
Modulation of the Microglial Nogo-A/NgR Signaling Pathway as a Therapeutic Target for Multiple Sclerosis

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Origin, Development and Microglia Cell Homeostasis
3. Microglial Cell Function in an Aging Population
4. The Role of Microglia in Propagating Neuroinflammation and Neurodegeneration in Multiple Sclerosis
5. Overview of Nogo-A and Nogo Receptor 1 Signaling Pathway in Neurodegenerative Disease—A Potential Role in Microglial Activation
6. Effects of Nogo-A/NgR1 on Microglia Activity
6.1. Microglia Migration
6.2. Microglia Phagocytosis
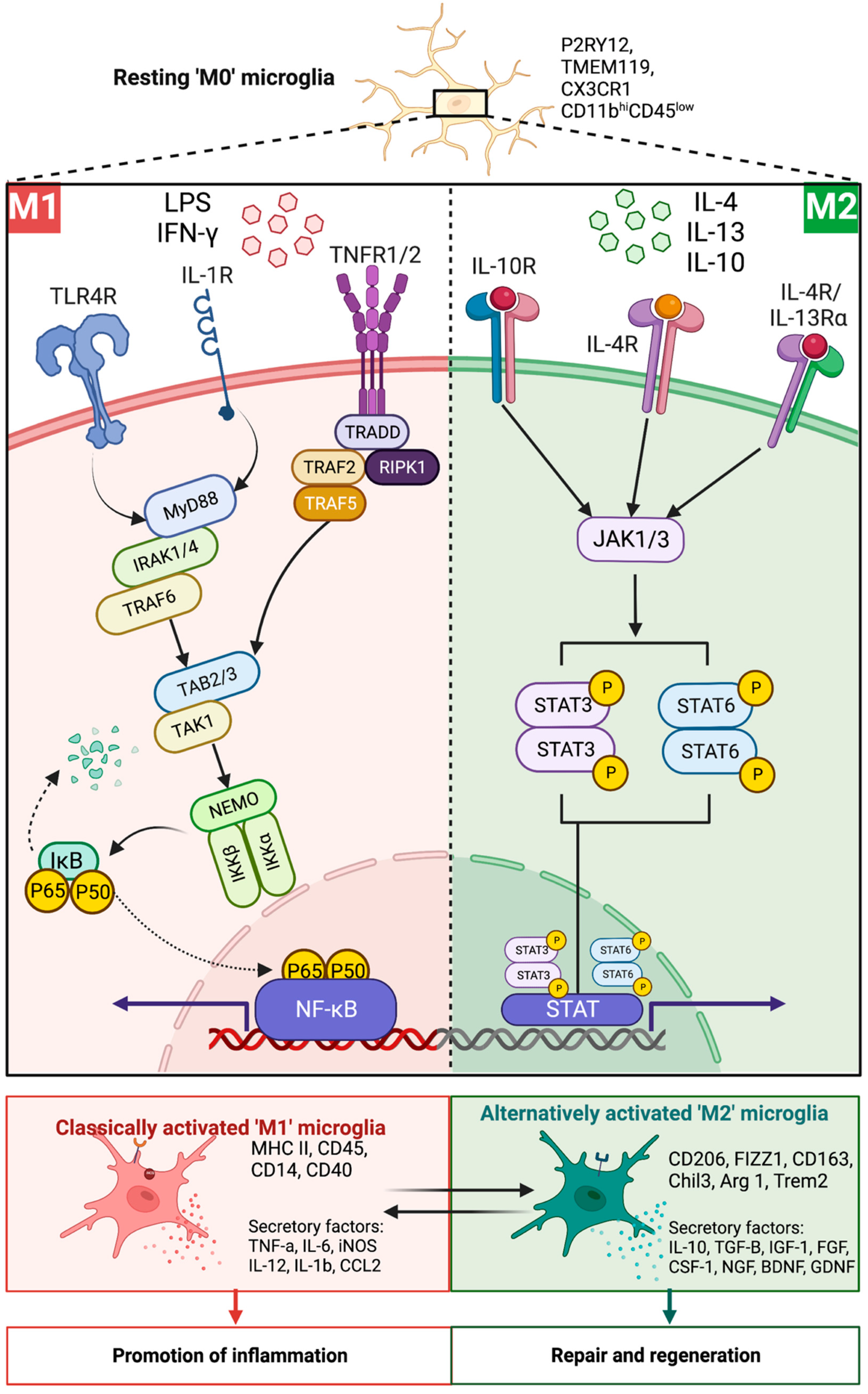
6.3. Microglia Polarization
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Popescu, B.F.G.; Pirko, I.; Lucchinetti, C.F. Pathology of multiple sclerosis: Where do we stand? Contin. Minneap Minn 2013, 19, 901–921. [Google Scholar] [CrossRef] [PubMed]
- Dubey, D.; Sguigna, P.; Stüve, O. Managing Disability in Progressive Multiple Sclerosis. Curr. Treat. Options Neurol. 2016, 18, 27. [Google Scholar] [CrossRef] [PubMed]
- Cadavid, D.; Cohen, J.A.; Freedman, M.S.; Goldman, M.D.; Hartung, H.P.; Havrdova, E.; Jeffery, D.; Kapoor, R.; Miller, A.; Sellebjerg, F.; et al. The EDSS-Plus, an improved endpoint for disability progression in secondary progressive multiple sclerosis. Mult. Scler. 2017, 23, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli Rosa, C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira Tiago, A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pencea, V.; Bingaman, K.D.; Wiegand, S.J.; Luskin, M.B. Infusion of Brain-Derived Neurotrophic Factor into the Lateral Ventricle of the Adult Rat Leads to New Neurons in the Parenchyma of the Striatum, Septum, Thalamus, and Hypothalamus. J. Neurosci. 2001, 21, 6706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., 3rd; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [Green Version]
- Olson, J.K.; Miller, S.D. Microglia Initiate Central Nervous System Innate and Adaptive Immune Responses through Multiple TLRs. J. Immunol. 2004, 173, 3916. [Google Scholar] [CrossRef] [Green Version]
- Guerrero, B.L.; Sicotte, N.L. Microglia in Multiple Sclerosis: Friend or Foe? Front. Immunol. 2020, 11, 374. [Google Scholar] [CrossRef]
- O’Loughlin, E.; Madore, C.; Lassmann, H.; Butovsky, O. Microglial Phenotypes and Functions in Multiple Sclerosis. Cold Spring Harb Perspect. Med. 2018, 8, a028993. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Lu, J.; Sivakumar, V.; Ling, E.A.; Kaur, C. Amoeboid Microglia in the Periventricular White Matter Induce Oligodendrocyte Damage through Expression of Proinflammatory Cytokines via MAP Kinase Signaling Pathway in Hypoxic Neonatal Rats. Brain Pathol. 2008, 18, 387–400. [Google Scholar] [CrossRef]
- Voet, S.; Prinz, M.; van Loo, G. Microglia in Central Nervous System Inflammation and Multiple Sclerosis Pathology. Trends Mol. Med. 2019, 25, 112–123. [Google Scholar] [CrossRef]
- Alrehaili, A.A.; Lee, J.Y.; Bakhuraysah, M.M.; Kim, M.J.; Aui, P.M.; Magee, K.A.; Petratos, S. Nogo receptor expression in microglia/macrophages during experimental autoimmune encephalomyelitis progression. Neural Regen Res. 2018, 13, 896–907. [Google Scholar] [PubMed]
- Lisi, L.; Ciotti, G.M.; Braun, D.; Kalinin, S.; Currò, D.; Dello Russo, C.; Coli, A.; Mangiola, A.; Anile, C.; Feinstein, D.L.; et al. Expression of iNOS, CD163 and ARG-1 taken as M1 and M2 markers of microglial polarization in human glioblastoma and the surrounding normal parenchyma. Neurosci. Lett. 2017, 645, 106–112. [Google Scholar] [CrossRef]
- Famenini, S.; Rigali, E.A.; Olivera-Perez, H.M.; Dang, J.; Chang, M.T.; Halder, R.; Rao, R.V.; Pellegrini, M.; Porter, V.; Bredesen, D.; et al. Increased intermediate M1–M2 macrophage polarization and improved cognition in mild cognitive impairment patients on ω-3 supplementation. FASEB J. 2017, 31, 148–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.; Imagama, S.; Ohgomori, T.; Hirano, K.; Uchimura, K.; Sakamoto, K.; Hirakawa, A.; Takeuchi, H.; Suzumura, A.; Ishiguro, N.; et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. 2013, 4, e525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bok, E.; Chung, Y.C.; Kim, K.-S.; Baik, H.H.; Shin, W.-H.; Jin, B.K. Modulation of M1/M2 polarization by capsaicin contributes to the survival of dopaminergic neurons in the lipopolysaccharide-lesioned substantia nigra in vivo. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Benson, M.J.; Manzanero, S.; Borges, K. Complex alterations in microglial M1/M2 markers during the development of epilepsy in two mouse models. Epilepsia 2015, 56, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Vogel, D.Y.S.; Vereyken, E.J.F.; Glim, J.E.; Heijnen, P.D.A.M.; Moeton, M.; van der Valk, P.; Amor, S.; Teunissen, C.E.; van Horssen, J.; Dijkstra, C.D. Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. J. Neuroinflamm. 2013, 10, 809. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.; Tourtellotte, W.W.; Rudick, R.; Trapp, B.D. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N. Engl. J. Med. 2002, 346, 165–173. [Google Scholar] [CrossRef]
- Kim, M.J.; Kang, J.H.; Theotokis, P.; Grigoriadis, N.; Petratos, S. Can We Design a Nogo Receptor-Dependent Cellular Therapy to Target MS? Cells 2018, 8, 1. [Google Scholar] [CrossRef]
- Fang, Y.; Wang, J.; Yao, L.; Li, C.; Wang, J.; Liu, Y.; Tao, X.; Sun, H.; Liao, H. The adhesion and migration of microglia to β-amyloid (Aβ) is decreased with aging and inhibited by Nogo/NgR pathway. J. Neuroinflamm. 2018, 15, 210. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Qin, X.; Sun, H.; He, M.; Lv, Q.; Gao, C.; He, X.; Liao, H. Nogo receptor impairs the clearance of fibril amyloid-β by microglia and accelerates Alzheimer’s-like disease progression. Aging Cell 2021, 20, e13515. [Google Scholar] [CrossRef] [PubMed]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Andjelkovic, A.V.; Nikolic, B.; Pachter, J.S.; Zecevic, N. Macrophages/microglial cells in human central nervous system during development: An immunohistochemical study. Brain Res. 1998, 814, 13–25. [Google Scholar] [CrossRef]
- Monier, A.; Evrard, P.; Gressens, P.; Verney, C. Distribution and differentiation of microglia in the human encephalon during the first two trimesters of gestation. J. Comp. Neurol. 2006, 499, 565–582. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler Mark, F.; Conway Simon, J.; Ng Lai, G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals that Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsters, C.M.; Nesan, D.; Far, R.; Klenin, N.; Pittman, Q.J.; Kurrasch, D.M. Embryonic microglia influence developing hypothalamic glial populations. J. Neuroinflamm. 2020, 17, 146. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.L.; Martínez-Cerdeño, V.; Noctor, S.C. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J. Neurosci 2013, 33, 4216–4233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, C.A.; Popescu, A.S.; Kitchener, E.J.A.; Allendorf, D.H.; Puigdellívol, M.; Brown, G.C. Microglial phagocytosis of neurons in neurodegeneration, and its regulation. J. Neurochem. 2021, 158, 621–639. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [Green Version]
- McMorris, F.A.; Dubois-Dalcq, M. Insulin-like growth factor I promotes cell proliferation and oligodendroglial commitment in rat glial progenitor cells developing in vitro. J. Neurosci. Res. 1988, 21, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Zöller, T.; Schneider, A.; Kleimeyer, C.; Masuda, T.; Potru, P.S.; Pfeifer, D.; Blank, T.; Prinz, M.; Spittau, B. Silencing of TGFβ signalling in microglia results in impaired homeostasis. Nat. Commun. 2018, 9, 4011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueno, M.; Fujita, Y.; Tanaka, T.; Nakamura, Y.; Kikuta, J.; Ishii, M.; Yamashita, T. Layer V cortical neurons require microglial support for survival during postnatal development. Nat. Neurosci. 2013, 16, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Dermitzakis, I.; Manthou, M.E.; Meditskou, S.; Miliaras, D.; Kesidou, E.; Boziki, M.; Petratos, S.; Grigoriadis, N.; Theotokis, P. Developmental Cues and Molecular Drivers in Myelinogenesis: Revisiting Early Life to Re-Evaluate the Integrity of CNS Myelin. Curr. Issues Mol. Biol. 2022, 44, 3208–3237. [Google Scholar] [CrossRef]
- Shigemoto-Mogami, Y.; Hoshikawa, K.; Goldman, J.E.; Sekino, Y.; Sato, K. Microglia Enhance Neurogenesis and Oligodendrogenesis in the Early Postnatal Subventricular Zone. J. Neurosci. 2014, 34, 2231–2243. [Google Scholar] [CrossRef] [Green Version]
- Sherafat, A.; Pfeiffer, F.; Reiss, A.M.; Wood, W.M.; Nishiyama, A. Microglial neuropilin-1 promotes oligodendrocyte expansion during development and remyelination by trans-activating platelet-derived growth factor receptor. Nat. Commun. 2021, 12, 2265. [Google Scholar] [CrossRef]
- Matarredona, E.R.; Talaverón, R.; Pastor, A.M. Interactions Between Neural Progenitor Cells and Microglia in the Subventricular Zone: Physiological Implications in the Neurogenic Niche and After Implantation in the Injured Brain. Front. Cell. Neurosci. 2018, 12, 268. [Google Scholar] [CrossRef]
- Talaverón, R.; Matarredona, E.R.; de la Cruz, R.R.; Macías, D.; Gálvez, V.; Pastor, A.M. Implanted neural progenitor cells regulate glial reaction to brain injury and establish gap junctions with host glial cells. Glia 2014, 62, 623–638. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grajchen, E.; Hendriks, J.J.A.; Bogie, J.F.J. The physiology of foamy phagocytes in multiple sclerosis. Acta Neuropathol. Commun. 2018, 6, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Askew, K.; Li, K.; Olmos-Alonso, A.; Garcia-Moreno, F.; Liang, Y.; Richardson, P.; Tipton, T.; Chapman, M.A.; Riecken, K.; Beccari, S.; et al. Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Rep. 2017, 18, 391–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Réu, P.; Khosravi, A.; Bernard, S.; Mold, J.E.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Druid, H.; et al. The Lifespan and Turnover of Microglia in the Human Brain. Cell Rep. 2017, 20, 779–784. [Google Scholar] [CrossRef] [Green Version]
- Moraga, A.; Pradillo, J.M.; García-Culebras, A.; Palma-Tortosa, S.; Ballesteros, I.; Hernández-Jiménez, M.; Moro, M.A.; Lizasoain, I. Aging increases microglial proliferation, delays cell migration, and decreases cortical neurogenesis after focal cerebral ischemia. J. Neuroinflamm. 2015, 12, 87. [Google Scholar] [CrossRef] [Green Version]
- Gebara, E.; Sultan, S.; Kocher-Braissant, J.; Toni, N. Adult hippocampal neurogenesis inversely correlates with microglia in conditions of voluntary running and aging. Front. Neurosci. 2013, 7, 145. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, M.; Zettel, M.L.; Ison, J.R.; Allen, P.D.; Majewska, A.K. Effects of aging and sensory loss on glial cells in mouse visual and auditory cortices. Glia 2012, 60, 541–558. [Google Scholar] [CrossRef] [Green Version]
- Ioannides, Z.A.; Csurhes, P.A.; Swayne, A.; Foubert, P.; Aftab, B.T.; Pender, M.P. Correlations between macrophage/microglial activation marker sTREM-2 and measures of T-cell activation, neuroaxonal damage and disease severity in multiple sclerosis. Mult. Scler. J.-Exp. Transl. Clin. 2021, 7, 20552173211019772. [Google Scholar] [CrossRef]
- Prineas, J.W.; Parratt, J.D.E. Multiple Sclerosis: Microglia, Monocytes, and Macrophage-Mediated Demyelination. J. Neuropathol. Exp. Neurol. 2021, 80, 975–996. [Google Scholar] [CrossRef]
- Nack, A.; Brendel, M.; Nedelcu, J.; Daerr, M.; Nyamoya, S.; Beyer, C.; Focke, C.; Deussing, M.; Hoornaert, C.; Ponsaerts, P.; et al. Expression of Translocator Protein and [18F]-GE180 Ligand Uptake in Multiple Sclerosis Animal Models. Cells 2019, 8, 94. [Google Scholar] [CrossRef]
- van Wageningen, T.A.; Vlaar, E.; Kooij, G.; Jongenelen, C.A.M.; Geurts, J.J.G.; van Dam, A.-M. Regulation of microglial TMEM119 and P2RY12 immunoreactivity in multiple sclerosis white and grey matter lesions is dependent on their inflammatory environment. Acta Neuropathol. Commun. 2019, 7, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zrzavy, T.; Hametner, S.; Wimmer, I.; Butovsky, O.; Weiner, H.L.; Lassmann, H. Loss of ‘homeostatic’ microglia and patterns of their activation in active multiple sclerosis. Brain 2017, 140, 1900–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peferoen, L.A.N.; Vogel, D.Y.S.; Ummenthum, K.; Breur, M.; Heijnen, P.D.A.M.; Gerritsen, W.H.; Peferoen-Baert, R.M.B.; van der Valk, P.; Dijkstra, C.D.; Amor, S. Activation Status of Human Microglia Is Dependent on Lesion Formation Stage and Remyelination in Multiple Sclerosis. J. Neuropathol. Exp. Neurol. 2015, 74, 48–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagan, N.; Kane, J.L.; Grover, D.; Woodworth, L.; Madore, C.; Saleh, J.; Sancho, J.; Liu, J.; Li, Y.; Proto, J.; et al. CSF1R signaling is a regulator of pathogenesis in progressive MS. Cell Death Dis. 2020, 11, 904. [Google Scholar] [CrossRef]
- Benedek, G.; Zhang, J.; Nguyen, H.; Kent, G.; Seifert, H.; Vandenbark, A.A.; Offner, H. Novel feedback loop between M2 macrophages/microglia and regulatory B cells in estrogen-protected EAE mice. J. Neuroimmunol. 2017, 305, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ma, J.; Ding, G.; Gong, Q.; Wang, Y.; Yu, H.; Cheng, X. Microglia Polarization from M1 toward M2 Phenotype Is Promoted by Astragalus Polysaccharides Mediated through Inhibition of miR-155 in Experimental Autoimmune Encephalomyelitis. Oxidative Med. Cell. Longev. 2021, 2021, 5753452. [Google Scholar] [CrossRef]
- Nissen, J.C.; Thompson, K.K.; West, B.L.; Tsirka, S.E. Csf1R inhibition attenuates experimental autoimmune encephalomyelitis and promotes recovery. Exp. Neurol. 2018, 307, 24–36. [Google Scholar] [CrossRef]
- Ponomarev, E.D.; Shriver, L.P.; Dittel, B.N. CD40 expression by microglial cells is required for their completion of a two-step activation process during central nervous system autoimmune inflammation. J. Immunol. 2006, 176, 1402–1410. [Google Scholar] [CrossRef] [Green Version]
- Ponomarev, E.D.; Shriver, L.P.; Maresz, K.; Dittel, B.N. Microglial cell activation and proliferation precedes the onset of CNS autoimmunity. J. Neurosci. Res. 2005, 81, 374–389. [Google Scholar] [CrossRef]
- Miron, V.E.; Boyd, A.; Zhao, J.-W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.M.; et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef]
- Zhang, Z.-J.; Zheng, X.-X.; Zhang, X.-Y.; Zhang, Y.; Huang, B.-Y.; Luo, T. Aging alters Hv1-mediated microglial polarization and enhances neuroinflammation after peripheral surgery. CNS Neurosci. Ther. 2020, 26, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Aryanpour, R.; Zibara, K.; Pasbakhsh, P.; Jame’ei, S.B.; Namjoo, Z.; Ghanbari, A.; Mahmoudi, R.; Amani, S.; Kashani, I.R. 17 β-Estradiol Reduces Demyelination in Cuprizone-fed Mice by Promoting M2 Microglia Polarity and Regulating NLRP3 Inflammasome. Neuroscience 2021, 463, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.; Golebiewska, A.; Poovathingal, S.K.; Kaoma, T.; Pires-Afonso, Y.; Martina, S.; Coowar, D.; Azuaje, F.; Skupin, A.; Balling, R.; et al. Single-cell transcriptomics reveals distinct inflammation-induced microglia signatures. EMBO Rep. 2018, 19, e46171. [Google Scholar] [CrossRef]
- Janova, H.; Böttcher, C.; Holtman, I.R.; Regen, T.; van Rossum, D.; Götz, A.; Ernst, A.S.; Fritsche, C.; Gertig, U.; Saiepour, N.; et al. CD14 is a key organizer of microglial responses to CNS infection and injury. Glia 2016, 64, 635–649. [Google Scholar] [CrossRef]
- Bennett Mariko, L.; Bennett, F.C.; Liddelow Shane, A.; Ajami, B.; Zamanian Jennifer, L.; Fernhoff Nathaniel, B.; Mulinyawe Sara, B.; Bohlen Christopher, J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, J.; Kino, Y.; Asahina, N.; Takitani, M.; Miyoshi, J.; Ishida, T.; Saito, Y. TMEM119 marks a subset of microglia in the human brain. Neuropathology 2016, 36, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Gómez Morillas, A.; Besson, V.C.; Lerouet, D. Microglia and Neuroinflammation: What Place for P2RY12? Int. J. Mol. Sci. 2021, 22, 1636. [Google Scholar] [CrossRef]
- Kenkhuis, B.; Somarakis, A.; Kleindouwel, L.R.T.; van Roon-Mom, W.M.C.; Höllt, T.; van der Weerd, L. Co-expression patterns of microglia markers Iba1, TMEM119 and P2RY12 in Alzheimer’s disease. Neurobiol. Dis. 2022, 167, 105684. [Google Scholar] [CrossRef]
- Gudi, V.; Gingele, S.; Skripuletz, T.; Stangel, M. Glial response during cuprizone-induced de- and remyelination in the CNS: Lessons learned. Front. Cell. Neurosci. 2014, 8, 73. [Google Scholar] [CrossRef] [Green Version]
- Vankriekelsvenne, E.; Chrzanowski, U.; Manzhula, K.; Greiner, T.; Wree, A.; Hawlitschka, A.; Llovera, G.; Zhan, J.; Joost, S.; Schmitz, C.; et al. Transmembrane protein 119 is neither a specific nor a reliable marker for microglia. Glia 2022, 70, 1170–1190. [Google Scholar] [CrossRef]
- Gensel, J.C.; Kopper, T.J.; Zhang, B.; Orr, M.B.; Bailey, W.M. Predictive screening of M1 and M2 macrophages reveals the immunomodulatory effectiveness of post spinal cord injury azithromycin treatment. Sci. Rep. 2017, 7, 40144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Absinta, M.; Maric, D.; Gharagozloo, M.; Garton, T.; Smith, M.D.; Jin, J.; Fitzgerald, K.C.; Song, A.; Liu, P.; Lin, J.-P.; et al. A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis. Nature 2021, 597, 709–714. [Google Scholar] [CrossRef] [PubMed]
- van Olst, L.; Rodriguez-Mogeda, C.; Picon, C.; Kiljan, S.; James, R.E.; Kamermans, A.; van der Pol, S.M.A.; Knoop, L.; Michailidou, I.; Drost, E.; et al. Meningeal inflammation in multiple sclerosis induces phenotypic changes in cortical microglia that differentially associate with neurodegeneration. Acta Neuropathol. 2021, 141, 881–899. [Google Scholar] [CrossRef] [PubMed]
- Ulland, T.K.; Song, W.M.; Huang, S.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663.e13. [Google Scholar] [CrossRef] [PubMed]
- Nugent, A.A.; Lin, K.; van Lengerich, B.; Lianoglou, S.; Przybyla, L.; Davis, S.S.; Llapashtica, C.; Wang, J.; Kim, D.J.; Xia, D.; et al. TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge. Neuron 2020, 105, 837–854.e9. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Song, N.; Jiang, H.; Wang, J.; Xie, J. Pro-inflammatory cytokines modulate iron regulatory protein 1 expression and iron transportation through reactive oxygen/nitrogen species production in ventral mesencephalic neurons. Biochim. Biophys. Acta 2013, 1832, 618–625. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, N.; Chiba, Y.; Hosono, M.; Fujii, M.; Kawamura, N.; Keino, H.; Yoshikawa, K.; Ishii, S.; Saitoh, Y.; Satoh, M.; et al. Involvement of pro-inflammatory cytokines and microglia in an age-associated neurodegeneration model, the SAMP10 mouse. Brain Res. 2007, 1185, 75–85. [Google Scholar] [CrossRef]
- Thomas, A.; Lehn, M.; Janssen, E.; Hildeman, D.; Chougnet, C. Naturally-aged microglia exhibit phagocytic dysfunction accompanied by gene expression changes reflective of underlying neurologic disease. Sci. Rep. 2022, 12, 19471. [Google Scholar] [CrossRef]
- Roser, A.-E.; Tönges, L.; Lingor, P. Modulation of Microglial Activity by Rho-Kinase (ROCK) Inhibition as Therapeutic Strategy in Parkinson’s Disease and Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2017, 9, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tischer, J.; Krueger, M.; Mueller, W.; Staszewski, O.; Prinz, M.; Streit, W.J.; Bechmann, I. Inhomogeneous distribution of Iba-1 characterizes microglial pathology in Alzheimer’s disease. Glia 2016, 64, 1562–1572. [Google Scholar] [CrossRef]
- Hefendehl, J.K.; Neher, J.J.; Sühs, R.B.; Kohsaka, S.; Skodras, A.; Jucker, M. Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell 2014, 13, 60–69. [Google Scholar] [CrossRef]
- Yanguas-Casás, N.; Crespo-Castrillo, A.; Arevalo, M.A.; Garcia-Segura, L.M. Aging and sex: Impact on microglia phagocytosis. Aging Cell 2020, 19, e13182. [Google Scholar] [CrossRef] [PubMed]
- Sim, F.J.; Zhao, C.; Penderis, J.; Franklin, R.J.M. The Age-Related Decrease in CNS Remyelination Efficiency Is Attributable to an Impairment of Both Oligodendrocyte Progenitor Recruitment and Differentiation. J. Neurosci. 2002, 22, 2451. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Li, W.-W.; Franklin, R.J.M. Differences in the early inflammatory responses to toxin-induced demyelination are associated with the age-related decline in CNS remyelination. Neurobiol. Aging 2006, 27, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Giaccone, G.; Orsi, L.; Cupidi, C.; Tagliavini, F. Lipofuscin Hypothesis of Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. Extra 2011, 1, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Jiang, H.; Xu, H.; Song, N.; Xie, J. Increased iron levels correlate with the selective nigral dopaminergic neuron degeneration in Parkinson’s disease. J. Neural Transm. 2011, 118, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Cantuti-Castelvetri, L.; Fitzner, D.; Bosch-Queralt, M.; Weil, M.-T.; Su, M.; Sen, P.; Ruhwedel, T.; Mitkovski, M.; Trendelenburg, G.; Lütjohann, D.; et al. Defective cholesterol clearance limits remyelination in the aged central nervous system. Science 2018, 359, 684–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, M.R.P.; Hohsfield, L.A.; Kramár, E.A.; Soreq, L.; Lee, R.J.; Pham, S.T.; Najafi, A.R.; Spangenberg, E.E.; Wood, M.A.; West, B.L.; et al. Replacement of microglia in the aged brain reverses cognitive, synaptic, and neuronal deficits in mice. Aging Cell 2018, 17, e12832. [Google Scholar] [CrossRef] [Green Version]
- O’Neil, S.M.; Witcher, K.G.; McKim, D.B.; Godbout, J.P. Forced turnover of aged microglia induces an intermediate phenotype but does not rebalance CNS environmental cues driving priming to immune challenge. Acta Neuropathol. Commun. 2018, 6, 129. [Google Scholar] [CrossRef]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Baillie, J.K.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 2016, 19, 504–516. [Google Scholar] [CrossRef]
- Frank, M.G.; Barrientos, R.M.; Biedenkapp, J.C.; Rudy, J.W.; Watkins, L.R.; Maier, S.F. mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiol. Aging 2006, 27, 717–722. [Google Scholar] [CrossRef]
- Chitnis, T.; Imitola, J.; Wang, Y.; Elyaman, W.; Chawla, P.; Sharuk, M.; Raddassi, K.; Bronson, R.T.; Khoury, S.J. Elevated neuronal expression of CD200 protects Wlds mice from inflammation-mediated neurodegeneration. Am. J. Pathol. 2007, 170, 1695–1712. [Google Scholar] [CrossRef] [Green Version]
- Watson, A.E.S.; de Almeida, M.M.A.; Dittmann, N.L.; Li, Y.; Torabi, P.; Footz, T.; Vetere, G.; Galleguillos, D.; Sipione, S.; Cardona, A.E.; et al. Fractalkine signaling regulates oligodendroglial cell genesis from SVZ precursor cells. Stem Cell Rep. 2021, 16, 1968–1984. [Google Scholar] [CrossRef] [PubMed]
- Wynne, A.M.; Henry, C.J.; Huang, Y.; Cleland, A.; Godbout, J.P. Protracted downregulation of CX3CR1 on microglia of aged mice after lipopolysaccharide challenge. Brain Behav. Immun. 2010, 24, 1190–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, F.F.; Carney, D.; Miller, A.-M.; Lynch, M.A. CD200 fusion protein decreases microglial activation in the hippocampus of aged rats. Brain Behav. Immun. 2012, 26, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Böttcher, C.; Amann, L.; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; Coenen, V.A. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef]
- Fenn, A.M.; Henry, C.J.; Huang, Y.; Dugan, A.; Godbout, J.P. Lipopolysaccharide-induced interleukin (IL)-4 receptor-α expression and corresponding sensitivity to the M2 promoting effects of IL-4 are impaired in microglia of aged mice. Brain Behav. Immun. 2012, 26, 766–777. [Google Scholar] [CrossRef] [Green Version]
- Caldeira, C.; Oliveira, A.F.; Cunha, C.; Vaz, A.R.; Falcão, A.S.; Fernandes, A.; Brites, D. Microglia change from a reactive to an age-like phenotype with the time in culture. Front. Cell. Neurosci. 2014, 8, 152. [Google Scholar] [CrossRef]
- Van Guilder, H.D.; Bixler, G.V.; Brucklacher, R.M.; Farley, J.A.; Yan, H.; Warrington, J.P.; Sonntag, W.E.; Freeman, W.M. Concurrent hippocampal induction of MHC II pathway components and glial activation with advanced aging is not correlated with cognitive impairment. J. Neuroinflamm. 2011, 8, 138. [Google Scholar] [CrossRef] [Green Version]
- Norden, D.M.; Godbout, J.P. Review: Microglia of the aged brain: Primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 2013, 39, 19–34. [Google Scholar] [CrossRef]
- Kuhlmann, T.; Ludwin, S.; Prat, A.; Antel, J.; Brück, W.; Lassmann, H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. 2017, 133, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Frischer, J.M.; Weigand, S.D.; Guo, Y.; Kale, N.; Parisi, J.E.; Pirko, I.; Mandrekar, J.; Bramow, S.; Metz, I.; Brück, W.; et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann. Neurol. 2015, 78, 710–721. [Google Scholar] [CrossRef] [Green Version]
- Magliozzi, R.; Howell, O.; Vora, A.; Serafini, B.; Nicholas, R.; Puopolo, M.; Reynolds, R.; Aloisi, F. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007, 130 Pt 4, 1089–1104. [Google Scholar] [CrossRef]
- Lovato, L.; Willis, S.N.; Rodig, S.J.; Caron, T.; Almendinger, S.E.; Howell, O.W.; Reynolds, R.; O’Connor, K.C.; Hafler, D.A. Related B cell clones populate the meninges and parenchyma of patients with multiple sclerosis. Brain 2011, 134 Pt 2, 534–541. [Google Scholar] [CrossRef]
- Howell, O.W.; Reeves, C.A.; Nicholas, R.; Carassiti, D.; Radotra, B.; Gentleman, S.M.; Serafini, B.; Aloisi, F.; Roncaroli, F.; Magliozzi, R.; et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 2011, 134 Pt 9, 2755–2771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touil, H.; Li, R.; Moore, C.; Antel, J.P.; Bar-Or, A. Cross-talk between human glial cells and B cells help propagation of CNS-compartmentalized in progressive MS. J. Immunol. 2017, 198 (Suppl. 1), 132–135. [Google Scholar]
- Bellver-Landete, V.; Bretheau, F.; Mailhot, B.; Vallières, N.; Lessard, M.; Janelle, M.E.; Vernoux, N.; Tremblay, M.; Fuehrmann, T.; Shoichet, M.S.; et al. Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat. Commun. 2019, 10, 518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanner, I.B.; Anderson, M.A.; Song, B.; Levine, J.; Fernandez, A.; Gray-Thompson, Z.; Ao, Y.; Sofroniew, M.V. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J. Neurosci. 2013, 33, 12870–12886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, E.G.; Kang, S.H.; Fukaya, M.; Bergles, D.E. Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat. Neurosci. 2013, 16, 668–676. [Google Scholar] [CrossRef] [Green Version]
- Voskuhl, R.R.; Peterson, R.S.; Song, B.; Ao, Y.; Morales, L.B.; Tiwari-Woodruff, S.; Sofroniew, M.V. Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J. Neurosci. 2009, 29, 11511–11522. [Google Scholar] [CrossRef] [Green Version]
- Lau, L.W.; Keough, M.B.; Haylock-Jacobs, S.; Cua, R.; Döring, A.; Sloka, S.; Stirling, D.P.; Rivest, S.; Yong, V.W. Chondroitin sulfate proteoglycans in demyelinated lesions impair remyelination. Ann. Neurol. 2012, 72, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Siebert, J.R.; Conta Steencken, A.; Osterhout, D.J. Chondroitin sulfate proteoglycans in the nervous system: Inhibitors to repair. Biomed Res. Int. 2014, 2014, 845323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geloso, M.C.; D’Ambrosi, N. Microglial Pruning: Relevance for Synaptic Dysfunction in Multiple Sclerosis and Related Experimental Models. Cells 2021, 10, 686. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.S.; Clarke, L.E.; Wang, G.X.; Stafford, B.K.; Sher, A.; Chakraborty, C.; Joung, J.; Foo, L.C.; Thompson, A.; Chen, C.; et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 2013, 504, 394–400. [Google Scholar] [CrossRef] [Green Version]
- Miron, V.E. Microglia-driven regulation of oligodendrocyte lineage cells, myelination, and remyelination. J. Leukoc. Biol. 2017, 101, 1103–1108. [Google Scholar] [CrossRef] [Green Version]
- Dehghan, S.; Aref, E.; Raoufy, M.R.; Javan, M. An optimized animal model of lysolecithin induced demyelination in optic nerve; more feasible, more reproducible, promising for studying the progressive forms of multiple sclerosis. J. Neurosci. Methods 2021, 352, 109088. [Google Scholar] [CrossRef]
- Rawji, K.S.; Kappen, J.; Tang, W.; Teo, W.; Plemel, J.R.; Stys, P.K.; Yong, V.W. Deficient Surveillance and Phagocytic Activity of Myeloid Cells Within Demyelinated Lesions in Aging Mice Visualized by Ex Vivo Live Multiphoton Imaging. J. Neurosci. 2018, 38, 1973–1988. [Google Scholar] [CrossRef] [Green Version]
- Lampron, A.; Larochelle, A.; Laflamme, N.; Préfontaine, P.; Plante, M.-M.; Sánchez, M.G.; Yong, V.W.; Stys, P.K.; Tremblay, M.-È.; Rivest, S. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J. Exp. Med. 2015, 212, 481–495. [Google Scholar] [CrossRef] [Green Version]
- Gyoneva, S.; Hosur, R.; Gosselin, D.; Zhang, B.; Ouyang, Z.; Cotleur, A.C.; Peterson, M.; Allaire, N.; Challa, R.; Cullen, P.; et al. Cx3cr1-deficient microglia exhibit a premature aging transcriptome. Life Sci. Alliance 2019, 2, e201900453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaels, N.J.; Lemmon, K.; Plemel, J.R.; Jensen, S.K.; Mishra, M.K.; Brown, D.; Rawji, K.S.; Koch, M.; Yong, V.W. Aging-Exacerbated Acute Axon and Myelin Injury Is Associated with Microglia-Derived Reactive Oxygen Species and Is Alleviated by the Generic Medication Indapamide. J. Neurosci. 2020, 40, 8587. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Stout, R.D.; Jiang, C.; Matta, B.; Tietzel, I.; Watkins, S.K.; Suttles, J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J. Immunol. 2005, 175, 342–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chávez-Galán, L.; Olleros, M.L.; Vesin, D.; Garcia, I. Much More than M1 and M2 Macrophages, There are also CD169 (+) and TCR (+) Macrophages. Front. Immunol. 2015, 6, 263. [Google Scholar]
- Chuang, Y.; Hung, M.E.; Cangelose, B.K.; Leonard, J.N. Regulation of the IL-10-driven macrophage phenotype under incoherent stimuli. Innate Immun. 2016, 22, 647–657. [Google Scholar] [CrossRef] [Green Version]
- Dhakal, M.; Hardaway, J.C.; Guloglu, F.B.; Miller, M.M.; Hoeman, C.M.; Zaghouani, A.A.; Wan, X.; Rowland, L.M.; Cascio, J.A.; Sherman, M.P.; et al. IL-13Rα1 is a surface marker for M2 macrophages influencing their differentiation and function. Eur. J. Immunol. 2014, 44, 842–855. [Google Scholar] [CrossRef] [Green Version]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage m1–m2 polarization balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Bronte, V. Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Investig. 2007, 117, 1155–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peferoen, L.; Kipp, M.; van der Valk, P.; van Noort, J.M.; Amor, S. Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology 2014, 141, 302–313. [Google Scholar] [CrossRef]
- Mikita, J.; Dubourdieu-Cassagno, N.; Deloire, M.S.; Vekris, A.; Biran, M.; Raffard, G.; Brochet, B.; Canron, M.-H.; Franconi, J.-M.; Boiziau, C. Altered M1/M2 activation patterns of monocytes in severe relapsing experimental rat model of multiple sclerosis. Amelioration of clinical status by M2 activated monocyte administration. Mult. Scler. J. 2011, 17, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Kotter, M.R.; Zhao, C.; van Rooijen, N.; Franklin, R.J. Macrophage-depletion induced impairment of experimental CNS remyelination is associated with a reduced oligodendrocyte progenitor cell response and altered growth factor expression. Neurobiol. Dis. 2005, 18, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Huber, J.D.; Egleton, R.D.; Davis, T.P. Molecular physiology and pathophysiology of tight junctions in the blood-brain barrier. Trends Neurosci. 2001, 24, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Schmandke, A.; Schmandke, A.; Schwab, M.E. Nogo-A: Multiple Roles in CNS Development, Maintenance, and Disease. Neuroscientist 2014, 20, 372–386. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Kim, J.A.; Sivasankaran, R.; Segal, R.; He, Z. P75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature 2002, 420, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Petratos, S. Multiple Sclerosis: Does Nogo Play a Role? Neurosci. A Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2013, 19, 394–408. [Google Scholar] [CrossRef]
- Fournier, A.E.; GrandPre, T.; Strittmatter, S.M. Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature 2001, 409, 341–346. [Google Scholar] [CrossRef]
- Filbin, M.T. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat. Rev. Neurosci. 2003, 4, 703–713. [Google Scholar] [CrossRef] [Green Version]
- Mi, S.; Lee, X.; Shao, Z.; Thill, G.; Ji, B.; Relton, J.; Levesque, M.; Allaire, N.; Perrin, S.; Sands, B.; et al. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat. Neurosci. 2004, 7, 221–228. [Google Scholar] [CrossRef]
- Park, J.B.; Yiu, G.; Kaneko, S.; Wang, J.; Chang, J.; He, X.L.; Garcia, K.C.; He, Z. A TNF receptor family member, TROY, is a coreceptor with Nogo receptor in mediating the inhibitory activity of myelin inhibitors. Neuron 2005, 45, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Gil, V.; Nicolas, O.; Mingorance, A.; Ureña, J.M.; Tang, B.L.; Hirata, T.; Sáez-Valero, J.; Ferrer, I.; Soriano, E.; del Río, J.A. Nogo-A Expression in the Human Hippocampus in Normal Aging and in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2006, 65, 433–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, J.-I.; Onoue, H.; Arima, K.; Yamamura, T. Nogo-A and Nogo Receptor Expression in Demyelinating Lesions of Multiple Sclerosis. J. Neuropathol. Exp. Neurol. 2005, 64, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Satoh, J.; Tabunoki, H.; Yamamura, T.; Arima, K.; Konno, H. TROY and LINGO-1 expression in astrocytes and macrophages/microglia in multiple sclerosis lesions. Neuropathol. Appl. Neurobiol. 2007, 33, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Petratos, S.; Ozturk, E.; Azari, M.F.; Kenny, R.; Lee, J.Y.; Magee, K.A.; Harvey, A.R.; McDonald, C.; Taghian, K.; Moussa, L.; et al. Limiting multiple sclerosis related axonopathy by blocking Nogo receptor and CRMP-2 phosphorylation. Brain A J. Neurol. 2012, 135 Pt 6, 1794–1818. [Google Scholar] [CrossRef] [PubMed]
- Grandpré, T.; Strittmatter, S.M. Nogo: A molecular determinant of axonal growth and regeneration. Neuroscientist 2001, 7, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chun, S.-J.; Treloar, H.; Vartanian, T.; Greer, C.A.; Strittmatter, S.M. Localization of Nogo-A and Nogo-66 receptor proteins at sites of axon-myelin and synaptic contact. J. Neurosci. 2002, 22, 5505–5515. [Google Scholar] [CrossRef] [Green Version]
- Dupuis, L.; Gonzalez de Aguilar, J.L.; di Scala, F.; Rene, F.; de Tapia, M.; Pradat, P.F.; Lacomblez, L.; Seihlan, D.; Prinjha, R.; Walsh, F.S.; et al. Nogo provides a molecular marker for diagnosis of amyotrophic lateral sclerosis. Neurobiol. Dis. 2002, 10, 358–365. [Google Scholar] [CrossRef] [Green Version]
- Jokic, N.; Gonzalez de Aguilar, J.-L.; Dimou, L.; Lin, S.; Fergani, A.; Ruegg, M.A.; Schwab, M.E.; Dupuis, L.; Loeffler, J.-P. The neurite outgrowth inhibitor Nogo-A promotes denervation in an amyotrophic lateral sclerosis model. EMBO Rep. 2006, 7, 1162–1167. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.S.; Harel, N.Y.; Strittmatter, S.M. Reticulon-4A (Nogo-A) redistributes protein disulfide isomerase to protect mice from SOD1-dependent amyotrophic lateral sclerosis. J. Neurosci. 2009, 29, 13850–13859. [Google Scholar] [CrossRef] [Green Version]
- Liebscher, T.; Schnell, L.; Schnell, D.; Scholl, J.; Schneider, R.; Gullo, M.; Fouad, K.; Mir, A.; Rausch, M.; Kindler, D.; et al. Nogo-A antibody improves regeneration and locomotion of spinal cord-injured rats. Ann. Neurol. 2005, 58, 706–719. [Google Scholar] [CrossRef] [PubMed]
- Zörner, B.; Schwab, M.E. Anti-Nogo on the go: From animal models to a clinical trial. Ann. N. Y. Acad. Sci. 2010, 1198 (Suppl. 1), E22–E34. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Kim, J.E.; Sivula, M.; Strittmatter, S.M. Nogo receptor antagonism promotes stroke recovery by enhancing axonal plasticity. J. Neurosci. 2004, 24, 6209–6217. [Google Scholar] [CrossRef] [Green Version]
- Wiessner, C.; Bareyre, F.M.; Allegrini, P.R.; Mir, A.K.; Frentzel, S.; Zurini, M.; Schnell, L.; Oertle, T.; Schwab, M.E. Anti-Nogo-A antibody infusion 24 h after experimental stroke improved behavioral outcome and corticospinal plasticity in normotensive and spontaneously hypertensive rats. J. Cereb. Blood Flow Metab. 2003, 23, 154–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rust, R.; Grönnert, L.; Gantner, C.; Enzler, A.; Mulders, G.; Weber, R.Z.; Siewert, A.; Limasale, Y.D.P.; Meinhardt, A.; Maurer, M.A.; et al. Nogo-A targeted therapy promotes vascular repair and functional recovery following stroke. Proc. Natl. Acad. Sci. USA 2019, 116, 14270–14279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Yan, J.; Li, C.; Zhou, X.; Yao, L.; Pang, T.; Yan, M.; Zhang, L.; Mao, L.; Liao, H. The Nogo/Nogo Receptor (NgR) Signal Is Involved in Neuroinflammation through the Regulation of Microglial Inflammatory Activation. J. Biol. Chem. 2015, 290, 28901–28914. [Google Scholar] [CrossRef] [Green Version]
- Theotokis, P.; Touloumi, O.; Lagoudaki, R.; Nousiopoulou, E.; Kesidou, E.; Siafis, S.; Tselios, T.; Lourbopoulos, A.; Karacostas, D.; Grigoriadis, N.; et al. Nogo receptor complex expression dynamics in the inflammatory foci of central nervous system experimental autoimmune demyelination. J. Neuroinflamm. 2016, 13, 265. [Google Scholar] [CrossRef] [Green Version]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. 2), 136–153. [Google Scholar] [CrossRef] [Green Version]
- Ridley, A.J.; Paterson, H.F.; Johnston, C.L.; Diekmann, D.; Hall, A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 1992, 70, 401–410. [Google Scholar] [CrossRef]
- Kozma, R.; Ahmed, S.; Best, A.; Lim, L. The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol. Cell Biol. 1995, 15, 1942–1952. [Google Scholar] [CrossRef] [Green Version]
- Jiang, R.; Wu, X.-F.; Wang, B.; Guan, R.-X.; Lv, L.-M.; Li, A.-P.; Lei, L.; Ma, Y.; Li, N.; Li, Q.-F.; et al. Reduction of NgR in perforant path decreases amyloid-β peptide production and ameliorates synaptic and cognitive deficits in APP/PS1 mice. Alzheimer’s Res. Ther. 2020, 12, 47. [Google Scholar] [CrossRef]
- Yan, J.; Zhou, X.; Guo, J.-J.; Mao, L.; Wang, Y.-J.; Sun, J.; Sun, L.-X.; Zhang, L.-Y.; Zhou, X.-F.; Liao, H. Nogo-66 inhibits adhesion and migration of microglia via GTPase Rho pathway in vitro. J. Neurochem. 2012, 120, 721–731. [Google Scholar] [CrossRef]
- Lindborg, J.A.; Tran, N.M.; Chenette, D.M.; DeLuca, K.; Foli, Y.; Kannan, R.; Sekine, Y.; Wang, X.; Wollan, M.; Kim, I.-J.; et al. Optic nerve regeneration screen identifies multiple genes restricting adult neural repair. Cell Rep. 2021, 34, 108777. [Google Scholar] [CrossRef]
- Liu, G.; Ni, J.; Mao, L.; Yan, M.; Pang, T.; Liao, H. Expression of Nogo receptor 1 in microglia during development and following traumatic brain injury. Brain Res. 2015, 1627, 41–51. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, T.; Maynard, G.D.; Terse, P.S.; Cafferty, W.B.; Kocsis, J.D.; Strittmatter, S.M. Nogo receptor decoy promotes recovery and corticospinal growth in non-human primate spinal cord injury. Brain 2020, 143, 1697–1713. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Shen, Q.; Xu, P.; Luo, J.J.; Tang, Y. Phagocytosis of Microglia in the Central Nervous System Diseases. Mol. Neurobiol. 2014, 49, 1422–1434. [Google Scholar] [CrossRef] [Green Version]
- Healy, L.M.; Perron, G.; Won, S.-Y.; Michell-Robinson, M.A.; Rezk, A.; Ludwin, S.K.; Moore, C.S.; Hall, J.A.; Bar-Or, A.; Antel, J.P. MerTK Is a Functional Regulator of Myelin Phagocytosis by Human Myeloid Cells. J. Immunol. 2016, 196, 1502562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brück, W.; Friede, R.L. Anti-macrophage CR3 antibody blocks myelin phagocytosis by macrophages in vitro. Acta Neuropathol. 1990, 80, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Reichert, F.; Rotshenker, S. Complement-receptor-3 and scavenger-receptor-AI/II mediated myelin phagocytosis in microglia and macrophages. Neurobiol. Dis. 2003, 12, 65–72. [Google Scholar] [CrossRef]
- Cignarella, F.; Filipello, F.; Bollman, B.; Cantoni, C.; Locca, A.; Mikesell, R.; Manis, M.; Ibrahim, A.; Deng, L.; Benitez, B.A.; et al. TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathol. 2020, 140, 513–534. [Google Scholar] [CrossRef]
- Gitik, M.; Reichert, F.; Rotshenker, S. Cytoskeleton plays a dual role of activation and inhibition in myelin and zymosan phagocytosis by microglia. FASEB J. 2010, 24, 2211–2221. [Google Scholar] [CrossRef]
- Scheiblich, H.; Bicker, G. Regulation of Microglial Phagocytosis by RhoA/ROCK-Inhibiting Drugs. Cell Mol. Neurobiol. 2017, 37, 461–473. [Google Scholar] [CrossRef]
- Durafourt, B.A.; Moore, C.S.; Zammit, D.A.; Johnson, T.A.; Zaguia, F.; Guiot, M.C.; Bar-Or, A.; Antel, J.P. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia 2012, 60, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, A.; David, S. Differences in the Phagocytic Response of Microglia and Peripheral Macrophages after Spinal Cord Injury and Its Effects on Cell Death. J. Neurosci. 2014, 34, 6316–6322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, D.; Yang, N.; Liu, Y.Y.; Zheng, J.; Ji, C.; Zuo, P.P. M2 Macrophage Transplantation Ameliorates Cognitive Dysfunction in Amyloid-β-Treated Rats Through Regulation of Microglial Polarization. J. Alzheimers Dis. 2016, 52, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Gimbel, D.A.; GrandPre, T.; Lee, J.-K.; Kim, J.-E.; Li, W.; Lee, D.H.S.; Strittmatter, S.M. Alzheimer precursor protein interaction with the Nogo-66 receptor reduces amyloid-beta plaque deposition. J. Neurosci. 2006, 26, 1386–1395. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Yao, L.; Li, C.; Wang, J.; Wang, J.; Chen, S.; Zhou, X.-F.; Liao, H. The blockage of the Nogo/NgR signal pathway in microglia alleviates the formation of Aβ plaques and tau phosphorylation in APP/PS1 transgenic mice. J. Neuroinflamm. 2016, 13, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Perez, A.I.; Borrajo, A.; Rodriguez-Pallares, J.; Guerra, M.J.; Labandeira-Garcia, J.L. Interaction between NADPH-oxidase and Rho-kinase in angiotensin II-induced microglial activation. Glia 2015, 63, 466–482. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Chen, X.; Wang, L.-Y.; Gao, W.; Zhu, M.-J. Rho Kinase Inhibitor Fasudil Protects against β-Amyloid-Induced Hippocampal Neurodegeneration in Rats. CNS Neurosci. Ther. 2013, 19, 603–610. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Y.; Yu, J.; Guo, M.; Meng, J.; Liu, C.; Xie, Y.; Feng, L.; Xiao, B.; Ma, C. Rho Kinase Inhibitor Fasudil Regulates Microglia Polarization and Function. Neuroimmunomodulation 2013, 20, 313–322. [Google Scholar] [CrossRef]
- Huang, M.; Li, Y.; Wu, K.; Yan, W.; Tian, T.; Wang, Y.; Yang, H. Paraquat modulates microglia M1/M2 polarization via activation of TLR4-mediated NF-κB signaling pathway. Chem.-Biol. Interact. 2019, 310, 108743. [Google Scholar] [CrossRef]
- Chen, C.; Li, Y.H.; Zhang, Q.; Yu, J.Z.; Zhao, Y.F.; Ma, C.G.; Xiao, B.G. Fasudil regulates T cell responses through polarization of BV-2 cells in mice experimental autoimmune encephalomyelitis. Acta Pharm. Sin. 2014, 35, 1428–1438. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wei, Y.-Z.; Wang, G.-Q.; Li, D.-D.; Shi, J.-S.; Zhang, F. Targeting MAPK Pathways by Naringenin Modulates Microglia M1/M2 Polarization in Lipopolysaccharide-Stimulated Cultures. Front. Cell. Neurosci. 2019, 12, 531. [Google Scholar] [CrossRef]
- Zheng, Z.V.; Chen, J.; Lyu, H.; Lam, S.Y.E.; Lu, G.; Chan, W.Y.; Wong, G.K.C. Novel role of STAT3 in microglia-dependent neuroinflammation after experimental subarachnoid haemorrhage. Stroke Vasc. Neurol. 2022, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Komohara, Y.; Horlad, H.; Ohnishi, K.; Ohta, K.; Makino, K.; Hondo, H.; Yamanaka, R.; Kajiwara, K.; Saito, T.; Kuratsu, J.; et al. M2 macrophage/microglial cells induce activation of Stat3 in primary central nervous system lymphoma. J. Clin. Exp. Hematop. 2011, 51, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Butturini, E.; Boriero, D.; Carcereri de Prati, A.; Mariotto, S. STAT1 drives M1 microglia activation and neuroinflammation under hypoxia. Arch. Biochem. Biophys. 2019, 669, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Tugal, D.; Liao, X.; Jain, M.K. Transcriptional control of macrophage polarization. Arter. Thromb Vasc. Biol. 2013, 33, 1135–1144. [Google Scholar] [CrossRef] [Green Version]
- Ghasemi, N.; Razavi, S.; Nikzad, E. Multiple Sclerosis: Pathogenesis, Symptoms, Diagnoses and Cell-Based Therapy. Cell J. 2017, 19, 1–10. [Google Scholar] [PubMed]
- Scalfari, A.; Knappertz, V.; Cutter, G.; Goodin, D.S.; Ashton, R.; Ebers, G.C. Mortality in patients with multiple sclerosis. Neurology 2013, 81, 184–192. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Markers | ||||
|---|---|---|---|---|
| Homeostatic Microglia | Classically Activated Microglia | Alternatively Active Microglia | Ref | |
| Multiple sclerosis | TMEM119 CX3CR1 P2RY12 Iba1 * CSF1R | MRP14+ * TSPO * MHC II * CD68+ * CD74 * CD40 * CD86 * CD80 * | CD206 * TREM2 CD163 * CD309 * CCL22 * | [48,49,50,51,52,53,54] |
| Animal models | ||||
| Models that include adaptive immune mechanisms (e.g., EAE) | TMEM119 P2RY12 Iba1 * CX3CR1 CD11b+/CD45− CSF1R CD68− | CD74 * CD40 * CD86 * CD80 * iNOS * TNF * IL-12 * | CD206 * TREM2 FIZZ1 * CD163 * Chil3 * Arginase 1 * | [53,55,56,57,58,59] |
| Innate immune toxin models (e.g., Cuprizone, lysolecithin) | TSPO * CD86 iNOS * MHC II * CCL2 * TNF * CD83 CD14 | CD206 * Arginase 1 * TREM2 MRC1 | [60,61,62,63,64] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nheu, D.; Ellen, O.; Ye, S.; Ozturk, E.; Pagnin, M.; Kertadjaja, S.; Theotokis, P.; Grigoriadis, N.; McLean, C.; Petratos, S. Modulation of the Microglial Nogo-A/NgR Signaling Pathway as a Therapeutic Target for Multiple Sclerosis. Cells 2022, 11, 3768. https://doi.org/10.3390/cells11233768
Nheu D, Ellen O, Ye S, Ozturk E, Pagnin M, Kertadjaja S, Theotokis P, Grigoriadis N, McLean C, Petratos S. Modulation of the Microglial Nogo-A/NgR Signaling Pathway as a Therapeutic Target for Multiple Sclerosis. Cells. 2022; 11(23):3768. https://doi.org/10.3390/cells11233768
Chicago/Turabian StyleNheu, Danica, Olivia Ellen, Sining Ye, Ezgi Ozturk, Maurice Pagnin, Stephen Kertadjaja, Paschalis Theotokis, Nikolaos Grigoriadis, Catriona McLean, and Steven Petratos. 2022. "Modulation of the Microglial Nogo-A/NgR Signaling Pathway as a Therapeutic Target for Multiple Sclerosis" Cells 11, no. 23: 3768. https://doi.org/10.3390/cells11233768
APA StyleNheu, D., Ellen, O., Ye, S., Ozturk, E., Pagnin, M., Kertadjaja, S., Theotokis, P., Grigoriadis, N., McLean, C., & Petratos, S. (2022). Modulation of the Microglial Nogo-A/NgR Signaling Pathway as a Therapeutic Target for Multiple Sclerosis. Cells, 11(23), 3768. https://doi.org/10.3390/cells11233768