9 gene editing in lps-stimulated bv-2 microglial cells. Cells 2021, 10, 1312. [Google Scholar] [CrossRef]
- Pouwels, S.D.; Wiersma, V.R.; Fokkema, I.E.; Berg, M.; ten Hacken, N.H.T.; van den Berge, M.; Heijink, I.; Faiz, A. Acute cigarette smoke-induced eQTL affects formyl peptide receptor expression and lung function. Respirology 2021, 26, 233–240. [Google Scholar] [CrossRef]
- Hahn, M.E.; Allan, L.L.; Sherr, D.H. Regulation of Constitutive and Inducible AHR Signaling: Complex Interactions Involving the AHR Repressor. Biochem. Pharmacol. 2009, 77, 485–497. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liang, X.; Yin, X.; Lv, J.; Tang, K.; Ma, J.; Ji, T.; Zhang, H.; Dong, W.; Jin, X.; et al. Blockade of IDO-kynurenine-AhR metabolic circuitry abrogates IFN-γ-induced immunologic dormancy of tumor-repopulating cells. Nat. Commun. 2017, 8, 15207. [Google Scholar] [CrossRef]
- Koliopanos, A.; Kleeff, J.; Xiao, Y.; Safe, S.; Zimmermann, A.; Büchler, M.W.; Friess, H. Increased arylhydrocarbon receptor expression offers a potential therapeutic target for pancreatic cancer. Oncogene 2002, 21, 6059–6070. [Google Scholar] [CrossRef] [Green Version]
- Kalpage, H.A.; Bazylianska, V.; Recanati, M.A.; Fite, A.; Liu, J.; Wan, J.; Mantena, N.; Malek, M.H.; Podgorski, I.; Heath, E.I.; et al. Tissue-specific regulation of cytochrome c by post-translational modifications: Respiration, the mitochondrial membrane potential, ROS, and apoptosis. FASEB J. 2019, 33, 1540–1553. [Google Scholar] [CrossRef]
- Takeuchi, F.; Takano, K.; Yamamoto, M.; Isono, M.; Miyake, W.; Mori, K.; Hara, H.; Hiroi, Y.; Kato, N. Clinical Implication of Smoking-Related Aryl-Hydrocarbon Receptor Repressor (AHRR) Hypomethylation in Japanese Adults. Circ. J. 2022, 86, 986–992. [Google Scholar] [CrossRef]
- Monick, M.M.; Beach, S.R.H.; Plume, J.; Sears, R.; Gerrard, M.; Brody, G.H.; Philibert, R.A. Coordinated changes in AHRR methylation in lymphoblasts and pulmonary macrophages from smokers. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159, 141–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Zhao, Y.; Zhang, Z.; Shi, L.; Fang, Y.; Chang, C. Aryl hydrocarbon receptor signaling pathway plays important roles in the proliferative and metabolic properties of bone marrow mesenchymal stromal cells. Acta Biochim. Biophys. Sin. 2021, 53, 1428–1439. [Google Scholar] [CrossRef]
- Milara, J.; Peiró, T.; Serrano, A.; Cortijo, J. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax 2013, 68, 410–420. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J.; Shapiro, S.D.; Pauwels, R.A. Chronic obstructive pulmonary disease: Molecular and cellular mechanisms. Eur. Respir. J. 2003, 22, 672–688. [Google Scholar] [CrossRef] [PubMed]
- Hackett, T.L.; Warner, S.M.; Stefanowicz, D.; Shaheen, F.; Pechkovsky, D.V.; Murray, L.A.; Argentieri, R.; Kicic, A.; Stick, S.M.; Bai, T.R.; et al. Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-β1. Am. J. Respir. Crit. Care Med. 2009, 180, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Kondrikov, D.; Elmansi, A.; Bragg, R.T.; Mobley, T.; Barrett, T.; Eisa, N.; Kondrikova, G.; Schoeinlein, P.; Aguilar-Perez, A.; Shi, X.M.; et al. Kynurenine inhibits autophagy and promotes senescence in aged bone marrow mesenchymal stem cells through the aryl hydrocarbon receptor pathway. Exp. Gerontol. 2020, 130, 110805. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.H.; Chang, C.C.; Yang, N.J.; Lee, Y.H.; Juan, S.H. RhoA-mediated inhibition of vascular endothelial cell mobility: Positive feedback through reduced cytosolic p21 and p27. J. Cell. Physiol. 2014, 229, 1455–1465. [Google Scholar] [CrossRef]
- Ren, R.; Fang, Y.; Sherchan, P.; Lu, Q.; Lenahan, C.; Zhang, J.H.; Zhang, J.; Tang, J. Kynurenine/Aryl Hydrocarbon Receptor Modulates Mitochondria-Mediated Oxidative Stress and Neuronal Apoptosis in Experimental Intracerebral Hemorrhage. Antioxid. Redox Signal. 2022; online ahead of print. [Google Scholar] [CrossRef]
- Lamas, B.; Natividad, J.M.; Sokol, H. Aryl hydrocarbon receptor and intestinal immunity. Mucosal Immunol. 2018, 11, 1024–1038. [Google Scholar] [CrossRef]
- Yuan, J.; Sun, X.; Che, S.; Zhang, L.; Ruan, Z.; Li, X.; Yang, J. AhR-mediated CYP1A1 and ROS overexpression are involved in hepatotoxicity of decabromodiphenyl ether (BDE-209). Toxicol. Lett. 2021, 352, 26–33. [Google Scholar] [CrossRef]
- Zajda, K.; Ptak, A.; Rak, A.; Fiedor, E.; Grochowalski, A.; Milewicz, T.; Gregoraszczuk, E.L. Effects of human blood levels of two PAH mixtures on the AHR signalling activation pathway and CYP1A1 and COMT target genes in granulosa non-tumor and granulosa tumor cell lines. Toxicology 2017, 389, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, Y.; Wang, M.; Lin, X.; Zhang, Y.; Laurent, I.; Zhong, Y.; Li, J. Ampelopsin Inhibits Breast Cancer Cell Growth through Mitochondrial Apoptosis Pathway. Biol. Pharm. Bull. 2021, 44, 1738–1745. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Guo, Y.; Xia, Y.; Li, C.; Xu, X.; Qi, T.; Zhang, F.; Fan, M.; Hu, G.; Zhao, H.; et al. FNDC5/Irisin attenuates diabetic cardiomyopathy in a type 2 diabetes mouse model by activation of integrin αV/β5-AKT signaling and reduction of oxidative/nitrosative stress. J. Mol. Cell. Cardiol. 2021, 160, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Mizumura, K.; Cloonan, S.M.; Nakahira, K.; Bhashyam, A.R.; Cervo, M.; Kitada, T.; Glass, K.; Owen, C.A.; Mahmood, A.; Washko, G.R.; et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Invest. 2014, 124, 3987–4003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraro, E.; Pulicati, A.; Cencioni, M.T.; Cozzolino, M.; Navoni, F.; Di Martino, S.; Nardacci, R.; Carrì, M.T.; Cecconi, F. Apoptosome-deficient Cells Lose Cytochrome c through Proteasomal Degradation but Survive by Autophagy-dependent Glycolysis. Mol. Biol. Cell 2008, 19, 3576. [Google Scholar] [CrossRef] [Green Version]
- Heijink, I.H.; De Bruin, H.G.; Van Den Berge, M.; Bennink, L.J.C.; Brandenburg, S.M.; Gosens, R.; Van Oosterhout, A.J.; Postma, D.S. Role of aberrant WNT signalling in the airway epithelial response to cigarette smoke in chronic obstructive pulmonary disease. Thorax 2013, 68, 709–716. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Never Smokers | Current Smokers | |||||
|---|---|---|---|---|---|---|
| All | Non-COPD | COPD | All | Non-COPD | COPD | |
| N | 903 | 587 | 316 | 658 | 379 | 279 |
| Male, N (%) * | 508 (56.3) | 364 (62) | 144 (45.6) | 375 (57) | 224 (59.1) | 151 (54.1) |
| Age (years) *,# median (min-max) | 46 (18–80) | 44 (18–79) | 50 (28–80) | 46 (22–79) | 44 (22–76) | 49 (26–79) |
| Pack-years ,# mean (min-max) | NA | NA | NA | 19 (5.05–100) | 16.4 (5.05–68.1) | 22.2 (5.25–100) |
| FEV1/FVC (%) *,# mean (SD) | 74.5 (8.22) | 79.5 (4.69) | 65.2 (4.38) | 71.7 (8.77) | 77.9 (4.75) | 63.3 (5.39) |
| FEV1 (%pred) *# mean (SD) | 101 (14.5) | 105 (12.6) | 92.6 (14.3) | 94.4 (14.9) | 101 (12) | 85.8 (14) |
| Subject | Non-COPD Control (n = 8) | COPD (n = 14) |
|---|---|---|
| Age (years), median (min-max) | 61 (52–63) | 56.5 (39–61) |
| Male (n/all) | 5/8 | 6/14 |
| Pack years, median (min-max) | 33 (17–64) | 39 (6–64) |
| % Predicted FEV1, median (min-max) * | 111.63 (102.46–123.95) | 47.69 (16.58–78.4) |
| Primer Name | Sequence (5′ to 3′) |
|---|---|
| Amplification primers | |
| FW_AHRR_cg05575921 | GGGGATTGTTTATTTTTGAGAGGGTAG |
| RV_AHRR_cg05575921 | ACCTATCCCCTACCTCCC |
| FW_AHRR_cg21161138 | ATTTTGTAGGGGTTTTGGTGGT |
| RV_AHRR_cg21161138 | CAACTCTAACCCCAAAATCTCT |
| Sequencing primers | |
| SEQ_AHRR_cg05575921 | ATTTTTGAGAGGGTAGT |
| SEQ_AHRR_cg21161138 | GGGTTTTGGTGGTTG |
| Gene Name | Cat. Number of Taqman Probs |
|---|---|
| B2M | Hs99999907_m1 |
| PPIA | Hs99999904_m1 |
| AHRR | Hs01005075_m1 |
| AHR | Hs00169233_m1 |
| P27 | Hs00153277_m1 |
| P21 | Hs00355782_m1 |
| BAX | Hs00180269_m1 |
| CYP1A1 | Hs01054796_g1 |
| HMOX1 | Hs00157965_m1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Q.; Nwozor, K.O.; van den Berge, M.; Slebos, D.-J.; Faiz, A.; Jonker, M.R.; Boezen, H.M.; Heijink, I.H.; de Vries, M. From Differential DNA Methylation in COPD to Mitochondria: Regulation of AHRR Expression Affects Airway Epithelial Response to Cigarette Smoke. Cells 2022, 11, 3423. https://doi.org/10.3390/cells11213423
Chen Q, Nwozor KO, van den Berge M, Slebos D-J, Faiz A, Jonker MR, Boezen HM, Heijink IH, de Vries M. From Differential DNA Methylation in COPD to Mitochondria: Regulation of AHRR Expression Affects Airway Epithelial Response to Cigarette Smoke. Cells. 2022; 11(21):3423. https://doi.org/10.3390/cells11213423
Chicago/Turabian StyleChen, Qing, Kingsley Okechukwu Nwozor, Maarten van den Berge, Dirk-Jan Slebos, Alen Faiz, Marnix R. Jonker, H. Marike Boezen, Irene H. Heijink, and Maaike de Vries. 2022. "From Differential DNA Methylation in COPD to Mitochondria: Regulation of AHRR Expression Affects Airway Epithelial Response to Cigarette Smoke" Cells 11, no. 21: 3423. https://doi.org/10.3390/cells11213423
APA StyleChen, Q., Nwozor, K. O., van den Berge, M., Slebos, D. -J., Faiz, A., Jonker, M. R., Boezen, H. M., Heijink, I. H., & de Vries, M. (2022). From Differential DNA Methylation in COPD to Mitochondria: Regulation of AHRR Expression Affects Airway Epithelial Response to Cigarette Smoke. Cells, 11(21), 3423. https://doi.org/10.3390/cells11213423