
Mitochondrial Cardiomyopathy: Molecular Epidemiology, Diagnosis, Models, and Therapeutic Management

 , ,
, ,
Abstract
:1. Introduction
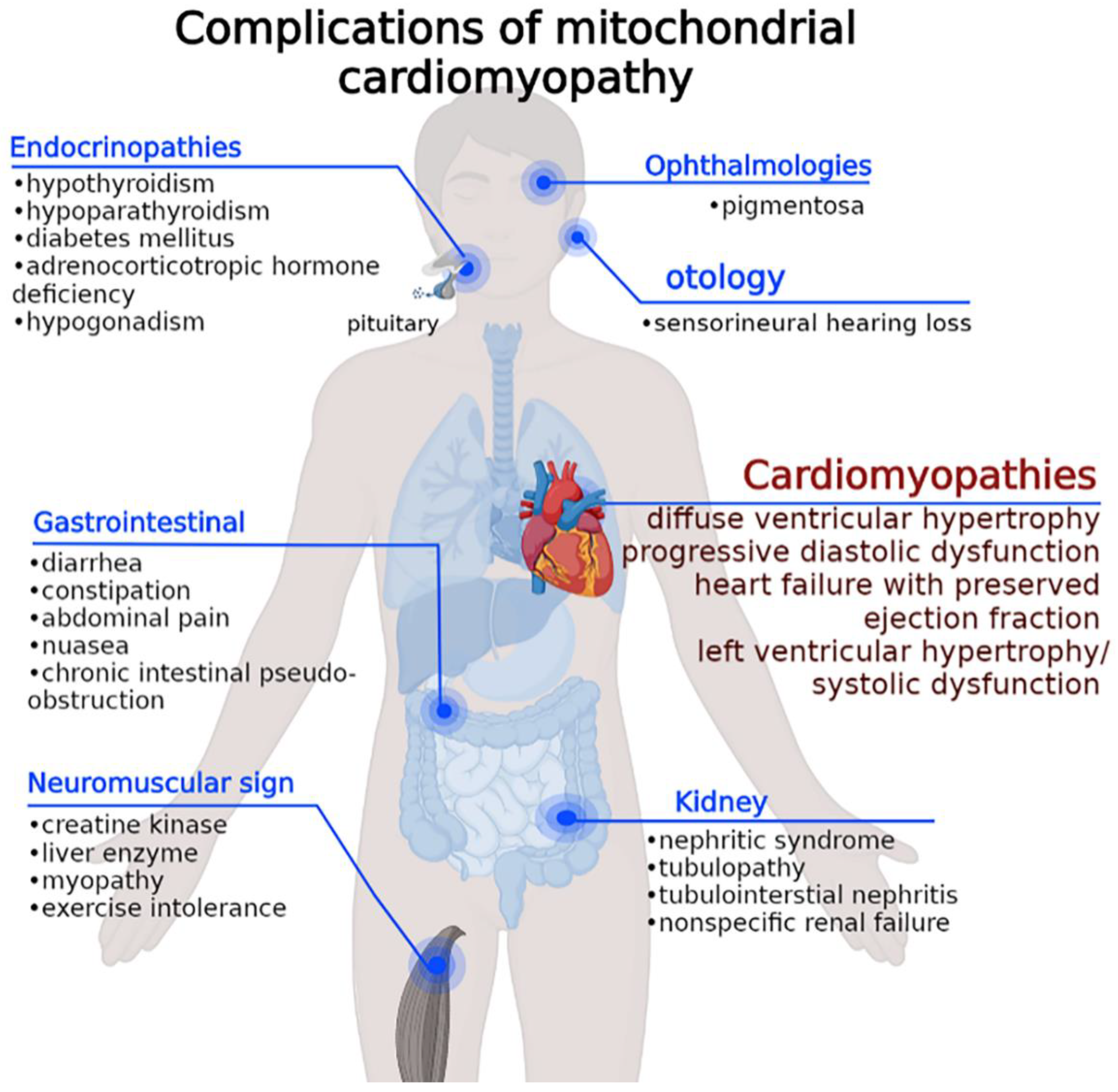
2. Molecular Epidemiology and Multiorgan Clinical Expression of MCM
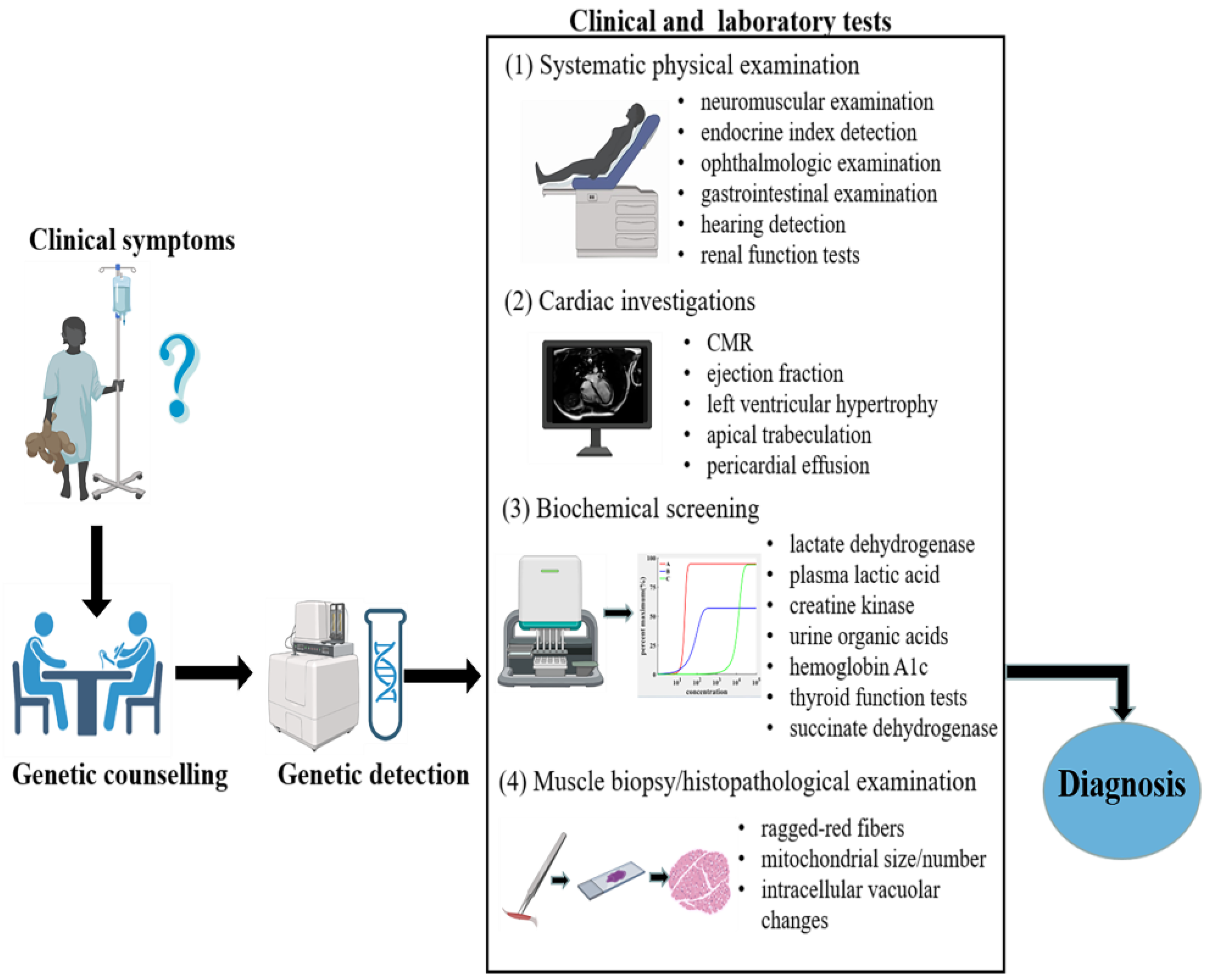
3. Diagnosis
3.1. Genetic Counselling and Detection
- Patients with clinical presentations consistent with mitochondrial myopathy, encephalopathy, lactic acidosis, or stroke-like episodes (MELAS) should undergo screening for common point mutations on mitochondrial DNA.
- Whole mitochondrial DNA screening [9].
- Investigations for mutations using a targeted gene panel of 241 genes known to cause mitochondrial diseases, as well as the whole mitochondrial genome.
- Whole-exome sequencing using next-generation sequencing for nuclear DNA mutations. Detailed procedures have been described previously.
- High-density oligonucleotide array for large chromosomal deletions, as previously described [10].
3.2. Cardiac Imaging Diagnosis
3.3. Muscle Biopsy and Histopathological Examination
3.4. Systematic Physical Examination
3.5. Biochemical Screening
4. Disease Modeling for MCM
4.1. Animal Models
4.2. Cellular Models
4.2.1. Immortalized Cells
4.2.2. Fibroblasts
4.2.3. Induced Pluripotent Stem Cells (iPSCs) and iPSC-Derived Cardiomyocytes (iPSC-CMs)
5. Advantages and Limitations of iPSC/iPSC-CM
6. Current and Novel Management in MCM
6.1. Pharmacological Strateges
6.2. Gene Therapy for MCM
6.3. Mitochondrial Replacement Therapy and AMT/T
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- El-Hattab, A.W.; Scaglia, F. Mitochondrial cardiomyopathies. Front. Cardiovasc. Med. 2016, 3, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandra, C.J.A.; Chua, J.; Cong, S.; Kp, M.M.J.; Shim, W.; Wu, J.C.; Hausenloy, D.J. Human-induced pluripotent stem cells for modelling metabolic perturbations and impaired bioenergetics underlying cardiomyopathies. Cardiovasc. Res. 2021, 117, 694–711. [Google Scholar] [CrossRef]
- Limongelli, G.; Masarone, D.; D’Alessandro, R.; Elliott, P.M. Long-term cardiac prognosis and risk stratification in 260 adults presenting with mitochondrial diseases. Future Cardiol. 2012, 8, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Meyers, D.E.; Basha, H.I.; Koenig, M.K. Mitochondrial cardiomyopathy: Pathophysiology, diagnosis, and management. Texas Heart Inst. J. 2013, 40, 385–394. [Google Scholar]
- Stojkovic, T.; Wahbi, K.; Bougouin, W.; Be, A.; Jardel, C.; Berber, N.; Mochel, F.; Eymard, B.; Duboc, D.; Lafore, P.; et al. Long-term cardiac prognosis and risk stratification in 260 adults presenting with mitochondrial diseases. Future Cardiol. 2015, 8, 2886–2893. [Google Scholar] [CrossRef] [Green Version]
- Bates, M.G.D.; Bourke, J.P.; Giordano, C.; Amati, G.; Turnbull, D.M.; Taylor, R.W. Clinical update cardiac involvement in mitochondrial DNA disease: Clinical spectrum, diagnosis, and management. Eur. Heart J. 2012, 33, 3023–3033. [Google Scholar] [CrossRef] [Green Version]
- Imai-okazaki, A.; Matsunaga, A.; Yatsuka, Y.; Nitta, K.R. Long-term prognosis and genetic background of cardiomyopathy in 223 pediatric mitochondrial disease patients. Int. J. Cardiol. 2021, 341, 48–55. [Google Scholar] [CrossRef]
- Brunel-Guitton, C.; Levtova, A.; Sasarman, F. Mitochondrial diseases and cardiomyopathies. Can. J. Cardiol. 2015, 31, 1360–1376. [Google Scholar] [CrossRef]
- Kohda, M.; Tokuzawa, Y.; Kishita, Y.; Nyuzuki, H.; Moriyama, Y.; Mizuno, Y.; Hirata, T.; Yatsuka, Y.; Yamashita-Sugahara, Y.; Nakachi, Y.; et al. A comprehensive genomic analysis reveals the genetic landscape of mitochondrial respiratory chain complex deficiencies. PLoS Genet. 2016, 12, e1005679. [Google Scholar] [CrossRef]
- Imai-Okazaki, A.; Kishita, Y.; Kohda, M.; Mizuno, Y.; Fushimi, T.; Matsunaga, A.; Yatsuka, Y.; Hirata, T.; Harashima, H.; Takeda, A.; et al. Cardiomyopathy in children with mitochondrial disease: Prognosis and genetic background. Int. J. Cardiol. 2019, 279, 115–121. [Google Scholar] [CrossRef]
- Jose, T.; Gdynia, H.-J.; Mahrholdt, H.; Vöhringer, M.; Klingel, K.; Kandolf, R.; Bornemann, A.; Yilmaz, A. CMR gives clue to “ragged red fibers” in the heart in a patient with mitochondrial myopathy. Int. J. Cardiol. 2011, 149, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Park, H.S.; Park, C.H.; Kim, K.-H.; Chung, H.; Kim, T.H.; Rim, S.-J.; Choi, E.-Y. Extracellular volume imaging and quantitative T2 mapping for the diagnosis of mitochondrial cardiomyopathy. Circulation 2014, 130, 1832–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Sampath, H. Mitochondrial DNA integrity: Role in health and disease. Cells 2019, 8, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardiomyopathy, M. Images in cardiovascular medicine:cardiac magnetic resonance aids in the diagnosis of mitochondrial cardiomyopathy. Circulation 2011, 123, 227–229. [Google Scholar] [CrossRef] [Green Version]
- Goldenthal, M.J. Mitochondrial involvement in myocyte death and heart failure. Heart Fail. Rev. 2016, 21, 137–155. [Google Scholar] [CrossRef]
- Cha, M.J.; Kim, C.; Park, C.H.; Hong, Y.J.; Shin, J.M.; Kim, T.H.; Cha, Y.J.; Park, C.H. Differential diagnosis of thick myocardium according to histologic features revealed by multiparametric cardiac magnetic resonance imaging. Korean J. Radiol. 2022, 23, 581–597. [Google Scholar] [CrossRef]
- Takeda, A.; Murayama, K.; Okazaki, Y.; Okazaki, A.I.; Ohtake, A.; Takakuwa, E.; Yamazawa, H.; Izumi, G.; Abe, J.; Nagai, A.; et al. Advanced pathological study for definite diagnosis of mitochondrial cardiomyopathy. J. Clin. pathol. 2020, 17, 365–371. [Google Scholar] [CrossRef]
- Finsterer, J.; Kothari, S. Cardiac manifestations of primary mitochondrial disorders. Int. J. Cardiol. 2014, 177, 754–763. [Google Scholar] [CrossRef]
- St-pierre, G.; Steinberg, C.; Dubois, M.; Sénéchal, M. What the cardiologist should know about mitochondrial cardiomyopathy? Can. J. Cardiol. 2019, 35, 221–224. [Google Scholar] [CrossRef]
- Parikh, S.; Goldstein, A.; Karaa, A.; Koenig, M.K.; Anselm, I.; Brunel-Guitton, C.; Christodoulou, J.; Cohen, B.H.; Dimmock, D.; Enns, G.M.; et al. Patient care standards for primary mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. Off. J. Am. Coll. Med. Genet. 2017, 19, 1380. [Google Scholar] [CrossRef] [Green Version]
- Graham, B.H.; Waymire, K.G.; Cottrell, B.; Trounce, I.A.; MacGregor, G.R.; Wallace, D.C. A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat. Genet. 1997, 16, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Narula, N.; Zaragoza, M.V.; Sengupta, P.P.; Li, P.; Haider, N.; Verjans, J.; Waymire, K.; Vannan, M.; Wallace, D.C. Adenine nucleotide translocase 1 deficiency results in dilated cardiomyopathy with defects in myocardial mechanics, histopathological alterations, and activation of apoptosis. JACC. Cardiovasc. Imaging 2011, 4, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richman, T.R.; Ermer, J.A.; Davies, S.M.K.; Perks, K.L.; Viola, H.M.; Shearwood, A.-M.J.; Hool, L.C.; Rackham, O.; Filipovska, A. Mutation in MRPS34 compromises protein synthesis and causes mitochondrial dysfunction. PLoS Genet. 2015, 11, e1005089. [Google Scholar] [CrossRef] [PubMed]
- Krebs, P.; Fan, W.; Chen, Y.-H.; Tobita, K.; Downes, M.R.; Wood, M.R.; Sun, L.; Li, X.; Xia, Y.; Ding, N.; et al. Lethal mitochondrial cardiomyopathy in a hypomorphic Med30 mouse mutant is ameliorated by ketogenic diet. Proc. Natl. Acad. Sci. USA 2011, 108, 19678–19682. [Google Scholar] [CrossRef] [Green Version]
- Ke, B.; Pepe, S.; Grubb, D.R.; Komen, J.C.; Laskowski, A.; Rodda, F.A. Tissue-specific splicing of an Ndufs6 gene-trap insertion generates a mitochondrial complex I deficiency-specific cardiomyopathy. Proc. Natl. Acad. Sci. USA 2012, 109, 6165–6170. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Li, Y.; Heims-waldron, D.; Bezzerides, V.; Guatimosim, S.; Guo, Y.; Gu, F.; Zhou, P.; Lin, Z.; Ma, Q.; et al. Mitochondrial cardiomyopathy caused by elevated reactive oxygen species and impaired cardiomyocyte proliferation. Circ. Res. 2018, 122, 74–87. [Google Scholar] [CrossRef]
- Hansson, A.; Hance, N.; Dufour, E.; Rantanen, A.; Hultenby, K.; Clayton, D.A.; Wibom, R.; Larsson, N.-G. A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain-deficient mouse hearts. Proc. Natl. Acad. Sci. USA 2004, 101, 3136–3141. [Google Scholar] [CrossRef] [Green Version]
- Sayles, N.M.; Southwell, N.; McAvoy, K.; Kim, K.; Pesini, A.; Anderson, C.J.; Quinzii, C.; Cloonan, S.; Kawamata, H.; Manfredi, G. Mutant CHCHD10 causes an extensive metabolic rewiring that precedes OXPHOS dysfunction in a murine model of mitochondrial cardiomyopathy. Cell Rep. 2022, 38, 110475. [Google Scholar] [CrossRef]
- Liu, Y.-T.; Huang, X.; Nguyen, D.; Shammas, M.K.; Wu, B.P.; Dombi, E.; Springer, D.A.; Poulton, J.; Sekine, S.; Narendra, D.P. Loss of CHCHD2 and CHCHD10 activates OMA1 peptidase to disrupt mitochondrial cristae phenocopying patient mutations. Hum. Mol. Genet. 2020, 29, 1547–1567. [Google Scholar] [CrossRef]
- Ling, Y.; Ma, J.; Qi, X.; Zhang, X.; Kong, Q.; Guan, F.; Dong, W.; Chen, W.; Gao, S.; Gao, X.; et al. Novel rat model of multiple mitochondrial dysfunction syndromes (MMDS) complicated with cardiomyopathy. Anim. Model. Exp. Med. 2021, 4, 381–390. [Google Scholar] [CrossRef]
- Meurs, K.M.; Friedenberg, S.G.; Olby, N.J.; Condit, J.; Weidman, J.; Rosenthal, S.; Shelton, G.D. A QIL1 variant associated with ventricular arrhythmias and sudden cardiac death in the juvenile rhodesian ridgeback dog. Genes 2019, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Zhang, Y.; Chen, R.; Gong, Y.; Zhang, M.; Guan, R.; Rotstein, O.D.; Liu, X.; Wen, X.-Y. ndufa7 plays a critical role in cardiac hypertrophy. J. Cell. Mol. Med. 2020, 24, 13151–13162. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Dong, W.-Q.; Kuryshev, Y.A.; Obejero-Paz, C.; Levy, M.N.; Brittenham, G.M.; Kiatchoosakun, S.; Kirkpatrick, D.; Hoit, B.D.; Brown, A.M. Bimodal cardiac dysfunction in an animal model of iron overload. J. Lab. Clin. Med. 2002, 140, 263–271. [Google Scholar] [CrossRef]
- Perdomini, M.; Belbellaa, B.; Monassier, L.; Reutenauer, L.; Messaddeq, N.; Cartier, N.; Crystal, R.G.; Aubourg, P.; Puccio, H. Prevention and reversal of severe mitochondrial cardiomyopathy by gene therapy in a mouse model of Friedreich’s ataxia. Nat. Med. 2014, 20, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, J.; Purhonen, J.; Tegelberg, S.; Smolander, O.-P.; Mörgelin, M.; Rozman, J.; Gailus-Durner, V.; Fuchs, H.; Hrabe de Angelis, M.; Auvinen, P.; et al. Alternative oxidase-mediated respiration prevents lethal mitochondrial cardiomyopathy. EMBO Mol. Med. 2019, 11, e9456. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Chen, Z.; Zhu, M.; Shen, Y.; Leon, L.J.; Chi, L.; Spinozzi, S.; Tan, C.; Gu, Y.; Nguyen, A.; et al. Cardiolipin remodeling defects impair mitochondrial architecture and function in a murine model of Barth syndrome cardiomyopathy. Circ. Heart Fail. 2021, 14, e008289. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Uchiumi, T.; Yagi, M.; Amamoto, R.; Setoyama, D.; Matsushima, Y.; Kang, D. Cardiomyocyte-specific loss of mitochondrial p32/C1qbp causes cardiomyopathy and activates stress responses. Cardiovasc. Res. 2017, 113, 1173–1185. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.; Lee, S.H.; Kwon, M.; Yang, D.K.; Seo, H.; Kim, J.; Kim, Y.; Im, S.; Abel, E.D.; Kim, K.; et al. Cardiomyocyte specific deletion of Crif1 causes mitochondrial cardiomyopathy in mice. PLoS ONE 2013, 8, e53577. [Google Scholar] [CrossRef]
- Davis, R.P.; van den Berg, C.W.; Casini, S.; Braam, S.R.; Mummery, C.L. Pluripotent stem cell models of cardiac disease and their implication for drug discovery and development. Trends Mol. Med. 2011, 17, 475–484. [Google Scholar] [CrossRef]
- Ruzzenente, B.; Rötig, A.; Metodiev, M.D. Mouse models for mitochondrial diseases. Hum. Mol. Genet. 2016, 25, R115–R122. [Google Scholar] [CrossRef] [Green Version]
- Karicheva, O.Z.; Kolesnikova, O.A.; Schirtz, T.; Vysokikh, M.Y.; Mager-Heckel, A.-M.; Lombès, A.; Boucheham, A.; Krasheninnikov, I.A.; Martin, R.P.; Entelis, N.; et al. Correction of the consequences of mitochondrial 3243A>G mutation in the MT-TL1 gene causing the MELAS syndrome by tRNA import into mitochondria. Nucleic Acids Res. 2011, 39, 8173–8186. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.-C.; Liou, C.-W.; Chen, S.-D.; Wang, P.-W.; Chuang, J.-H.; Tiao, M.-M.; Hsu, T.-Y.; Lin, H.-Y.; Lin, T.-K. Mitochondrial transfer from wharton’s jelly mesenchymal stem cell to merrf cybrid reduces oxidative stress and improves mitochondrial bioenergetics. Oxid. Med. Cell. Longev. 2017, 2017, 5691215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, W.; Reynolds, C.A.; Li, Y.; Liu, J.; Hüttemann, M.; Schlame, M.; Stevenson, D.; Strathdee, D.; Greenberg, M.L. Loss of tafazzin results in decreased myoblast differentiation in C2C12 cells: A myoblast model of Barth syndrome and cardiolipin deficiency. Biochim. Biophys. Acta Mol. Cell Biol. Lipid 2018, 1863, 857–865. [Google Scholar] [CrossRef]
- James, A.M.; Wei, Y.H.; Pang, C.Y.; Murphy, M.P. Altered mitochondrial function in fibroblasts containing MELAS or MERRF mitochondrial DNA mutations. Biochem. J. 1996, 318, 401–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlame, M.; Kelley, R.I.; Feigenbaum, A.; Towbin, J.A.; Heerdt, P.M.; Schieble, T.; Wanders, R.J.A.; DiMauro, S.; Blanck, T.J.J. Phospholipid abnormalities in children with Barth syndrome. J. Am. Coll. Cardiol. 2003, 42, 1994–1999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, A.; Bellin, M.; Welling, A.; Jung, C.B.; Lam, J.T.; Bott-Flügel, L.; Dorn, T.; Goedel, A.; Höhnke, C.; Hofmann, F.; et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N. Engl. J. Med. 2010, 363, 1397–1409. [Google Scholar] [CrossRef] [Green Version]
- Kodaira, M.; Hatakeyama, H.; Yuasa, S.; Seki, T.; Egashira, T.; Tohyama, S.; Kuroda, Y.; Tanaka, A.; Okata, S.; Hashimoto, H.; et al. Impaired respiratory function in MELAS-induced pluripotent stem cells with high heteroplasmy levels. FEBS Open Bio 2015, 5, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Yokota, M.; Hatakeyama, H.; Ono, Y.; Kanazawa, M.; Goto, Y.-I. Mitochondrial respiratory dysfunction disturbs neuronal and cardiac lineage commitment of human iPSCs. Cell Death Dis. 2017, 8, e2551. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; McCain, M.L.; Yang, L.; He, A.; Pasqualini, F.S.; Agarwal, A.; Yuan, H.; Jiang, D.; Zhang, D.; Zangi, L.; et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 2014, 20, 616–623. [Google Scholar] [CrossRef]
- Wu, H.; Yang, H.; Rhee, J.; Zhang, J.Z.; Lam, C.K.; Sallam, K.; Chang, A.C.Y.; Ma, N.; Lee, J.; Zhang, H.; et al. Modelling diastolic dysfunction in induced pluripotent stem cell-derived cardiomyocytes from hypertrophic cardiomyopathy patients. Eur. Heart J. 2019, 40, 3685–3695. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-K.; Ho, P.W.-L.; Schick, R.; Lau, Y.-M.; Lai, W.-H.; Zhou, T.; Li, Y.; Ng, K.-M.; Ho, S.-L.; Esteban, M.A.; et al. Modeling of friedreich ataxia-related iron overloading cardiomyopathy using patient-specific-induced pluripotent stem cells. Pflugers Arch. 2014, 466, 1831–1844. [Google Scholar] [CrossRef] [PubMed]
- Hick, A.; Wattenhofer-Donzé, M.; Chintawar, S.; Tropel, P.; Simard, J.P.; Vaucamps, N.; Gall, D.; Lambot, L.; André, C.; Reutenauer, L.; et al. Neurons and cardiomyocytes derived from induced pluripotent stem cells as a model for mitochondrial defects in Friedreich’s ataxia. Dis. Model. Mech. 2013, 6, 608–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Pan, H.; Tan, C.; Sun, Y.; Song, Y.; Zhang, X.; Yang, W. Mitochondrial dysfunctions contribute to hypertrophic cardiomyopathy in patient iPSC-derived cardiomyocytes with MT-RNR2 mutation. Stem Cell Rep. 2018, 10, 808–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, J.; Zhou, Y.; Li, H.; Li, A.; Tan, X.; Wang, G.; Lei, M. Generation of two induced pluripotent stem cell lines (XACHi0010-A, XACHi0011-A)from a Chinese family with combined oxidative phosphorylation deficiency carrying homozygous and heterozygous C1QBP-L275F mutation. Stem Cell Res. 2020, 47, 101912. [Google Scholar] [CrossRef] [PubMed]
- Rohani, L.; Machiraju, P.; Sabouny, R.; Meng, G.; Liu, S.; Zhao, T.; Iqbal, F.; Ravandi, A.; Wu, J.C.; Khan, A.; et al. Reversible mitochondrial fragmentation in ipsc-derived cardiomyocytes from children with DCMA, a mitochondrial cardiomyopathy. Can. J. Cardiol. 2019, 36, 554–563. [Google Scholar] [CrossRef]
- Shao, K.; Koch, C.; Gupta, M.K.; Lin, Q.; Lenz, M.; Laufs, S.; Denecke, B.; Schmidt, M.; Linke, M.; Hennies, H.C.; et al. Induced pluripotent mesenchymal stromal cell clones retain donor-derived differences in DNA methylation profiles. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 240–250. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Tellez, N.; Greenway, S.C. Cellular models for human cardiomyopathy: What is the best option? World J. Cardiol. 2019, 11, 221–235. [Google Scholar] [CrossRef]
- Duran, A.G.; Reidell, O.; Stachelscheid, H.; Klose, K.; Gossen, M.; Falk, V.; Röll, W.; Stamm, C. Regenerative medicine/cardiac cell therapy: Pluripotent stem cells. Thorac. Cardiovasc. Surg. 2018, 66, 53–62. [Google Scholar] [CrossRef]
- Pavez-Giani, M.G.; Cyganek, L. Recent Advances in Modeling Mitochondrial Cardiomyopathy Using Human Induced Pluripotent Stem Cells. Front. Cell. Dev. Biol. 2021, 9, 800529. [Google Scholar] [CrossRef]
- Dambrot, C.; Braam, S.R.; Tertoolen, L.G.J.; Birket, M.; Atsma, D.E.; Mummery, C.L. Serum supplemented culture medium masks hypertrophic phenotypes in human pluripotent stem cell derived cardiomyocytes. J. Cell. Mol. Med. 2014, 18, 1509–1518. [Google Scholar] [CrossRef]
- Mekhoubad, S.; Bock, C.; de Boer, A.S.; Kiskinis, E.; Meissner, A.; Eggan, K. Erosion of dosage compensation impacts human iPSC disease modeling. Cell Stem Cell 2012, 10, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-velasco, A.; Bui, T.; Collins, L.; Wang, X. Mitochondrial function in the heart: The insight into mechanisms and therapeutic potentials. Br. J. Pharmacol. 2019, 176, 4302–4318. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.F.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diguet, N.; Trammell, S.A.J.; Tannous, C.; Deloux, R.; Piquereau, J.; Mougenot, N.; Gouge, A.; Gressette, M.; Manoury, B.; Blanc, J.; et al. Nicotinamide riboside preserves cardiac function in a mouse model of dilated cardiomyopathy. Circulation 2018, 137, 2256–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Xu, Y.; Chen, W.; Qiu, Z. Stomatin-like protein-2: A potential target to treat mitochondrial cardiomyopathy. Heart Lung Circ. 2021, 30, 1449–1455. [Google Scholar] [CrossRef]
- Ribeiro Junior, R.F.; Dabkowski, E.R.; Shekar, K.C.; O Connell, K.A.; Hecker, P.A.; Murphy, M.P. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic. Biol. Med. 2018, 117, 18–29. [Google Scholar] [CrossRef]
- Rossman, M.J.; Santos-Parker, J.R.; Steward, C.A.C.; Bispham, N.Z.; Cuevas, L.M.; Rosenberg, H.L.; Woodward, K.A.; Chonchol, M.; Gioscia-Ryan, R.A.; Murphy, M.P.; et al. Chronic supplementation with a mitochondrial antioxidant (MitoQ) improves vascular function in healthy older adults. Hypertension 2018, 71, 1056–1063. [Google Scholar] [CrossRef]
- Himori, K.; Abe, M.; Tatebayashi, D.; Lee, J.; Westerblad, H.; Lanner, J.T.; Yamada, T. Superoxide dismutase/catalase mimetic EUK-134 prevents diaphragm muscle weakness in monocrotalin-induced pulmonary hypertension. PLoS ONE 2017, 12, e0169146. [Google Scholar] [CrossRef] [Green Version]
- Javadov, S.; Jang, S.; Rodriguez-Reyes, N.; Rodriguez-Zayas, A.E.; Soto Hernandez, J.; Krainz, T.; Wipf, P.; Frontera, W. Mitochondria-targeted antioxidant preserves contractile properties and mitochondrial function of skeletal muscle in aged rats. Oncotarget 2015, 6, 39469–39481. [Google Scholar] [CrossRef] [Green Version]
- Machiraju, P.; Wang, X.; Sabouny, R.; Huang, J.; Zhao, T. SS-31 peptide reverses the mitochondrial fragmentation present in fibroblasts from patients with DCMA, a mitochondrial cardiomyopathy. Front. Cardiovasc. Med. 2019, 6, 167. [Google Scholar] [CrossRef]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Clarke, S.J.; Javadov, S.A. Mitochondrial permeability transition pore opening during myocardial reperfusion-a target for cardioprotection. Cardiovasc. Res. 2004, 61, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Sharov, V.G.; Todor, A.; Khanal, S.; Imai, M.; Sabbah, H.N. Cyclosporine A attenuates mitochondrial permeability transition and improves mitochondrial respiratory function in cardiomyocytes isolated from dogs with heart failure. J. Mol. Cell. Cardiol. 2007, 42, 150–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Ruiz-Velasco, A.; Wang, S.; Khan, S.; Zi, M.; Jungmann, A.; Dolores Camacho-Muñoz, M.; Guo, J.; Du, G.; Xie, L.; et al. Metabolic stress-induced cardiomyopathy is caused by mitochondrial dysfunction due to attenuated Erk5 signaling. Nat. Commun. 2017, 8, 494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, P.K.; Craven, L.; Hoogewijs, K.; Russell, O.M.; Lightowlers, R.N. Advances in methods for reducing mitochondrial DNA disease by replacing or manipulating the mitochondrial genome. Essays Biochem. 2018, 62, 455–465. [Google Scholar] [CrossRef]
- Hyslop, L.A.; Blakeley, P.; Craven, L.; Richardson, J.; Fogarty, N.M.E.; Fragouli, E.; Lamb, M.; Wamaitha, S.E.; Prathalingam, N.; Zhang, Q.; et al. Towards clinical application of pronuclear transfer to prevent mitochondrial DNA disease. Nature 2016, 534, 383–386. [Google Scholar] [CrossRef] [Green Version]
- Tachibana, M.; Amato, P.; Sparman, M.; Woodward, J.; Sanchis, D.M.; Ma, H.; Gutierrez, N.M.; Tippner-Hedges, R.; Kang, E.; Lee, H.-S.; et al. Towards germline gene therapy of inherited mitochondrial diseases. Nature 2013, 493, 627–631. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Fan, X.L.; Jiang, D.; Zhang, Y.; Li, X.; Xu, Z.B.; Fang, S.B.; Chiu, S.; Tse, H.F.; Lian, Q.; et al. Connexin 43-mediated mitochondrial transfer of iPSC-MSCs alleviates asthma inflammation. Stem Cell Rep. 2018, 11, 1120–1135. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Cheung, M.K.H.; Han, S.; Zhang, Z.; Chen, L.; Chen, J.; Zeng, H.; Qiu, J. Mesenchymal stem cells and their mitochondrial transfer: A double-edged sword. Biosci. Rep. 2019, 39, BSR20182417. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Sun, Y.; Qi, Z.; Cao, L.; Ding, S. Mitochondrial transfer/transplantation: An emerging therapeutic approach for multiple diseases. Cell Biosci. 2022, 12, 66. [Google Scholar] [CrossRef]
- Blitzer, D.; Guariento, A.; Doulamis, I.P.; Shin, B.; Moskowitzova, K.; Barbieri, G.R.; Orfany, A.; Del Nido, P.J.; McCully, J.D. Delayed transplantation of autologous mitochondria for cardioprotection in a porcine model. Ann. Thorac. Surg. 2020, 109, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Doulamis, I.P.; Guariento, A.; Duignan, T.; Orfany, A.; Kido, T.; Zurakowski, D.; Del Nido, P.J.; McCully, J.D. Mitochondrial transplantation for myocardial protection in diabetic hearts. Eur. J. Cardio-Thorac. Surg. 2020, 57, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.; Shi, X.; Zhang, H.; Fu, B. Mitotherapy for fatty liver by intravenous administration of exogenous mitochondria in male mice. Front. Pharmacol. 2017, 8, 241. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.-J.; Kuo, C.-C.; Lee, H.-C.; Shen, C.-I.; Cheng, F.-C.; Wu, S.-F.; Chang, J.-C.; Pan, H.-C.; Lin, S.-Z.; Liu, C.-S.; et al. Transferring xenogenic mitochondria provides neural protection against ischemic stress in ischemic rat brains. Cell Transpl. 2016, 25, 913–927. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Xiong, G.; Feng, H.; Zhang, Z.; Chen, P.; Yan, B.; Chen, L.; Gandhervin, K.; Ma, C.; Li, C.; et al. Donation of mitochondria by iPSC-derived mesenchymal stem cells protects retinal ganglion cells against mitochondrial complex I defect-induced degeneration. Theranostics 2019, 9, 2395–2410. [Google Scholar] [CrossRef]
- Jiang, D.; Chen, F.; Zhou, H.; Lu, Y.; Tan, H.; Yu, S.; Yuan, J.; Liu, H. Theranostics bioenergetic crosstalk between mesenchymal stem cells and various ocular cells through the intercellular trafficking of mitochondria. Theranostics 2020, 10, 7260–7271. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.-H.; Sagullo, E.; Case, D.; Zheng, X.; Li, Y.; Hong, J.S.; TeSlaa, T.; Patananan, A.N.; McCaffery, J.M.; Niazi, K.; et al. Mitochondrial transfer by photothermal nanoblade restores metabolite profile in mammalian cells. Cell Metab. 2016, 23, 921–929. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, Y.; Yeung, S.C.; Liang, Y.; Liang, X.; Ding, Y.; Ip, M.S.M.; Tse, H.F.; Mak, J.C.W.; Lian, Q. Mitochondrial transfer of induced pluripotent stem cell-derived mesenchymal stem cells to airway epithelial cells attenuates cigarette smoke-induced damage. Am. J. Respir. Cell Mol. Biol. 2014, 51, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, J.; Lu, Y.; Huang, S.; Xiao, R.; Zeng, X.; Zhang, X.; Li, J.; Wang, T.; Li, T.; et al. Mitochondrial transplantation attenuates hypoxic pulmonary vasoconstriction. Oncotarget 2016, 7, 31284–31298. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y.; Park, J.-H.; Hayakawa, K. Therapeutic use of extracellular mitochondria in CNS injury and disease. Exp. Neurol. 2020, 324, 113114. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.-C.; Wu, S.-L.; Liu, K.-H.; Chen, Y.-H.; Chuang, C.-S.; Cheng, F.-C.; Su, H.-L.; Wei, Y.-H.; Kuo, S.-J.; Liu, C.-S. Allogeneic/xenogeneic transplantation of peptide-labeled mitochondria in Parkinson’s disease: Restoration of mitochondria functions and attenuation of 6-hydroxydopamine-induced neurotoxicity. Transl. Res. 2016, 170, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Orfany, A.; Arriola, G.; Doulamis, I.P.; Guariento, A. Mitochondrial transplantation ameliorates acute limb ischemia. J. Vasc. Surg. 2020, 71, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Ali Pour, P.; Kenney, M.C.; Kheradvar, A. Bioenergetics consequences of mitochondrial transplantation in cardiomyocytes. J. Am. Heart Assoc. 2020, 9, e014501. [Google Scholar] [CrossRef] [PubMed]
- Ali Pour, P.; Hosseinian, S.; Kheradvar, A. Mitochondrial transplantation in cardiomyocytes: Foundation, methods, and outcomes. Am. J. Physiol. Cell Physiol. 2021, 321, C489–C503. [Google Scholar] [CrossRef] [PubMed]
- Louwagie, E.J.; Larsen, T.D.; Wachal, A.L.; Gandy, T.C.T.; Baack, M.L. Mitochondrial transfer improves cardiomyocyte bioenergetics and viability in male rats exposed to pregestational diabetes. Int. J. Mol. Sci. 2021, 22, 2382. [Google Scholar] [CrossRef]
- Steven, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Kuntic, M.; Bayo Jimenez, M.T.; Vujacic-Mirski, K.; Helmstädter, J.; Kröller-Schön, S.; Münzel, T.; et al. Vascular inflammation and oxidative stress: Major triggers for cardiovascular disease. Oxid. Med. Cell. Longev. 2019, 2019, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yu, Z.; Jiang, D.; Liang, X.; Liao, S.; Zhang, Z.; Yue, W.; Li, X.; Chiu, S.M.; Chai, Y.H.; et al. iPSC-MSCs with high intrinsic Miro1 and sensitivity to TNF-α yield efficacious mitochondrial transfer to rescue anthracycline-induced cardiomyopathy. Stem Cell Rep. 2016, 7, 749–763. [Google Scholar] [CrossRef] [Green Version]
- Emani, S.M.; Piekarski, B.L.; Harrild, D.; Del Nido, P.J.; McCully, J.D. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 2017, 154, 286–289. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Jiang, X.; Yang, Q.; Zhao, J.; Zhou, Q.; Zhou, Y. The functions, methods, and mobility of mitochondrial transfer between cells. Front. Oncol. 2021, 11, 1630. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| a. Gene Mutations in Mitochondrial DNA and Mitochondrial Disorder | |||
|---|---|---|---|
| Genes | Amino Acid Change | Cardiological Phenotype | Other Disorder |
| MT-ND1: m. 3481G > A | p. Glu59Lys | HCM, LVNC | LHON |
| MT-ND4: m. 11778G > A | p. Arg340His | DCM | LHON, progressive dystonia |
| MTND5: m. 12338T > C | p. Met1Thr | HCM. WPW | Leigh syndrome |
| MT-ATP6/8: m. 8528 T > C | p. Pro10Ser | HCM | Subacute encephalopathy |
| MT-ATP6: m. 8851 T > C | p. Trp109Arg | HCM | NARP, Leigh disease |
| MT-ND6: m. 14453G > A | p. tRNALeu | HCM | LHON, MELAS |
| MT-ND6: m. 8528 T > C | Syn | DCM | LHON, MELAS |
| MT-CYB: m. 14849T > C | p. Ser35Pro | HCM | Septo-optic dysplasia |
| MT-TL1: m. 3260A > G | p. tRNALeu (UUR) | HCM, RCM, LVNC | MELAS, Leigh syndrome |
| MT-TI: m. 4300A > G | - | HCM, DCM | MERRF, Leigh syndrome |
| MT-TV: m. 1644G > A | - | HCM | MERRF, Leigh syndrome |
| MT-TK: m. 8344A > G | p. tRNALys | HCM | MERRF, Leigh syndrome |
| MT-RNR1: m. 1555A > G | - | RCM | Maternally inherited deafness |
| b. Genes Mutations in Nuclear DNA and Mitochondrial Disorder | |||
| Genes | Amino acid change | Cardiological phenotype | Other disorder |
| NDUFS2: c. 208 + 5G > A | p. Pro229Gln | HCM | Mitochondrial CI deficiency |
| NDUFV2: c. 669_670insG | p. Ser224fs | HCM | Mitochondrial CI deficiency |
| NDUFA11: c. 99 C + 5G> A | p. Ala132Pro | HCM | Mitochondrial CI deficiency and/or encephalocardiomyopathy |
| NDUFB11: c. 136_142dup | p. Arg134Ser | LVNC, WPW | Mitochondrial CI deficiency |
| SDHD: c. 275A > G | p. Asp92Gly | DCM, LVNC | Mitochondrial CII deficiency |
| NDUFAF1: c. 631C > T | p. Arg211Cys | HCM | Mitochondrial CI deficiency |
| ACAD9: c. 797G > A | p. Arg266Gln | HCM | Mitochondrial CI deficiency |
| SCO2: c. 418G > A | p. Glu140Lys | HCM | Cytochrome C oxidase deficiency |
| COX10: c. 610A > G | p. Asn204Asp | HCM | Mitochondrial CIV deficiency |
| COX15: c. 1129A > T | p. Lys377x | HCM | Cytochrome C oxidase deficiency |
| COA6: c. 196 T > C | p. Trp66Arg | HCM | Mitochondrial CIV deficiency |
| COX6B1: c. 58C > T | p. Arg20Cys | HCM | MELAS, MERRF |
| TEME70: c. 366A > T | p. Tyr112Ter | HCM | Mitochondrial CV deficiency |
| TEME70: c. 317-2A > G | - | HCM | Mitochondrial CV deficiency |
| AARS2: c. 1774C > T | p. Arg958 * | HCM | COXPD 8 |
| MRPS22: c. 644T > C | p. Leu215Pro | HCM | COXPD 8 |
| MRPL3: c. 950C > G | p. Pro317Arg | HCM | COXPD9 |
| MRPL3: c. 49delC | Arg17Aspfs * 57 | HCM | COXPD9 |
| MRPL44: c. 467T > G | p. Leu156Arg | HCM | Mitochondrial CIV deficiency |
| TSFM: c. 355G > C | p. Val119Leu | HCM, DCM | COXPD 3 |
| GTPB3: c. 1291dupC; | p. Pro430Argfs * 86 | HCM, DCM | COXPD23, Encephalopathy |
| GTPB3: c. 1375G > A | p. Glu459Lys | HCM, DCM | COXPD23 |
| GTPB3: c. 476A > T | p. Glu159Val | HCM, DCM | lactic acidosis, leukodystrophy |
| GTPB3: c. 964G > C | p. Ala322Pro | HCM, DCM | lactic acidosis, leukodystrophy |
| MTO1: c. 1282G > A | p. Ala428Thr | HCM | COXPD10 |
| MTO1: c. 1858dup | p. Arg620Lysfs * 8 | HCM | COXPD10 |
| ELAC2: c. 631C > T | p. Arg211 * | HCM | COXPD17 |
| ELAC2: c. 1559C > T | p. Thr520Ile | HCM | COXPD17 |
| ELAC2: c. 460T > C | p. Phe154Leu | MELAS | Cardiac failure |
| ELAC2: c. 1267C > T | p. Leu423Phe | DCM | Cardiac failure, COX deficiency |
| TAZ: c. 527A > G | p. His176Arg | DCM, LVNC | BTHS |
| AGK: c. 306T > G | p. Tyr102Ter | HCM | Sengers syndrome |
| SLC22A5: c. 12C > G | p. Tyr4 * | HCM, DCM | Systemic primary carnitine deficiency |
| ACADVL: c. 104delC | p. P35Lfs * 26 | HCM, DCM | VLCAD deficiency |
| ACADVL: c. 848T > C | p. V283A | HCM | VLCAD deficiency |
| ACADVL: c. 1141_1143del GAG | p. E381del | HCM | VLCAD deficiency |
| ACAD9: c. 555-2A > G | p. Ala390Thr | HCM | MTP deficiency with myopathy and neuropathy |
| ATAD3A-C: c. 1064G > A | p. G355D | HCM | Hereditary spastic paraplegia, axonal neuropathy |
| SLC25A4: c. 239G > A | p. Arg80His | HCM | Mitochondrial DNA depletion syndrome-12 |
| SLC25A4: c. 703C > G | p. Arg235Gly | HCM | Mitochondrial DNA depletion syndrome-12 |
| QRSL1: c. 398G > T | p. G133V | HCM | COXPD40 |
| KARS: c. 1343 T > A: | p. V448D | HCM, DCM, MC | Infantile-onset progressive leukoencephalopathy /or deafness |
| KARS: c. 953 T > C | p. I318T | HCM, DCM, MC | Mitochondrial cytopathy |
| TOP3A: c. 298A > G | p. Met100Val | DCM | adult-onset mitochondrial disorder |
| TOP3: c. 403C > T | p. Arg135Ter | DCM | Adult-onset mitochondrial disorder |
| FXN: GAA repeat expansion | - | HCM | Friedreich ataxia, MELAS, MERRF |
| BOLA3: c. 287A > G | p. H96R | HCM | Multiple mitochondrial dysfunctions syndrome-2 with hyperglycinemia |
| CoQ4: c. 718C > T | p. R240C | HCM | Coenzyme Q10 deficiency 7 |
| CoQ4: c. 421C > T | p. R141X | HCM | Lethal infantile mitochondrial disorder |
| DNAJC19: IVS3-1G > C | - | DCM, LVNC | 3-methylglutaconic aciduria type V |
| Animal | Mutations | Cardiological Phenotype | Clinical Manifestations | Year | Ref |
|---|---|---|---|---|---|
| Mice | Ant1 | mitochondrial myopathy/MCM | ragged-red muscle fibers, dramatic proliferation of mitochondria, cardiac hypertrophy with mitochondrial proliferation, severe defects in coupled respiration, metabolic acidosis, exercise intolerance. | 1997 | [21] |
| Mice | Ant1 | DMC | substantial myocardial hypertrophy/ventricular dilation, cardiac function declining in early age, LV circumferential, radial, rotational mechanics reduced, myocyte hypertrophy, fibrosis, calcification. | 2011 | [22] |
| Mice | MRPS34 | progressive cardiomyopathy | fractional shortening of the heart, pronounced liver dysfunction, inhibition of mitochondrial translation, decreased oxygen consumption and respiratory complex activity. | 2015 | [23] |
| Mice | Med30zg | MCM/cardiac failure | changes in transcription of cardiac genes for OXPHOS and mitochondrial integrity precipitous lethality 2–3 weeks after weaning. | 2011 | [24] |
| Mice | Ndufs6 | CI deficiency-specific MCM | LV systolic function, cardiac output, and functional work capacity markedly reduced, at increased risk of cardiac failure and death after 4 months in males and 8 in females, ATP synthesis decreased, hydroxyacylcarnitine increased. | 2012 | [25] |
| Mice | TFAM | progressive, lethal DCM | elevated ROS production, activated DNA damage response pathway, decreased cardiomyocyte proliferation. | 2018 | [26] |
| Mice | TFAM | MCM | critical enzymes in fatty acid oxidation show decreased expression, glycolytic enzymes show increased expression. | 2004 | [27] |
| Mice | CHCHD10-S55L | MCM | typical ISRmt, mitochondrial architecture and function altered in the heart, metabolic pathway changed from oxidative to glycolytic. | 2022 | [28] |
| Mice | CHCHD2/CHCHD10 | MCM | C2/C10 DKO mice have disrupted mitochondrial cristae, cleavage of the l-OPA1, activation of the ISRmt and development of cardiomyopathy. | 2020 | [29] |
| Rat | Isca1 | MMDS with cardiomyopathy; MCM | Isca1 HET rats exhibit thin-walled ventricles, larger chambers, cardiac dysfunction and myocardium fibrosis, damaged mitochondrial morphology, enzyme activity and ATP production. | 2021 | [30] |
| Dog | QIL1 | MCM | cristae abnormalities and cardiac arrhythmias, hyperplastic mitochondrial, cristae rearrangement, electron dense inclusions, lipid bodies in muscle. | 2019 | [31] |
| Zebrafish | ndufa7/hhatla | HCM | cardiac functional defects, associated with increased expression of pathological hypertrophy biomarkers ANP and BNP. | 2020 | [32] |
| Mongolian gerbils | iron overload | Iron-overload cardiomyopathy | cardiac hypertrophy, increased cardiac output, and normal exercise tolerance at shorter durations, concentric cardiac hypertrophy, cardiac output and exercise capacity were impaired at longer duration. | 2002 | [33] |
| Mice | FXN | FRDA- cardiomyopathy | impaired mitochondrial OXPHOS, bioenergetics imbalance, deficit of Fe-S cluster enzymes and mitochondrial iron overload, recapitulated most features of FRDA cardiomyopathy. | 2014 | [34] |
| Mice | Bcs1lp.S78G | MCM | GRACILE syndrome, growth failure, progressive hepatopathy and kidney tubulopathy, | 2019 | [35] |
| Mice | TAZ | BTHS cardiomyopathy | significantly enlarged hearts, ventricular. dilation at 16-weeks of age, lower total CL concentration, abnormal CL fatty acyl composition. | 2021 | [36] |
| Mice | C1QBP | MCM | increased oxidative stress, embryonic lethality with the embryonic fibroblast, cardiomyocyte dysfunction. | 2017 | [37] |
| Mice | Crif1 | MCM | mice suffered from progressive hypertrophy and died from heart failure; mutant mice died within 2 weeks postnatal, showing cardiac hypertrophy associated with mitochondrial dysfunction. | 2013 | [38] |
| Cell Type | Gene | Variants | Disease | Phenotype | Year | Ref. |
|---|---|---|---|---|---|---|
| Immortalized cells | MT-TL1 | m. 3243A > G | MELAS syndrome | defective protein synthesis, reduced activities of MRC. | 2011 | [41] |
| Immortalized cells | tRNALys | m. 8344A > G mutation | MERRF | increase ROS, oxidative stress, impaired mitochondrial bioenergetics. | 2017 | [42] |
| C2C12 | TAZ | TAZ-KO | BTHS | mitochondrial deficits, accumulation of MLCL, ROS, production increased, mitochondrial respiration decreased, myocyte differentiation impaired. | 2018 | [43] |
| Fibroblasts | tRNA(Leu/tRNA(Lys) | tRNA(Lys) | MELAS/MERRF | mitochondrial membrane potential and respiration rate decreased, incompetent mitochondria assembly, cell volume occupied by secondary lysosomes and residual bodies. | 1996 | [44] |
| Fibroblasts | TAZ | Positive-TAZ mutation | BTHS | cardiolipin, phosphatidylcholine, and phosphatidylethanolamine abnormalities in all tissues. | 2003 | [45] |
| iPSCs | KCNQ1 | R190Q | Long-QT Syndrome | susceptibility to catecholamine-induced tachyarrhythmia, beta-blockade attenuated duration of the action potential prolonged, reduction in I(Ks) current and altered channel activation and deactivation increased. | 2010 | [46] |
| iPSCs | mtDNA mutation | m. 3243A > G | MELAS | MELAS-iPSC-derived fibroblasts with high heteroplasmy levels showed defective CI activity, with low heteroplasmy levels showed normal CI activity. | 2015 | [47] |
| iPSCs | mtDNA mutation | m. 3243A > G | HMC | neuronal and cardiac maturation defects in iPSC line carrying a quite high proportion of m. 3243A > G, defective mitochondrial respiratory inhibits maturation of iPSC. | 2017 | [48] |
| iPSC-CMs | TAZ | c. 328T > C | MCM-BTHS | abnormal metabolic, structural and functional, assembled sparse and irregular sarcomeres. | 2014 | [49] |
| iPSC-CMs | HCM-mutation | MYH7/MYBPC3/TNNT2 | DD-HCM | impaired diastolic function, prolonged relaxation time, decreased relaxation rate and sarcomere length. | 2019 | [50] |
| iPSC-CMs | FXN | rs137854888 | FRDA- MCM | disorganized mitochondrial network and (mtDNA) depletion, α-actinin-positive cell sizes and BNP gene expression increased, intracellular iron accumulated, energy synthesis dynamics, ATP production rate impaired. | 2014 | [51] |
| iPSC-CMs | FXN | expanded GAA | FRDA | no biochemical phenotype, decreased membrane potential in neurons and progressive mitochondrial degeneration in cardiomyocytes, increased BNP expression and disrupted iron homeostasis. | 2013 | [52] |
| iPSC-CMs | MT-RNR2 A | m. 2336T > C | HCM | mitochondrial dysfunctions and ultrastructure defects, ATP/ADP ratio and membrane potential reduced, intracellular Ca2+ elevated, electrophysiological abnormalities. | 2018 | [53] |
| PBMC-iPSC | C1QBP | c. 823C > T | COXPD | iPSCs express pluripotent markers, have trilineage differentiation potential, carry C1QBP-L275F mutation, and have a normal karyotype. | 2014 | [54] |
| iPSC-CMs | DNAJC19 | rs137854888 | DCMA | highly fragmented and abnormally shaped mitochondria associated with imbalanced isoform ratio of OPA1. | 2020 | [55] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Chen, S.; Duan, F.; Wang, X.; Zhang, X.; Lian, B.; Kou, M.; Chiang, Z.; Li, Z.; Lian, Q. Mitochondrial Cardiomyopathy: Molecular Epidemiology, Diagnosis, Models, and Therapeutic Management. Cells 2022, 11, 3511. https://doi.org/10.3390/cells11213511
Yang J, Chen S, Duan F, Wang X, Zhang X, Lian B, Kou M, Chiang Z, Li Z, Lian Q. Mitochondrial Cardiomyopathy: Molecular Epidemiology, Diagnosis, Models, and Therapeutic Management. Cells. 2022; 11(21):3511. https://doi.org/10.3390/cells11213511
Chicago/Turabian StyleYang, Jinjuan, Shaoxiang Chen, Fuyu Duan, Xiuxiu Wang, Xiaoxian Zhang, Boonxuan Lian, Meng Kou, Zhixin Chiang, Ziyue Li, and Qizhou Lian. 2022. "Mitochondrial Cardiomyopathy: Molecular Epidemiology, Diagnosis, Models, and Therapeutic Management" Cells 11, no. 21: 3511. https://doi.org/10.3390/cells11213511
APA StyleYang, J., Chen, S., Duan, F., Wang, X., Zhang, X., Lian, B., Kou, M., Chiang, Z., Li, Z., & Lian, Q. (2022). Mitochondrial Cardiomyopathy: Molecular Epidemiology, Diagnosis, Models, and Therapeutic Management. Cells, 11(21), 3511. https://doi.org/10.3390/cells11213511