
Dynamic Changes in the Extracellular Matrix in Primary, Metastatic, and Recurrent Ovarian Cancers
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
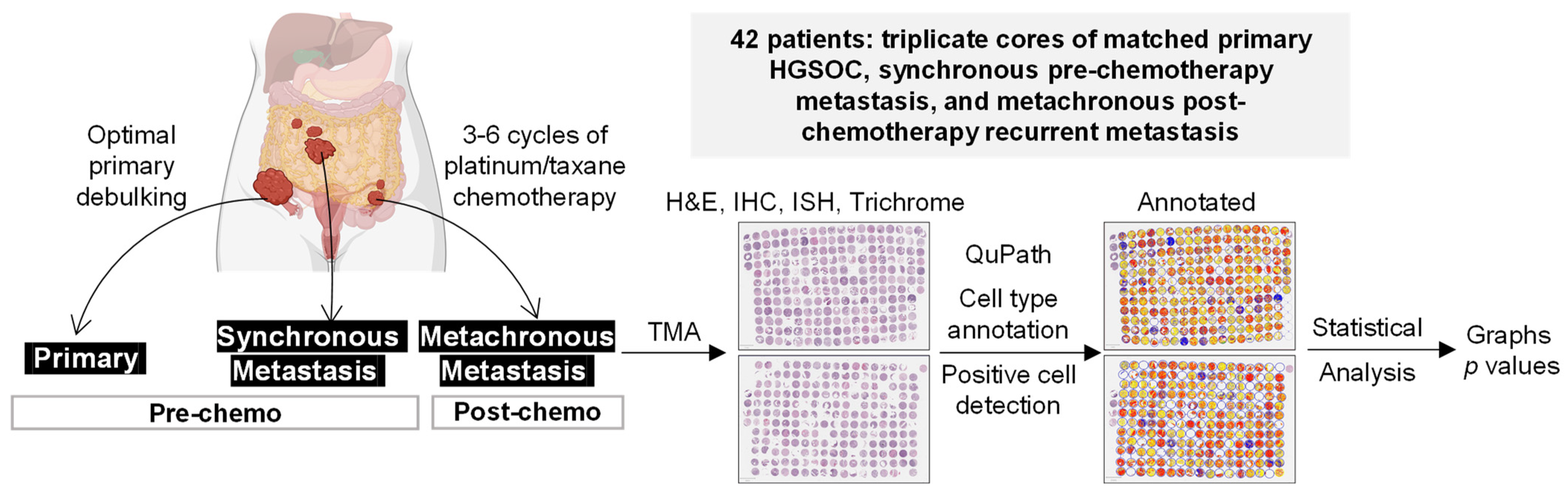
2. Materials and Methods
2.1. Tissue Microarray (TMA)
2.2. Immunohistochemistry (IHC), In Situ Hybridization (ISH), Masson’s Trichrome Staining, and Hematoxylin and Eosin (H&E) Staining
2.3. Cell Type Annotation by Computational Image Analysis
2.4. Masson’s Trichrome Image Feature Extraction and Analysis
2.5. Identification and Validation of Computational Image Features Associated with COL11A1 Positivity
2.6. Statistical Analyses
3. Results
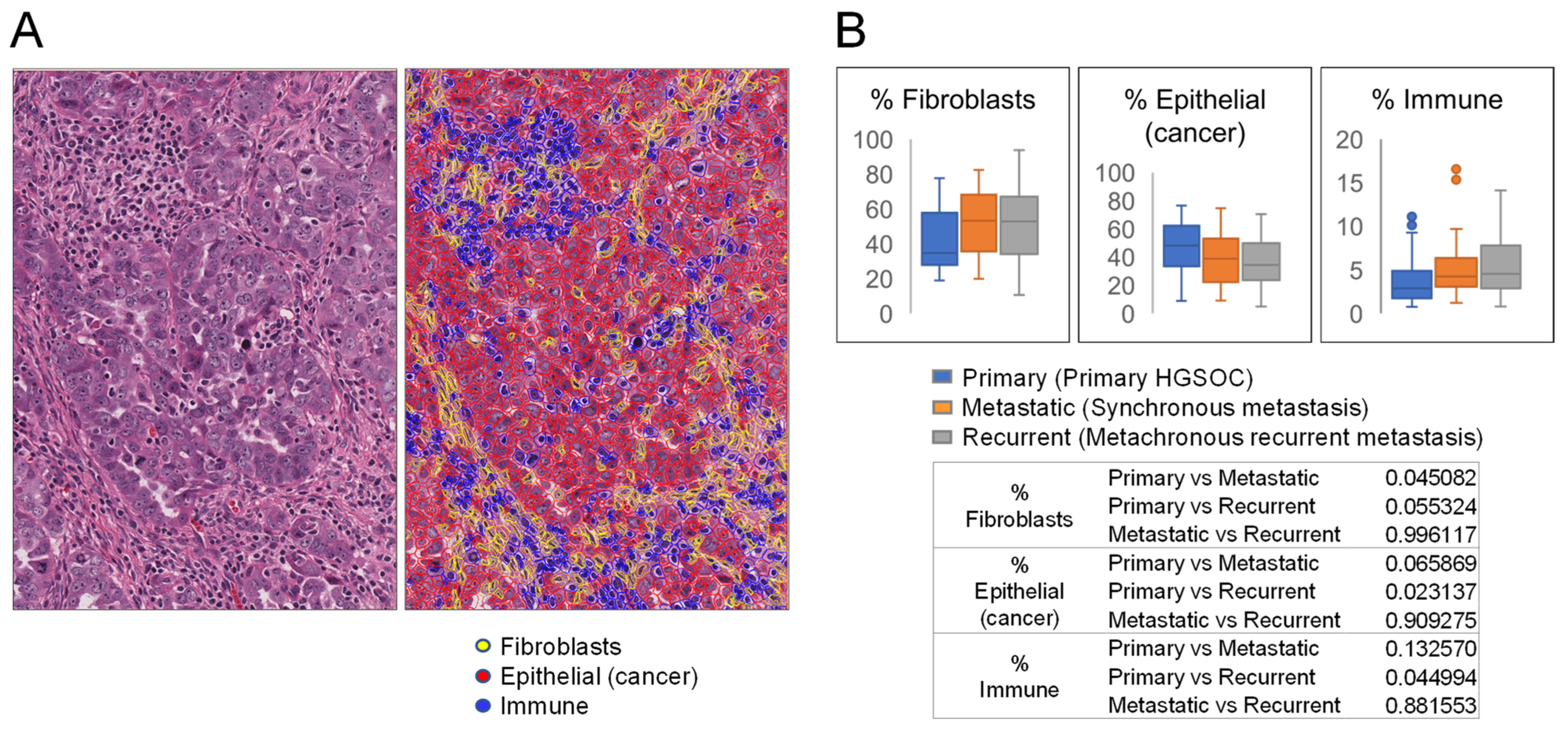
3.1. Primary Tumors and Metastases Have Different Percentages of Fibroblasts, Epithelial Cancer Cells, and Immune Cells
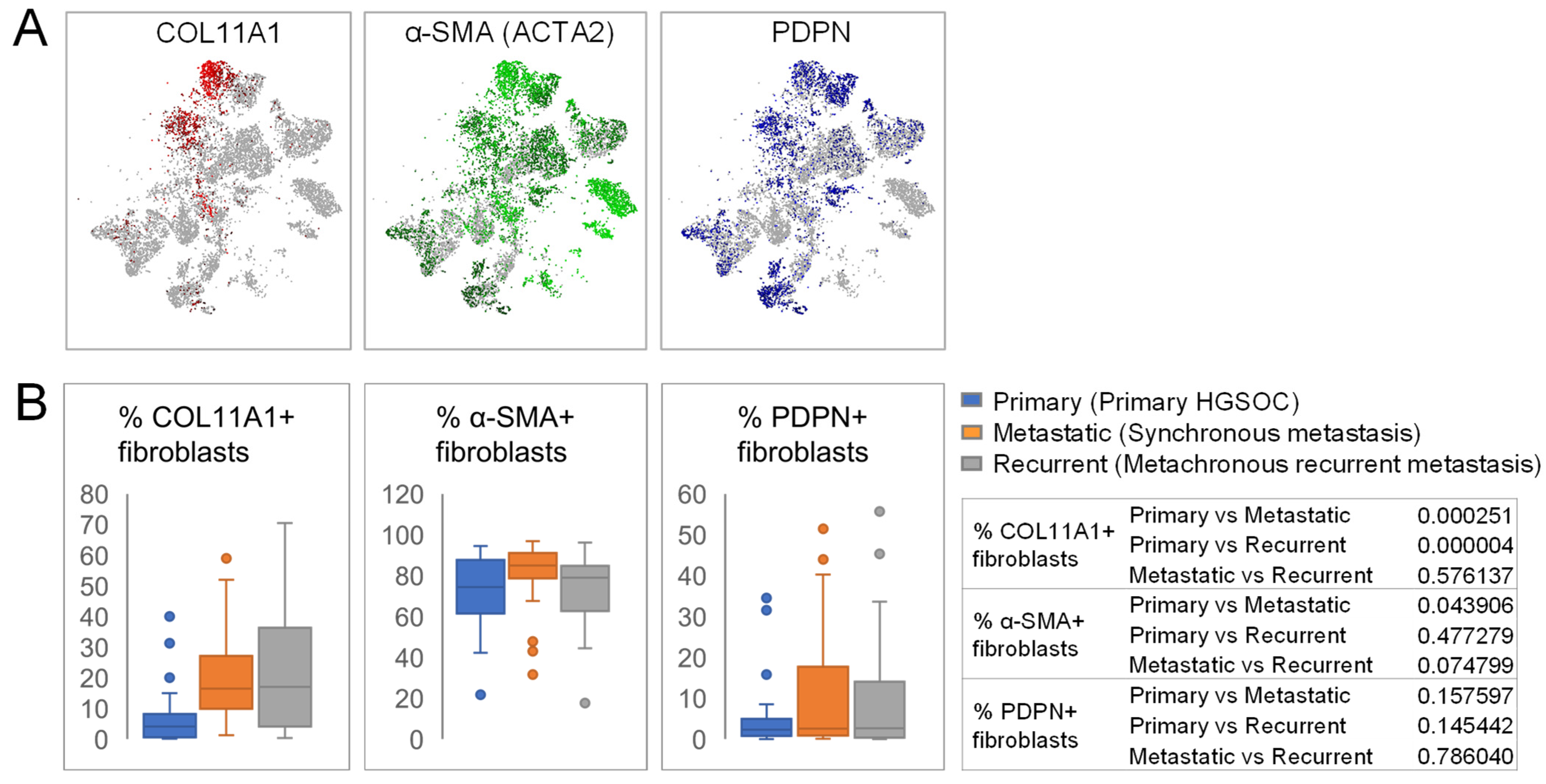
3.2. COL11A1+, α-SMA+, and PDPN+ CAF Subsets Show Differential Distribution during HGSOC Progression
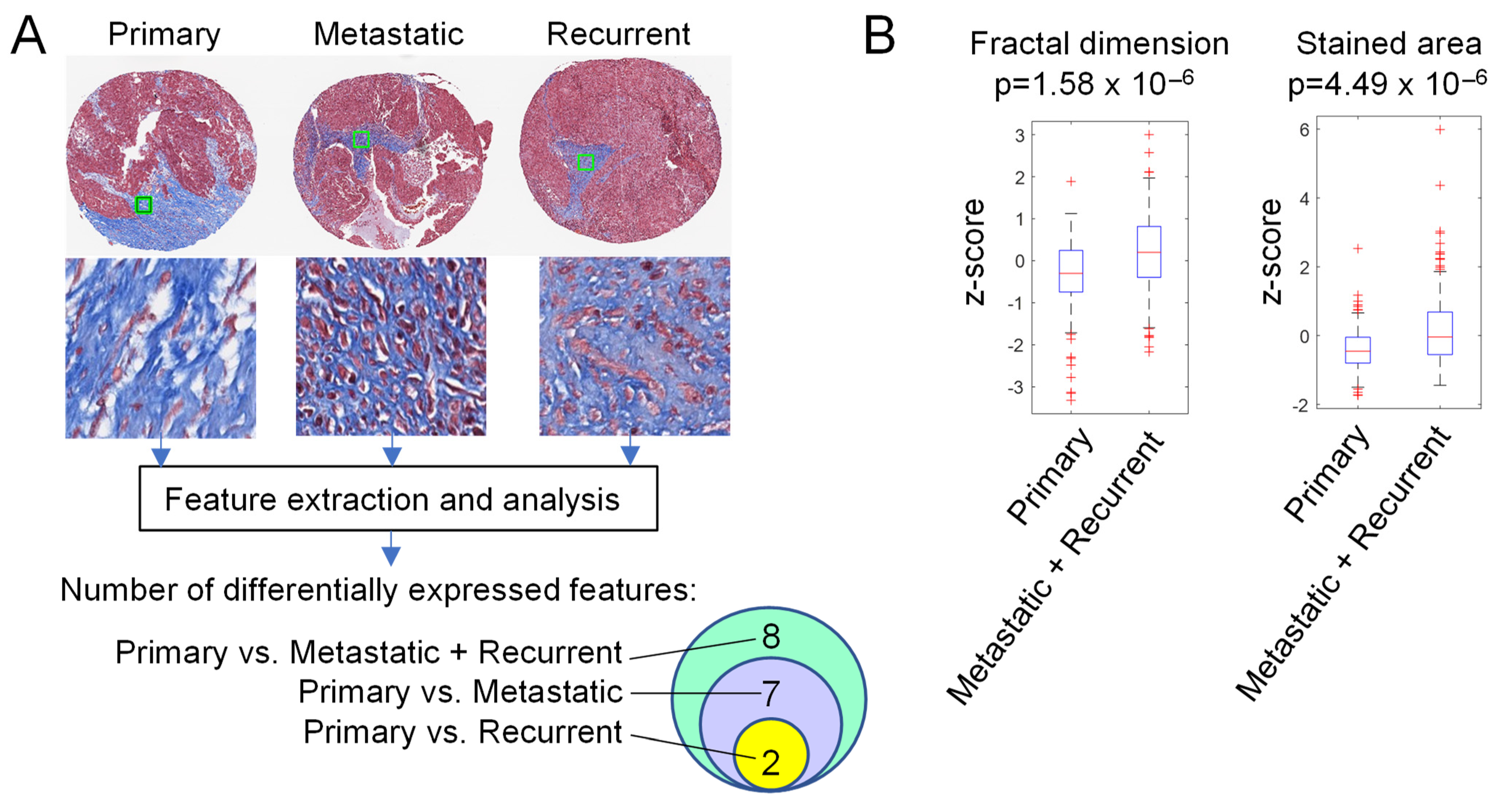
3.3. Extracellular Matrix Texture and Pattern Differ in Primary HGSOC and Metastases
3.4. ECM Texture and Pattern Are Altered in Tumor Areas Positive for COL11A1
3.5. HGSOC Metastases Have an Increased CD8+ T Cell Infiltration, However, Not All CD8+ T Cells Are Reaching the Tumor Parenchyma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Desbois, M.; Udyavar, A.R.; Ryner, L.; Kozlowski, C.; Guan, Y.; Dürrbaum, M.; Lu, S.; Fortin, J.P.; Koeppen, H.; Ziai, J.; et al. Integrated digital pathology and transcriptome analysis identifies molecular mediators of T-cell exclusion in ovarian cancer. Nat. Commun. 2020, 11, 5583. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel Iii, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Canellas, A.; Hernando-Momblona, X.; et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grauel, A.L.; Nguyen, B.; Ruddy, D.; Laszewski, T.; Schwartz, S.; Chang, J.; Chen, J.; Piquet, M.; Pelletier, M.; Yan, Z.; et al. TGFβ-blockade uncovers stromal plasticity in tumors by revealing the existence of a subset of interferon-licensed fibroblasts. Nat. Commun. 2020, 11, 6315. [Google Scholar] [CrossRef]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Bowtell, D.D.; Bohm, S.; Ahmed, A.A.; Aspuria, P.J.; Bast, R.C., Jr.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [Green Version]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Hegde, P.S.; Clynes, R.; Foukas, P.G.; Harari, A.; Kleen, T.O.; Kvistborg, P.; Maccalli, C.; Maecker, H.T.; Page, D.B.; et al. Novel technologies and emerging biomarkers for personalized cancer immunotherapy. J. Immunother. Cancer 2016, 4, 3. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Kryczek, I.; Dostal, L.; Lin, H.; Tan, L.; Zhao, L.; Lu, F.; Wei, S.; Maj, T.; Peng, D.; et al. Effector T Cells Abrogate Stroma-Mediated Chemoresistance in Ovarian Cancer. Cell 2016, 165, 1092–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fucikova, J.; Coosemans, A.; Orsulic, S.; Cibula, D.; Vergote, I.; Galluzzi, L.; Spisek, R. Immunological configuration of ovarian carcinoma: Features and impact on disease outcome. J. Immunother. Cancer 2021, 9, e002873. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernandez, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [Green Version]
- Ruifrok, A.C.; Johnston, D.A. Quantification of histochemical staining by color deconvolution. Anal. Quant. Cytol. Histol. 2001, 23, 291–299. [Google Scholar]
- Wu, J.; Raz, Y.; Recouvreux, M.S.; Diniz, M.A.; Lester, J.; Karlan, B.Y.; Walts, A.E.; Gertych, A.; Orsulic, S. Focal Serous Tubal Intra-Epithelial Carcinoma Lesions Are Associated with Global Changes in the Fallopian Tube Epithelia and Stroma. Front. Oncol. 2022, 12, 853755. [Google Scholar] [CrossRef]
- Miller, E.; Salemi, H.; Klimov, S.; Lewis, M.; Garraway, I.P.; Knudsen, B.S.; Gertych, A. PD71-09 Quantitative digital image analysis and machine learning accurately classifies primary prostate tumors of bone metastatic disease based on histomorphometric features in diagnostic prostate needle biopsies. J. Urol. 2017, 197, e1358–e1359. [Google Scholar] [CrossRef]
- Costa, A.F.; Humpire-Mamani, G.; Traina, A.J.M. An Efficient Algorithm for Fractal Analysis of Textures. In Proceedings of the 2012 25th SIBGRAPI Conference on Graphics, Patterns and Images, Ouro Preto, Brazil, 22–25 August 2012; pp. 39–46. [Google Scholar]
- Fogel, I.; Sagi, D. Gabor filters as texture discriminator. Biol. Cybern. 1989, 61, 103–113. [Google Scholar] [CrossRef]
- Jia, D.; Liu, Z.; Deng, N.; Tan, T.Z.; Huang, R.Y.; Taylor-Harding, B.; Cheon, D.J.; Lawrenson, K.; Wiedemeyer, W.R.; Walts, A.E.; et al. A COL11A1-correlated pan-cancer gene signature of activated fibroblasts for the prioritization of therapeutic targets. Cancer Lett. 2016, 382, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Cheon, D.J.; Tong, Y.; Sim, M.S.; Dering, J.; Berel, D.; Cui, X.; Lester, J.; Beach, J.A.; Tighiouart, M.; Walts, A.E.; et al. A collagen-remodeling gene signature regulated by TGF-beta signaling is associated with metastasis and poor survival in serous ovarian cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Pula, B.; Witkiewicz, W.; Dziegiel, P.; Podhorska-Okolow, M. Significance of podoplanin expression in cancer-associated fibroblasts: A comprehensive review. Int. J. Oncol. 2013, 42, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Olbrecht, S.; Boeckx, B.; Vos, H.; Laoui, D.; Etlioglu, E.; Wauters, E.; Pomella, V.; Verbandt, S.; Busschaert, P.; et al. A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res. 2020, 30, 745–762. [Google Scholar] [CrossRef] [PubMed]
- Gelse, K.; Poschl, E.; Aigner, T. Collagens--structure, function, and biosynthesis. Adv. Drug Deliv. Rev. 2003, 55, 1531–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thom, J.R.; Morris, N.P. Biosynthesis and proteolytic processing of type XI collagen in embryonic chick sterna. J. Biol. Chem. 1991, 266, 7262–7269. [Google Scholar] [CrossRef] [PubMed]
- Birk, D.E.; Fitch, J.M.; Babiarz, J.P.; Doane, K.J.; Linsenmayer, T.F. Collagen fibrillogenesis in vitro: Interaction of types I and V collagen regulates fibril diameter. J. Cell Sci. 1990, 95, 649–657. [Google Scholar] [CrossRef]
- Marchant, J.K.; Hahn, R.A.; Linsenmayer, T.F.; Birk, D.E. Reduction of type V collagen using a dominant-negative strategy alters the regulation of fibrillogenesis and results in the loss of corneal-specific fibril morphology. J. Cell Biol. 1996, 135, 1415–1426. [Google Scholar] [CrossRef]
- Davies, G.B.; Oxford, J.T.; Hausafus, L.C.; Smoody, B.F.; Morris, N.P. Temporal and spatial expression of alternative splice-forms of the alpha1(XI) collagen gene in fetal rat cartilage. Dev. Dyn. 1998, 213, 12–26. [Google Scholar] [CrossRef]
- Gregory, K.E.; Oxford, J.T.; Chen, Y.; Gambee, J.E.; Gygi, S.P.; Aebersold, R.; Neame, P.J.; Mechling, D.E.; Bachinger, H.P.; Morris, N.P. Structural organization of distinct domains within the non-collagenous N-terminal region of collagen type XI. J. Biol. Chem. 2000, 275, 11498–11506. [Google Scholar] [CrossRef] [Green Version]
- Van der Rest, M.; Garrone, R. Collagen family of proteins. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1991, 5, 2814–2823. [Google Scholar] [CrossRef] [Green Version]
- Linsenmayer, T.F.; Gibney, E.; Igoe, F.; Gordon, M.K.; Fitch, J.M.; Fessler, L.I.; Birk, D.E. Type V collagen: Molecular structure and fibrillar organization of the chicken alpha 1(V) NH2-terminal domain, a putative regulator of corneal fibrillogenesis. J. Cell Biol. 1993, 121, 1181–1189. [Google Scholar] [CrossRef] [Green Version]
- Dongre, H.; Costea, D.E. Tumor-Fibroblast Interactions in Carcinomas. In Biomarkers of the Tumor Microenvironment; Akslen, L.A., Watnick, R.S., Eds.; Springer International Publishing: Cham, Switzerland, 2022; pp. 109–124. [Google Scholar] [CrossRef]
- Conklin, M.W.; Eickhoff, J.C.; Riching, K.M.; Pehlke, C.A.; Eliceiri, K.W.; Provenzano, P.P.; Friedl, A.; Keely, P.J. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am. J. Pathol. 2011, 178, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Arnold, T.D.; Katamura, Y.; Giacomini, M.M.; Rodriguez, J.D.; McCarty, J.H.; Pellicoro, A.; Raschperger, E.; Betsholtz, C.; Ruminski, P.G.; et al. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 2013, 19, 1617–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamanishi, J.; Mandai, M.; Iwasaki, M.; Okazaki, T.; Tanaka, Y.; Yamaguchi, K.; Higuchi, T.; Yagi, H.; Takakura, K.; Minato, N.; et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 3360–3365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milne, K.; Kobel, M.; Kalloger, S.E.; Barnes, R.O.; Gao, D.; Gilks, C.B.; Watson, P.H.; Nelson, B.H. Systematic analysis of immune infiltrates in high-grade serous ovarian cancer reveals CD20, FoxP3 and TIA-1 as positive prognostic factors. PLoS ONE 2009, 4, e6412. [Google Scholar] [CrossRef] [Green Version]
- Hwang, W.T.; Adams, S.F.; Tahirovic, E.; Hagemann, I.S.; Coukos, G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2012, 124, 192–198. [Google Scholar] [CrossRef] [Green Version]
- Hamanishi, J.; Mandai, M.; Ikeda, T.; Minami, M.; Kawaguchi, A.; Murayama, T.; Kanai, M.; Mori, Y.; Matsumoto, S.; Chikuma, S.; et al. Safety and antitumor activity of anti-PD-1 antibody, Nivolumab, in patients with platinum-resistant ovarian cancer. J. Clin. Oncol. 2015, 33, 4015–4022. [Google Scholar] [CrossRef]
- Callahan, M.J.; Nagymanyoki, Z.; Bonome, T.; Johnson, M.E.; Litkouhi, B.; Sullivan, E.H.; Hirsch, M.S.; Matulonis, U.A.; Liu, J.; Birrer, M.J.; et al. Increased HLA-DMB expression in the tumor epithelium is associated with increased CTL infiltration and improved prognosis in advanced-stage serous ovarian cancer. Clin. Cancer Res. 2008, 14, 7667–7673. [Google Scholar] [CrossRef] [Green Version]
- Han, L.Y.; Fletcher, M.S.; Urbauer, D.L.; Mueller, P.; Landen, C.N.; Kamat, A.A.; Lin, Y.G.; Merritt, W.M.; Spannuth, W.A.; Deavers, M.T.; et al. HLA class I antigen processing machinery component expression and intratumoral T-cell infiltrate as independent prognostic markers in ovarian carcinoma. Clin. Cancer Res. 2008, 14, 3372–3379. [Google Scholar] [CrossRef]
- Clarke, B.; Tinker, A.V.; Lee, C.H.; Subramanian, S.; van de Rijn, M.; Turbin, D.; Kalloger, S.; Han, G.; Ceballos, K.; Cadungog, M.G.; et al. Intraepithelial T cells and prognosis in ovarian carcinoma: Novel associations with stage, tumor type, and BRCA1 loss. Mod. Pathol. 2009, 22, 393–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preston, C.C.; Maurer, M.J.; Oberg, A.L.; Visscher, D.W.; Kalli, K.R.; Hartmann, L.C.; Goode, E.L.; Knutson, K.L. The ratios of CD8+ T cells to CD4+CD25+ FOXP3+ and FOXP3- T cells correlate with poor clinical outcome in human serous ovarian cancer. PLoS ONE 2013, 8, e80063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goode, E.L.; Block, M.S.; Kalli, K.R.; Vierkant, R.A.; Chen, W.; Fogarty, Z.C.; Gentry-Maharaj, A.; Tołoczko, A.; Hein, A.; Bouligny, A.L.; et al. Dose-Response Association of CD8+ Tumor-Infiltrating Lymphocytes and Survival Time in High-Grade Serous Ovarian Cancer. JAMA Oncol. 2017, 3, e173290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeffler, C.M.L.; Ortiz Bruechle, N.; Jung, M.; Seillier, L.; Rose, M.; Laleh, N.G.; Knuechel, R.; Brinker, T.J.; Trautwein, C.; Gaisa, N.T.; et al. Artificial Intelligence-based Detection of FGFR3 Mutational Status Directly from Routine Histology in Bladder Cancer: A Possible Preselection for Molecular Testing? Eur. Urol. Focus 2021, 8, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Mlynska, A.; Vaišnorė, R.; Rafanavičius, V.; Jocys, S.; Janeiko, J.; Petrauskytė, M.; Bijeikis, S.; Cimmperman, P.; Intaitė, B.; Žilionytė, K.; et al. A gene signature for immune subtyping of desert, excluded, and inflamed ovarian tumors. Am. J. Reprod. Immunol. 2020, 84, e13244. [Google Scholar] [CrossRef] [PubMed]
- Kather, J.N.; Suarez-Carmona, M.; Charoentong, P.; Weis, C.A.; Hirsch, D.; Bankhead, P.; Horning, M.; Ferber, D.; Kel, I.; Herpel, E.; et al. Topography of cancer-associated immune cells in human solid tumors. eLife 2018, 7, e36967. [Google Scholar] [CrossRef]
- Pietilä, E.A.; Gonzalez-Molina, J.; Moyano-Galceran, L.; Jamalzadeh, S.; Zhang, K.; Lehtinen, L.; Turunen, S.P.; Martins, T.A.; Gultekin, O.; Lamminen, T.; et al. Co-evolution of matrisome and adaptive adhesion dynamics drives ovarian cancer chemoresistance. Nat. Commun. 2021, 12, 3904. [Google Scholar] [CrossRef]
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. J. Int. Soc. Matrix Biol. 2016, 49, 10–24. [Google Scholar] [CrossRef]
- Kreuzinger, C.; Geroldinger, A.; Smeets, D.; Braicu, E.I.; Sehouli, J.; Koller, J.; Wolf, A.; Darb-Esfahani, S.; Joehrens, K.; Vergote, I.; et al. A Complex Network of Tumor Microenvironment in Human High-Grade Serous Ovarian Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 7621–7632. [Google Scholar] [CrossRef] [Green Version]
- Westergaard, M.C.W.; Milne, K.; Pedersen, M.; Hasselager, T.; Olsen, L.R.; Anglesio, M.S.; Borch, T.H.; Kennedy, M.; Briggs, G.; Ledoux, S.; et al. Changes in the Tumor Immune Microenvironment during Disease Progression in Patients with Ovarian Cancer. Cancers 2020, 12, 3828. [Google Scholar] [CrossRef]
- Wang, J.; Cheng, F.H.C.; Tedrow, J.; Chang, W.; Zhang, C.; Mitra, A.K. Modulation of Immune Infiltration of Ovarian Cancer Tumor Microenvironment by Specific Subpopulations of Fibroblasts. Cancers 2020, 12, 3184. [Google Scholar] [CrossRef] [PubMed]
- Pearce, O.M.T.; Delaine-Smith, R.M.; Maniati, E.; Nichols, S.; Wang, J.; Böhm, S.; Rajeeve, V.; Ullah, D.; Chakravarty, P.; Jones, R.R.; et al. Deconstruction of a Metastatic Tumor Microenvironment Reveals a Common Matrix Response in Human Cancers. Cancer Discov. 2018, 8, 304–319. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [Green Version]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.G.; Tamayo, P.; Yang, J.Y.; Hubbard, D.; Zhang, H.; Creighton, C.J.; Fereday, S.; Lawrence, M.; Carter, S.L.; Mermel, C.H.; et al. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J. Clin. Investig. 2013, 123, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.Z.; Heong, V.; Ye, J.; Lim, D.; Low, J.; Choolani, M.; Scott, C.; Tan, D.S.P.; Huang, R.Y. Decoding transcriptomic intra-tumour heterogeneity to guide personalised medicine in ovarian cancer. J. Pathol. 2019, 247, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Lindemann, K.; Anderson, L.; Fereday, S.; Hung, J.; Alsop, K.; Tothill, R.W.; Gebski, V.; Kennedy, C.; Balleine, R.L.; et al. Serous ovarian and primary peritoneal cancers: A comparative analysis of clinico-pathological features, molecular subtypes and treatment outcome. Gynecol. Oncol. 2016, 142, 458–464. [Google Scholar] [CrossRef]
- Hu, Y.; Taylor-Harding, B.; Raz, Y.; Haro, M.; Recouvreux, M.S.; Taylan, E.; Lester, J.; Millstein, J.; Walts, A.E.; Karlan, B.Y.; et al. Are Epithelial Ovarian Cancers of the Mesenchymal Subtype Actually Intraperitoneal Metastases to the Ovary? Front. Cell Dev. Biol. 2020, 8, 647. [Google Scholar] [CrossRef]
- Eckert, M.A.; Pan, S.; Hernandez, K.M.; Loth, R.M.; Andrade, J.; Volchenboum, S.L.; Faber, P.; Montag, A.; Lastra, R.; Peter, M.E.; et al. Genomics of Ovarian Cancer Progression Reveals Diverse Metastatic Trajectories Including Intraepithelial Metastasis to the Fallopian Tube. Cancer Discov. 2016, 6, 1342–1351. [Google Scholar] [CrossRef] [Green Version]
- McPherson, A.; Roth, A.; Laks, E.; Masud, T.; Bashashati, A.; Zhang, A.W.; Ha, G.; Biele, J.; Yap, D.; Wan, A.; et al. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat. Genet. 2016, 48, 758–767. [Google Scholar] [CrossRef]
- Teng, P.N.; Wang, G.; Hood, B.L.; Conrads, K.A.; Hamilton, C.A.; Maxwell, G.L.; Darcy, K.M.; Conrads, T.P. Identification of candidate circulating cisplatin-resistant biomarkers from epithelial ovarian carcinoma cell secretomes. Br. J. Cancer 2014, 110, 123–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.H.; Chang, T.H.; Huang, Y.F.; Huang, H.D.; Chou, C.Y. COL11A1 promotes tumor progression and predicts poor clinical outcome in ovarian cancer. Oncogene 2014, 33, 3432–3440. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.H.; Chang, T.H.; Huang, Y.F.; Chen, C.C.; Chou, C.Y. COL11A1 confers chemoresistance on ovarian cancer cells through the activation of Akt/c/EBPbeta pathway and PDK1 stabilization. Oncotarget 2015, 6, 23748–23763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheon, D.J.; Orsulic, S. Ten-gene biomarker panel: A new hope for ovarian cancer? Biomark. Med. 2014, 8, 523–526. [Google Scholar] [CrossRef] [Green Version]
- Rada, M.; Nallanthighal, S.; Cha, J.; Ryan, K.; Sage, J.; Eldred, C.; Ullo, M.; Orsulic, S.; Cheon, D.J. Inhibitor of apoptosis proteins (IAPs) mediate collagen type XI alpha 1-driven cisplatin resistance in ovarian cancer. Oncogene 2018, 37, 4809–4820. [Google Scholar] [CrossRef]
- Boyd, N.F.; Martin, L.J.; Stone, J.; Greenberg, C.; Minkin, S.; Yaffe, M.J. Mammographic densities as a marker of human breast cancer risk and their use in chemoprevention. Curr. Oncol. Rep. 2001, 3, 314–321. [Google Scholar] [CrossRef]
- Egeblad, M.; Rasch, M.G.; Weaver, V.M. Dynamic interplay between the collagen scaffold and tumor evolution. Curr. Opin. Cell Biol. 2010, 22, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Ursin, G.; Hovanessian-Larsen, L.; Parisky, Y.R.; Pike, M.C.; Wu, A.H. Greatly increased occurrence of breast cancers in areas of mammographically dense tissue. Breast Cancer Res. BCR 2005, 7, R605–R608. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Bera, K.; Toro, P.; Fu, P.; Zhang, Z.; Lu, C.; Feldman, M.; Ganesan, S.; Goldstein, L.J.; Davidson, N.E.; et al. Collagen fiber orientation disorder from H&E images is prognostic for early stage breast cancer: Clinical trial validation. NPJ Breast Cancer 2021, 7, 104. [Google Scholar] [CrossRef]
- Adur, J.; Pelegati, V.B.; de Thomaz, A.A.; Baratti, M.O.; Andrade, L.A.; Carvalho, H.F.; Bottcher-Luiz, F.; Cesar, C.L. Second harmonic generation microscopy as a powerful diagnostic imaging modality for human ovarian cancer. J. Biophotonics 2014, 7, 37–48. [Google Scholar] [CrossRef]
- Pouli, D.; Genega, E.M.; Sullivan, T.B.; Rieger-Christ, K.M.; Wright, V.; Georgakoudi, I.; Schnelldorfer, T. Two-photon images reveal unique texture features for label-free identification of ovarian cancer peritoneal metastases. Biomed. Opt. Express 2019, 10, 4479–4488. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gertych, A.; Walts, A.E.; Cheng, K.; Liu, M.; John, J.; Lester, J.; Karlan, B.Y.; Orsulic, S. Dynamic Changes in the Extracellular Matrix in Primary, Metastatic, and Recurrent Ovarian Cancers. Cells 2022, 11, 3769. https://doi.org/10.3390/cells11233769
Gertych A, Walts AE, Cheng K, Liu M, John J, Lester J, Karlan BY, Orsulic S. Dynamic Changes in the Extracellular Matrix in Primary, Metastatic, and Recurrent Ovarian Cancers. Cells. 2022; 11(23):3769. https://doi.org/10.3390/cells11233769
Chicago/Turabian StyleGertych, Arkadiusz, Ann E. Walts, Keyi Cheng, Manyun Liu, Joshi John, Jenny Lester, Beth Y. Karlan, and Sandra Orsulic. 2022. "Dynamic Changes in the Extracellular Matrix in Primary, Metastatic, and Recurrent Ovarian Cancers" Cells 11, no. 23: 3769. https://doi.org/10.3390/cells11233769
APA StyleGertych, A., Walts, A. E., Cheng, K., Liu, M., John, J., Lester, J., Karlan, B. Y., & Orsulic, S. (2022). Dynamic Changes in the Extracellular Matrix in Primary, Metastatic, and Recurrent Ovarian Cancers. Cells, 11(23), 3769. https://doi.org/10.3390/cells11233769