


Divergent Molecular and Cellular Responses to Low and High-Dose Ionizing Radiation

 , , ,
, , ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Protein Labelling and Irradiation
2.2. Phosphoproteomics and Mass Spectrometry Measurements
2.3. MS Bioinformatics and Data Analyses
2.4. Cloud-Enabled High-Performance Computing (cHPC)
2.5. BrU-Seq Sample Preparation
2.6. BrU-Seq Bioinformatics and Data Analysis
2.7. Pathway, Motif and Enrichment Analyses
2.8. High-Throughput (HM) Microscopy Experiments
2.9. High Content-Analysis (HCA)
2.10. High-Throughput Cellular Bioenergetics (HCB)
2.11. Proliferation Assays
2.12. Gene Editing Using CRISPR-Cas9
3. Results
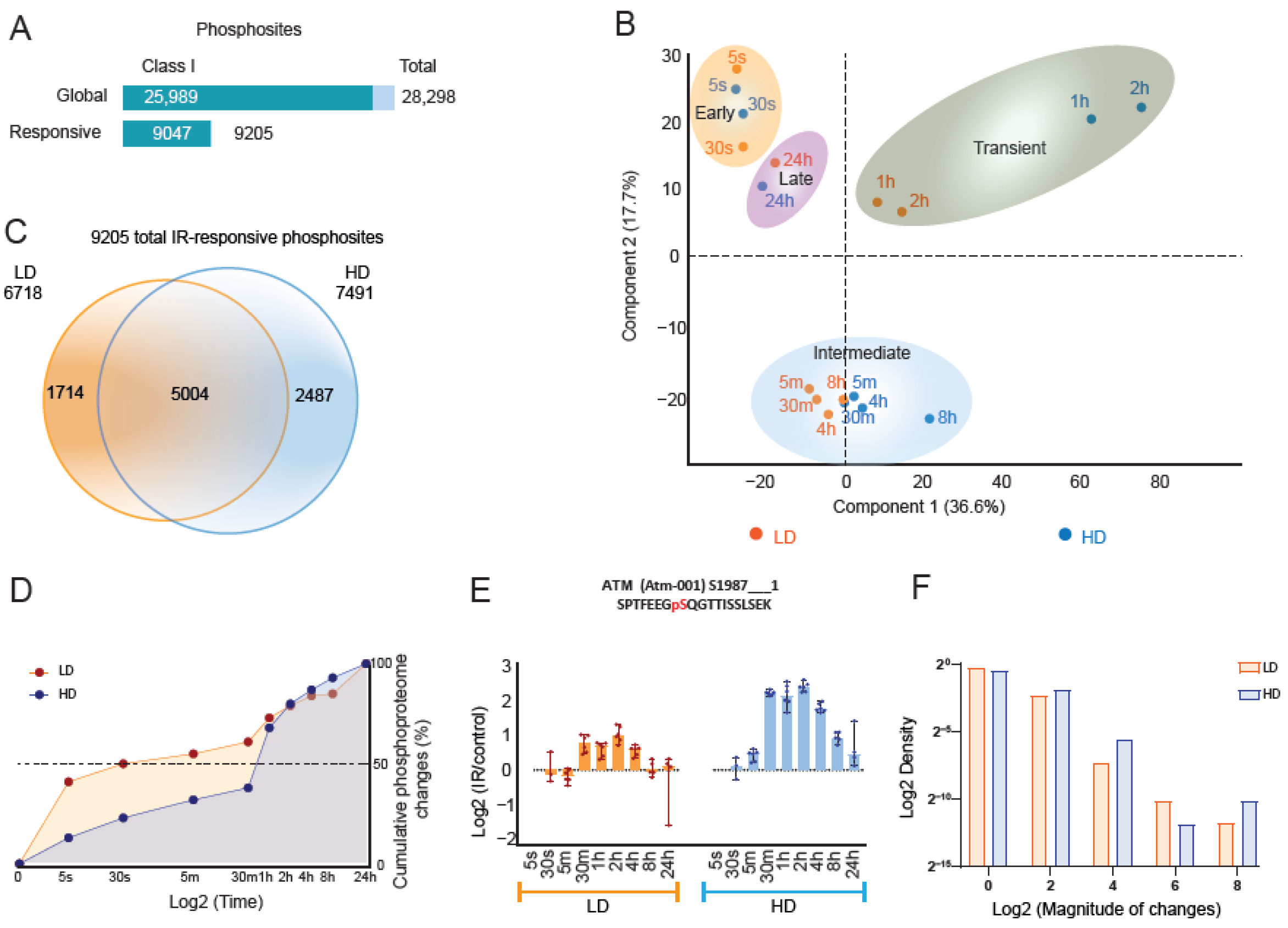
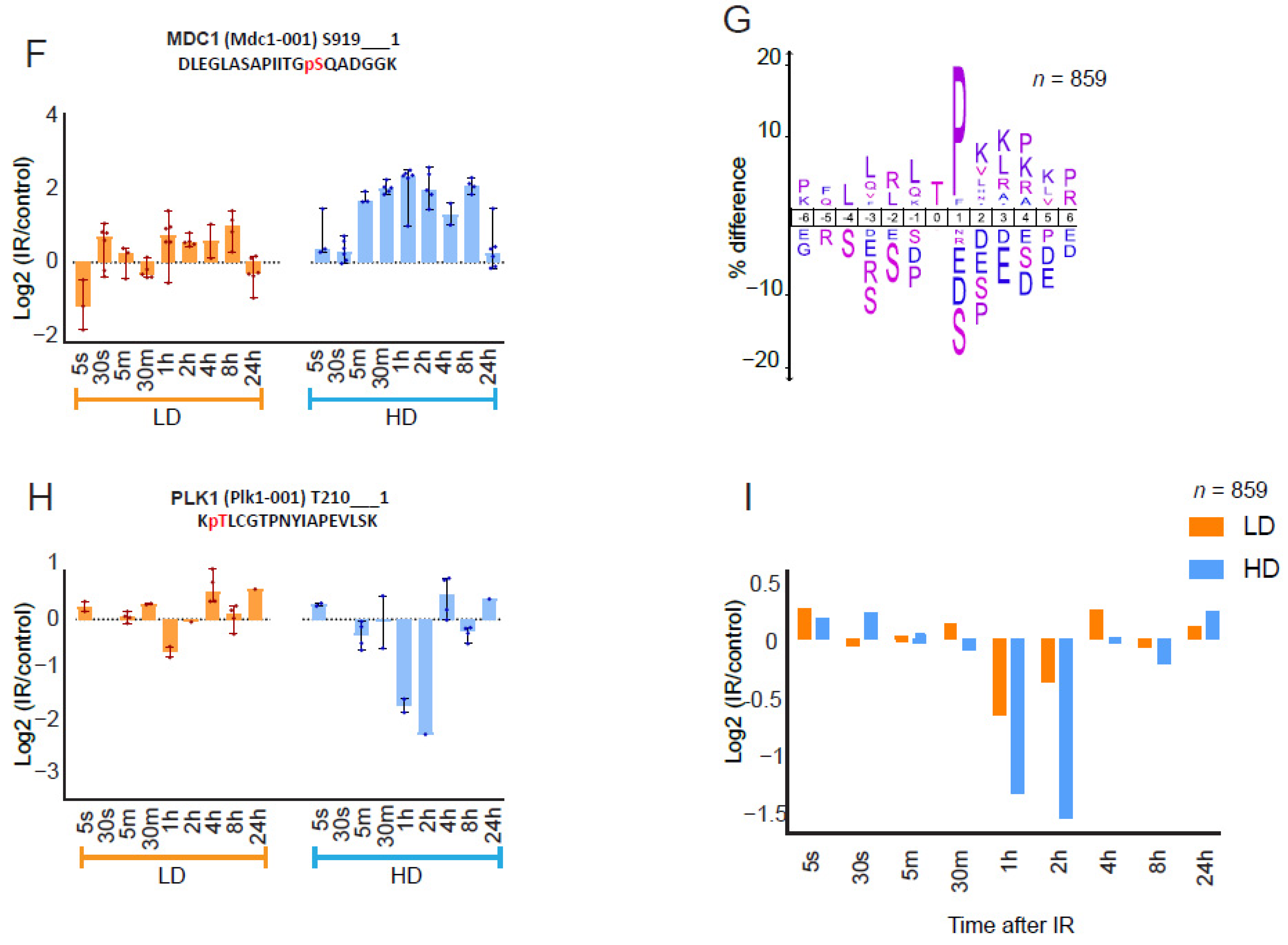
3.1. High-Temporal-Resolution Map of the Dose-Dependent Phosphoproteome after IR
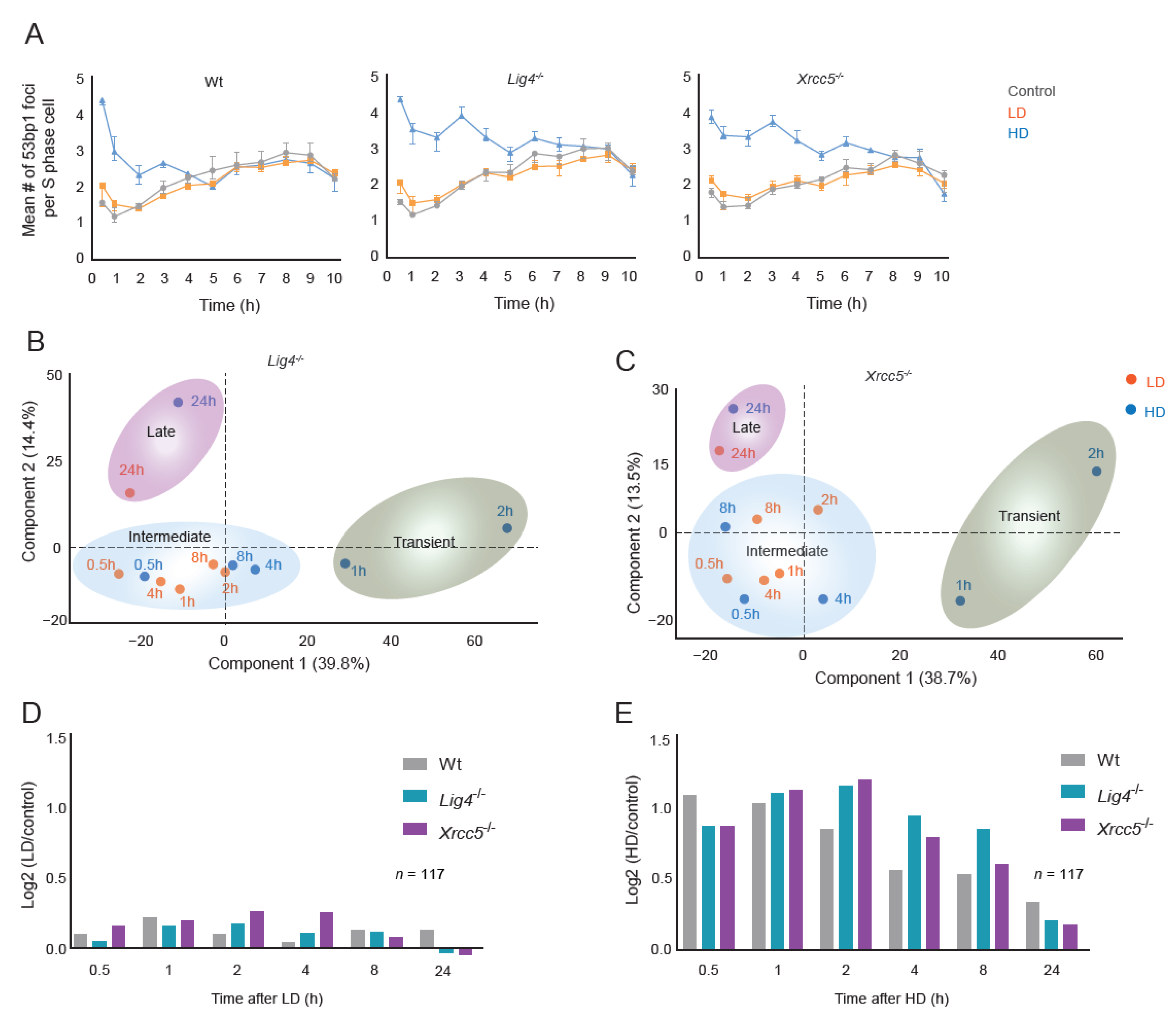
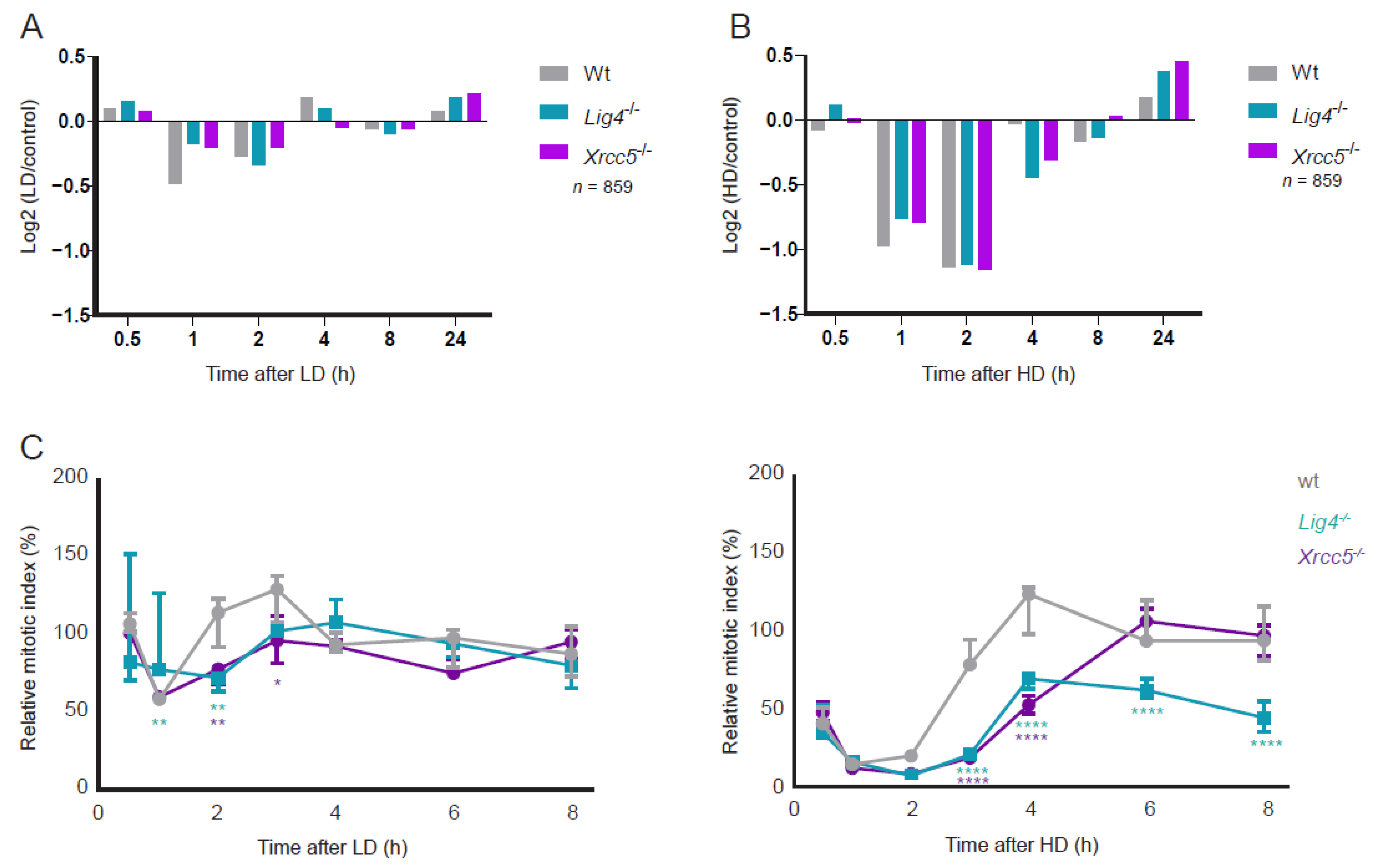
3.2. DSB Repair, Signaling Dynamics and G2/M Checkpoint Activation after LD and HD
3.3. Multi-Site Dephosphorylation of Cell Cycle Proteins Activates the G2/M Checkpoint in mESC
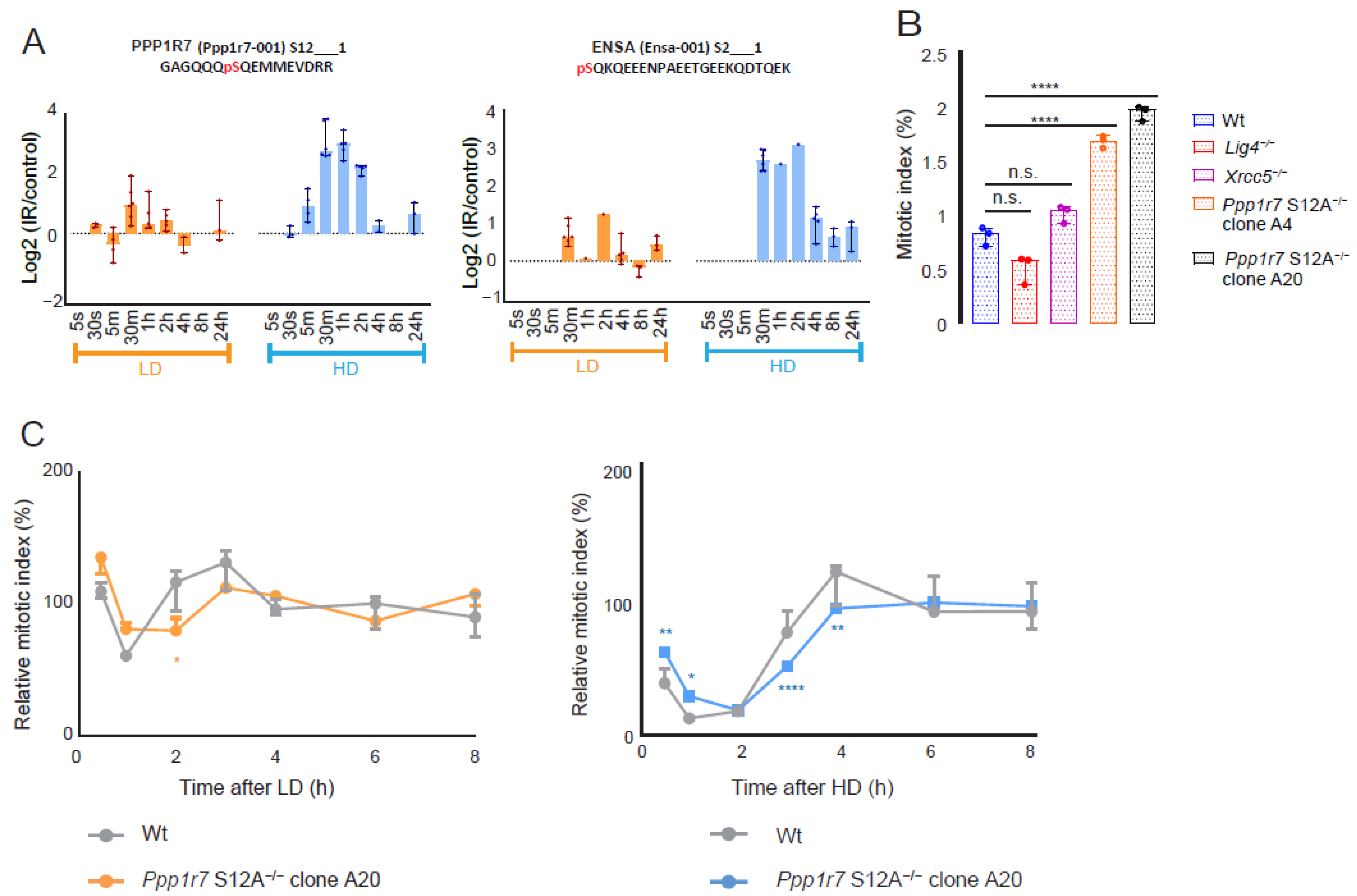
3.4. Phospho-Signaling via PPP1R7-S12 Is Involved in G2/M Checkpoint Activation
3.5. Proliferation Responses after LD and HD
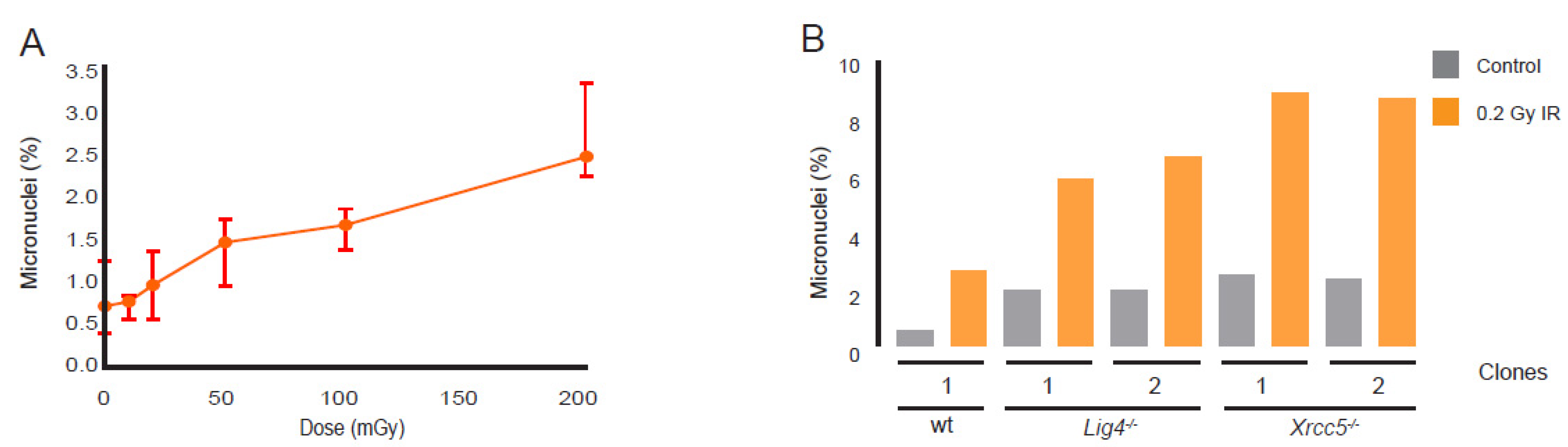
3.6. Micronuclei Induction after LD Exposure
3.7. Impaired DSB Repair Dynamically Rewires Crucial Signaling Pathways after IR
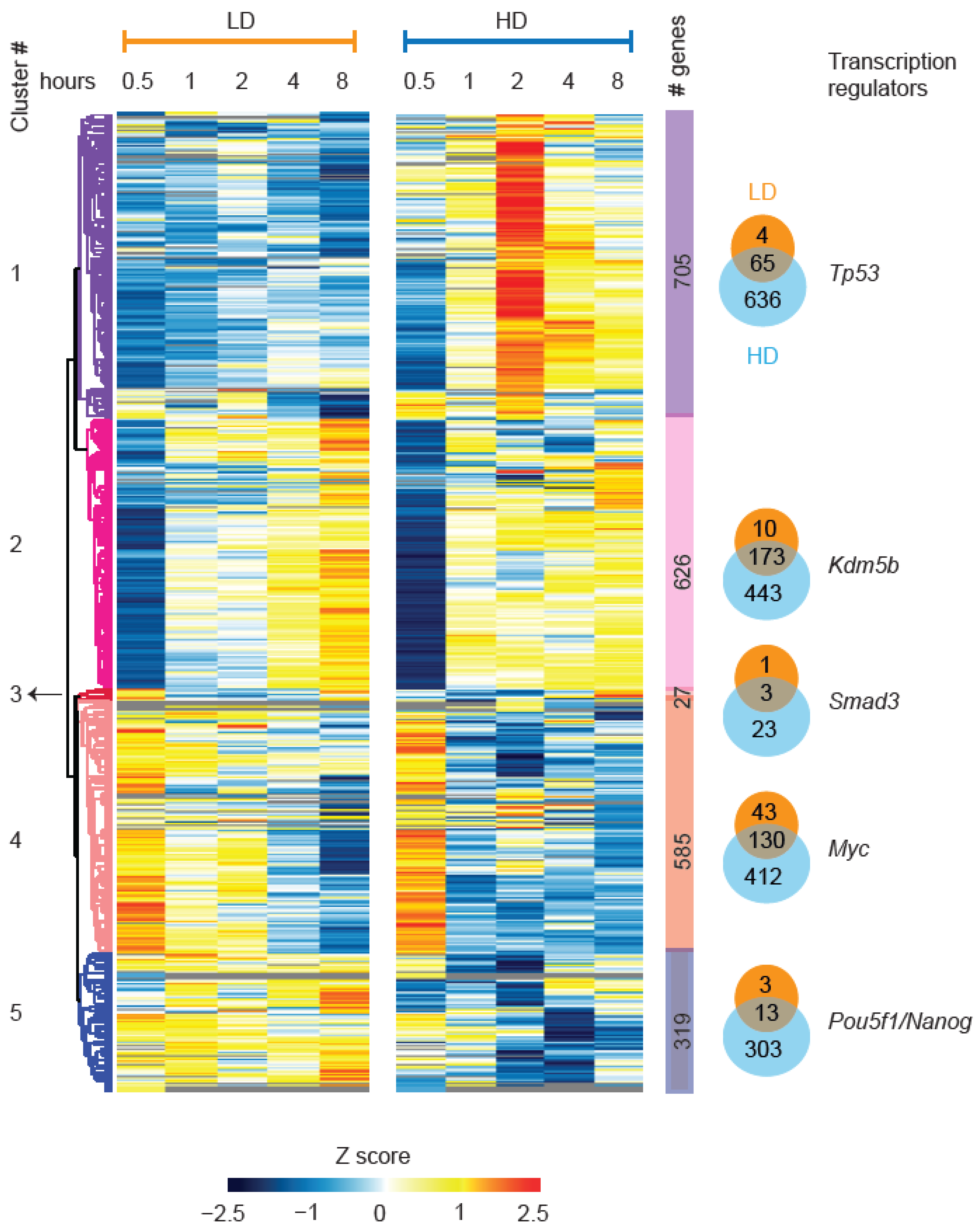
3.8. Temporal Dynamics of Gene Expression, and Metabolic Control Programs after LD and HD
4. Discussion
4.1. Phosphoproteome and Transcriptome Responses after LD and HD
4.2. DDR and Mitotic Signaling after LD and HD
4.3. P53 and Distinct Gene Expression Programs after LD and HD
4.4. mTOR/ERK Signaling after LD and HD
4.5. Mitochondrial Function Alterations after LD and HD
4.6. Persistent DSB and Phospho-Signaling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kamiya, K.; Ozasa, K.; Akiba, S.; Niwa, O.; Kodama, K.; Takamura, N.; Zaharieva, E.K.; Kimura, Y.; Wakeford, R. Long-term effects of radiation exposure on health. Lancet 2015, 386, 469–478. [Google Scholar] [CrossRef] [PubMed]
- McLean, A.R.; Adlen, E.K.; Cardis, E.; Elliott, A.; Goodhead, D.T.; Harms-Ringdahl, M.; Hendry, J.H.; Hoskin, P.; Jeggo, P.A.; Mackay, D.J.C.; et al. A restatement of the natural science evidence base concerning the health effects of low-level ionizing radiation. Proc. R. Soc. B Biol. Sci. 2017, 284, 20171070. [Google Scholar] [CrossRef] [PubMed]
- Pearce, M.S.; A Salotti, J.; Little, M.P.; McHugh, K.; Lee, C.; Kim, K.P.; Howe, N.L.; Ronckers, C.M.; Rajaraman, P.; Craft, A.W.; et al. Radiation exposure from CT scans in childhood and subsequent risk of leukaemia and brain tumours: A retrospective cohort study. Lancet 2012, 380, 499–505. [Google Scholar] [CrossRef] [Green Version]
- Brenner, D.J. Medical imaging in the 21st century—Getting the best bang for the rad. N. Engl. J. Med. 2010, 362, 943–945. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.J.; Shuryak, I.; Jozsef, G.; DeWyngaert, K.J.; Formenti, S.C. Risk and risk reduction of major coronary events associated with contemporary breast radiotherapy. JAMA Intern. Med. 2014, 174, 158. [Google Scholar] [CrossRef] [Green Version]
- Ruano-Ravina, A.; Wakeford, R. The increasing exposure of the global population to ionizing radiation. Epidemiology 2020, 31, 155–159. [Google Scholar] [CrossRef]
- Robison, L.L.; Hudson, M.M. Survivors of childhood and adolescent cancer: Life-long risks and responsibilities. Nat. Rev. Cancer 2014, 14, 61–70. [Google Scholar] [CrossRef]
- De Ruysscher, D.; Niedermann, G.; Burnet, N.G.; Siva, S.; Lee, A.W.; Hegi-Johnson, F. Radiotherapy toxicity. Nat. Rev. Dis. Primers 2019, 5, 13. [Google Scholar] [CrossRef]
- Goodhead, D.T. Fifth Warren K. Sinclair keynote address: Issues in quantifying the effects of low-level radiation. Health Phys. 2009, 97, 394–406. [Google Scholar] [CrossRef]
- Lall, R.; Ganapathy, S.; Yang, M.; Xiao, S.; Xu, T.; Su, H.; Shadfan, M.; Asara, J.; Ha, C.; Ben-Sahra, I.; et al. Low-dose radiation exposure induces a HIF-1-mediated adaptive and protective metabolic response. Cell Death Differ. 2014, 21, 836. [Google Scholar] [CrossRef]
- Prise, K.M.; Saran, A. Concise review: Stem cell effects in radiation risk. Stem Cells 2011, 29, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Barazzuol, L.; Hopkins, S.R.; Ju, L.; Jeggo, P.A. Distinct response of adult neural stem cells to low versus high dose ionising radiation. DNA Repair 2019, 76, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Zelensky, A.N.; Schoonakker, M.; Brandsma, I.; Tijsterman, M.; van Gent, D.C.; Essers, J.; Kanaar, R. Low dose ionizing radiation strongly stimulates insertional mutagenesis in a γH2AX dependent manner. PLoS Genet. 2020, 16, e1008550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagle, P.W.; Hosper, N.A.; Barazzuol, L.; Jellema, A.L.; Baanstra, M.; van Goethem, M.-J.; Brandenburg, S.; Giesen, U.; Langendijk, J.A.; van Luijk, P.; et al. Lack of DNA damage response at low radiation doses in adult stem cells contributes to organ dysfunction. Clin. Cancer Res. 2018, 24, 6583–6593. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues-Moreira, S.; Moreno, S.; Ghinatti, G.; Lewandowski, D.; Hoffschir, F.; Ferri, F.; Gallouet, A.-S.; Gay, D.; Motohashi, H.; Yamamoto, M.; et al. Low-dose irradiation promotes persistent oxidative stress and decreases self-renewal in hematopoietic stem cells. Cell Rep. 2017, 20, 3199–3211. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Antoran, D.; Piedrafita, G.; Murai, K.; Ong, S.H.; Herms, A.; Frezza, C.; Jones, P.H. Outcompeting p53-mutant cells in the normal esophagus by redox manipulation. Cell Stem Cell 2019, 25, 329–341.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Wang, M.; Staudt, C.; Liakis, G. Post-irradiation chemical processing of DNA damage generates double-strand breaks in cells already engaged in repair. Nucleic Acids Res. 2011, 39, 8416–8429. [Google Scholar] [CrossRef]
- Asaithamby, A.; Chen, D.J. Cellular responses to DNA double-strand breaks after low-dose γ-irradiation. Nucleic Acids Res. 2009, 37, 3912–3923. [Google Scholar] [CrossRef] [Green Version]
- Henry, E.; Souissi-Sahraoui, I.; Deynoux, M.; Lefèvre, A.; Barroca, V.; Campalans, A.; Ménard, V.; Calvo, J.; Pflumio, F.; Arcangeli, M.-L. Human hematopoietic stem/progenitor cells display reactive oxygen species-dependent long-term hematopoietic defects after exposure to low doses of ionizing radiations. Haematologica 2020, 105, 2044–2055. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, K.; Qi, F.; Kobayashi, J. Potential relationship between the biological effects of low-dose irradiation and mitochondrial ROS production. J. Radiat. Res. 2018, 59, ii91–ii97. [Google Scholar] [CrossRef]
- Konkova, M.; Abramova, M.; Kalianov, A.; Ershova, E.; Dolgikh, O.; Umriukhin, P.; Izhevskaya, V.; Kutsev, S.; Veiko, N.; Kostyuk, S. Mesenchymal stem cells early response to low-dose ionizing radiation. Front. Cell Dev. Biol. 2020, 8, 584497. [Google Scholar] [CrossRef] [PubMed]
- United Nations Scientific Committee on the Effects of Atomic Radiation. Sources, and effects of Ionizing Radiation. United Nations Scientific Committee on the Effects of Atomic Radiation UNSCEAR 1993 Report to the General Assembly, with Scientific Annexes; United Nations: New York, NY, USA, 1993. [Google Scholar]
- United Nations Scientific Committee on the Effects of Atomic Radiation. Sources, Effects, and Risks of Ionizing Radiation. United Nations Scientific Committee on the Effects of Atomic Radiation UNSCEAR 2013 Report to the General Assembly, with Scientific Annexes; United Nations: New York, NY, USA, 2013. [Google Scholar]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., III; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Bennetzen, M.V.; Larsen, D.H.; Bunkenborg, J.; Bartek, J.; Lukas, J.; Andersen, J.S. Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol. Cell Proteom. 2010, 9, 1314–1323. [Google Scholar] [CrossRef] [Green Version]
- Roitinger, E.; Hofer, M.; Köcher, T.; Pichler, P.; Novatchkova, M.; Yang, J.; Schlögelhofer, P.; Mechtler, K. Quantitative phosphoproteomics of the ataxia telangiectasia-mutated (ATM) and ataxia telangiectasia-mutated and rad3-related (ATR) dependent DNA damage response in Arabidopsis thaliana. Mol. Cell Proteom. 2015, 14, 556–571. [Google Scholar] [CrossRef] [Green Version]
- Beli, P.; Lukashchuk, N.; Wagner, S.; Weinert, B.; Olsen, J.; Baskcomb, L.; Mann, M.; Jackson, S.; Choudhary, C. Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell 2012, 46, 212–225. [Google Scholar] [CrossRef] [Green Version]
- Bensimon, A.; Schmidt, A.; Ziv, Y.; Elkon, R.; Wang, S.-Y.Y.; Chen, D.J.; Aebersold, R.; Shiloh, Y. ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage. Sci. Signal. 2010, 3, rs3. [Google Scholar] [CrossRef]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef]
- Rashi-Elkeles, S.; Warnatz, H.-J.; Elkon, R.; Kupershtein, A.; Chobod, Y.; Paz, A.; Amstislavskiy, V.; Sultan, M.; Safer, H.; Nietfeld, W.; et al. Parallel profiling of the transcriptome, cistrome, and epigenome in the cellular response to ionizing radiation. Sci. Signal. 2014, 7, rs3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, J.R.; Fisher, B.E.; Batchelor, E. p53 pulses diversify target gene expression dynamics in an mRNA half-life-dependent manner and delineate co-regulated target gene subnetworks. Cell Syst. 2016, 2, 272–282. [Google Scholar] [CrossRef] [Green Version]
- Porter, J.R.; Fisher, B.E.; Baranello, L.; Liu, J.C.; Kambach, D.M.; Nie, Z.; Koh, W.S.; Luo, J.; Stommel, J.M.; Levens, D.; et al. Global inhibition with specific activation: How p53 and MYC redistribute the transcriptome in the DNA double-strand break response. Mol. Cell 2017, 67, 1013–1025.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafner, A.; Stewart-Ornstein, J.; Purvis, J.E.; Forrester, W.C.; Bulyk, M.L.; Lahav, G. p53 pulses lead to distinct patterns of gene expression albeit similar DNA-binding dynamics. Nat. Struct. Mol. Biol. 2017, 24, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Venkata Narayanan, I.; Paulsen, M.T.; Bedi, K.; Berg, N.; Ljungman, E.A.; Francia, S.; Veloso, A.; Magnuson, B.; di Fagagna, F.; d’Adda, D.; et al. Transcriptional and post-transcriptional regulation of the ionizing radiation response by ATM and p53. Sci. Rep. 2017, 7, 43598. [Google Scholar] [CrossRef]
- Jaehnig, E.J.; Kuo, D.; Hombauer, H.; Ideker, T.G.; Kolodner, R.D. Checkpoint kinases regulate a global network of transcription factors in response to DNA damage. Cell Rep. 2013, 4, 174–188. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Waters, K.; Miller, J.H.; Gritsenko, M.A.; Zhao, R.; Du, X.; Livesay, E.A.; Purvine, S.; Monroe, M.E.; Wang, Y.; et al. Phosphoproteomics profiling of human skin fibroblast cells reveals pathways and proteins affected by low doses of ionizing radiation. PLoS ONE 2010, 5, e14152. [Google Scholar] [CrossRef]
- Mullenders, L.; Atkinson, M.; Paretzke, H.; Sabatier, L.; Bouffler, S. Assessing cancer risks of low-dose radiation. Nat. Rev. Cancer 2009, 9, 596–604. [Google Scholar] [CrossRef]
- Thompson, L.H.; Limoli, C.L. Origin, recognition, signalling and repair of DNA double-strand breaks in mammalian cells. In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2012; pp. 2000–2013. [Google Scholar]
- Schimmel, J.; Kool, H.; van Schendel, R.; Tijsterman, M. Mutational signatures of non-homologous and polymerase theta-mediated end-joining in embryonic stem cells. EMBO J. 2017, 36, 3634–3649. [Google Scholar] [CrossRef]
- Zelensky, A.N.; Schimmel, J.; Kool, H.; Kanaar, R.; Tijsterman, M. Inactivation of Pol θ and C-NHEJ eliminates off-target integration of exogenous DNA. Nat. Commun. 2017, 8, 66. [Google Scholar] [CrossRef]
- Sampadi, B.; Pines, A.; Munk, S.; Mišovic, B.; de Groot, A.J.; van de Water, B.; Olsen, J.V.; Mullenders, L.H.F.; Vrieling, H. Quantitative phosphoproteomics to unravel the cellular response to chemical stressors with different modes of action. Arch. Toxicol. 2020, 94, 1655–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampadi, B.; Mullenders, L.H.F.; Vrieling, H. Phosphoproteomics sample preparation impacts biological interpretation of phosphorylation signaling outcomes. Cells 2021, 10, 3407. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, S.J.; Azimifar, S.B.; Mann, M. High-throughput phosphoproteomics reveals in vivo insulin signalling dynamics. Nat. Biotechnol. 2015, 33, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Robles, M.S.; Humphrey, S.J.; Mann, M. Phosphorylation is a central mechanism for circadian control of metabolism and physiology. Cell Metab. 2017, 25, 118–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacco, F.; Humphrey, S.J.; Cox, J.; Mischnik, M.; Schulte, A.; Klabunde, T.; Schäfer, M.; Mann, M. Glucose-regulated and drug-perturbed phosphoproteome reveals molecular mechanisms controlling insulin secretion. Nat. Commun. 2016, 7, 13250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulsen, M.T.; Veloso, A.; Prasad, J.; Bedi, K.; Ljungman, E.A.; Tsan, Y.-C.; Chang, C.-W.; Tarrier, B.; Washburn, J.G.; Lyons, R.; et al. Coordinated regulation of synthesis and stability of RNA during the acute TNF-induced proinflammatory response. Proc. Natl. Acad. Sci. USA. 2013, 110, 2240–2245. [Google Scholar] [CrossRef] [Green Version]
- Chepelev, I.; Wei, G.; Tang, Q.; Zhao, K. Detection of single nucleotide variations in expressed exons of the human genome using RNA-Seq. Nucleic Acids Res. 2009, 37, e106. [Google Scholar] [CrossRef] [Green Version]
- Colaert, N.; Helsens, K.; Martens, L.; Vandekerckhove, J.; Gevaert, K. Improved visualization of protein consensus sequences by IceLogo. Nat. Methods 2009, 6, 786–787. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Pelgrom, L.R.; van der Ham, A.J.; Everts, B. Analysis of TLR-induced metabolic changes in dendritic cells using the seahorse XFe96 extracellular flux analyzer. Methods Mol. Biol. 2016, 1390, 273–285. [Google Scholar]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Hartlerode, A.; Morgan, M.; Wu, Y.; Buis, J.; Ferguson, D. Recruitment and activation of the ATM kinase in the absence of DNA-damage sensors. Nat. Struct. Mol. Biol. 2015, 22, 736–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, R.S.; Castella, M.; Abeyta, A.; Gafken, P.R.; Tucker, N.; Taniguchi, T. Ubiquitination-linked phosphorylation of the FANCI S/TQ cluster contributes to activation of the fanconi anemia I/D2 complex. Cell Rep. 2017, 19, 2432–2440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, W.; van Twest, S.; Murphy, V.J.; Deans, A.J. ATR-mediated FANCI phosphorylation regulates both ubiquitination and deubiquitination of FANCD2. Front. Cell Dev. Biol. 2020, 8, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Técher, H.; Koundrioukoff, S.; Nicolas, A.; Debatisse, M. The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat. Rev. Genet. 2017, 18, 535–550. [Google Scholar] [CrossRef]
- Swaffer, M.P.; Jones, A.W.; Flynn, H.R.; Snijders, A.P.; Nurse, P. CDK substrate phosphorylation and ordering the cell cycle. Cell 2016, 167, 1750–1761.e16. [Google Scholar] [CrossRef] [Green Version]
- McCloy, R.A.; Parker, B.L.; Rogers, S.; Chaudhuri, R.; Gayevskiy, V.; Hoffman, N.J.; Ali, N.; Watkins, D.N.; Daly, R.J.; James, D.E.; et al. Global phosphoproteomic mapping of early mitotic exit in human cells identifies novel substrate dephosphorylation motifs. Mol. Cell Proteom. 2015, 14, 2194–2212. [Google Scholar] [CrossRef] [Green Version]
- Godfrey, M.; Touati, S.A.; Kataria, M.; Jones, A.; Snijders, A.P.; Uhlmann, F. PP2ACdc55 phosphatase imposes ordered cell-cycle phosphorylation by opposing threonine phosphorylation. Mol. Cell 2017, 65, 393–402.e3. [Google Scholar] [CrossRef] [Green Version]
- Swaffer, M.P.; Jones, A.W.; Flynn, H.R.; Snijders, A.P.; Nurse, P. Quantitative phosphoproteomics reveals the signalling dynamics of cell-cycle kinases in the fission yeast Schizosaccharomyces pombe. Cell Rep. 2018, 24, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Joukov, V.; De Nicolo, A. Aurora-PLK1 cascades as key signalling modules in the regulation of mitosis. Sci. Signal. 2018, 11, eaar4195. [Google Scholar] [CrossRef] [Green Version]
- Pomerening, J.R.; Sontag, E.D.; Ferrell, J.E. Building a cell cycle oscillator: Hysteresis and bistability in the activation of Cdc2. Nat. Cell Biol. 2003, 5, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Mochida, S.; Rata, S.; Hino, H.; Nagai, T.; Novák, B. Two bistable switches govern M phase entry. Curr. Biol. 2016, 26, 3361–3367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrell, J.E.; Ha, S.H. Ultrasensitivity part II: Multisite phosphorylation, stoichiometric inhibitors, and positive feedback. Trends Biochem. Sci. 2014, 39, 556–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Songyang, Z.; Blechner, S.; Hoagland, N.; Hoekstra, M.F.; Piwnica-Worms, H.; Cantley, L.C. Use of an oriented peptide library to determine the optimal substrates of protein kinases. Curr. Biol. 1994, 4, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.; Fey, D.; McCloy, R.A.; Parker, B.L.; Mitchell, N.J.; Payne, R.J.; Daly, R.J.; James, D.E.; Caldon, C.E.; Watkins, D.N.; et al. PP1 initiates the dephosphorylation of MASTL, triggering mitotic exit and bistability in human cells. J. Cell Sci. 2016, 129, 1340–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Vigneron, S.; Robert, P.; Strub, J.; Cianferani, S.; Castro, A.; Lorca, T. Greatwall dephosphorylation and inactivation upon mitotic exit is triggered by PP1. J. Cell Sci. 2016, 129, 1329–1339. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, N.; Lekomtsev, S.; Jananji, S.; Kriston-Vizi, J.; Hickson, G.; Baum, B. Kinetochore-localized PP1–Sds22 couples chromosome segregation to polar relaxation. Nature 2015, 524, 489. [Google Scholar] [CrossRef] [Green Version]
- Mochida, S.; Maslen, S.L.; Skehel, M.; Hunt, T. Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science 2010, 330, 1670–1673. [Google Scholar] [CrossRef]
- Mendoza, M.; Er, E.; Blenis, J. The RAS-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.P.; Kang, S.A.; Rameseder, J.; Zhang, Y.; Ottina, K.A.; Lim, D.; Peterson, T.R.; Choi, Y.; Gray, N.S.; Yaffe, M.B.; et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signalling. Science 2011, 332, 1317–1322. [Google Scholar] [CrossRef] [Green Version]
- Nett, I.R.; Mulas, C.; Gatto, L.; Lilley, K.S.; Smith, A. Negative feedback via RSK modulates Erk-dependent progression from naïve pluripotency. EMBO Rep. 2018, 19, e45642. [Google Scholar] [CrossRef] [PubMed]
- Sampadi, B.; Mullenders, L.H.F.; Vrieling, H. Low and high doses of ionising radiation evoke discrete global (phospho)proteome responses. DNA Repair 2022, 113, 103305. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cunningham, M.; Zhang, X.; Tokarz, S.; Laraway, B.; Troxell, M.; Sears, R.C. Phosphorylation regulates c-Myc’s Oncogenic activity in the mammary gland. Cancer Res. 2011, 71, 925–936. [Google Scholar] [CrossRef] [Green Version]
- Herold, S.; Kalb, J.; Büchel, G.; Ade, C.P.; Baluapuri, A.; Xu, J.; Koster, J.; Solvie, D.; Carstensen, A.; Klotz, C.; et al. Recruitment of BRCA1 limits MYCN-driven accumulation of stalled RNA polymerase. Nature 2019, 567, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Baluapuri, A.; Wolf, E.; Eilers, M. Target gene-independent functions of MYC oncoproteins. Nat. Rev. Mol. Cell Biol. 2020, 21, 255–267. [Google Scholar] [CrossRef]
- Tanner, L.B.; Goglia, A.G.; Wei, M.H.; Sehgal, T.; Parsons, L.R.; Park, J.O.; White, E.; Toettcher, J.E.; Rabinowitz, J.D. Four key steps control glycolytic flux in mammalian cells. Cell Syst. 2018, 7, 49–62.e8. [Google Scholar] [CrossRef]
- Carey, B.W.; Finley, L.; Cross, J.R.; Nature, A.-C. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef] [Green Version]
- Bell, E.; Klimova, T.; Eisenbart, J.; Moraes, C.; Murphy, M.; Budinger, G.R.; Chandel, N. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signalling via reactive oxygen species production. J. Cell Biol. 2007, 177, 1029–1036. [Google Scholar] [CrossRef] [Green Version]
- Pines, A.; Perrone, L.; Bivi, N.; Romanello, M.; Damante, G.; Gulisano, M.; Kelley, M.R.; Quadrifoglio, F.; Tell, G. Activation of APE1/Ref-1 is dependent on reactive oxygen species generated after purinergic receptor stimulation by ATP. Nucleic Acids Res. 2005, 33, 4379–4394. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Ren, J.; Lu, W.; Harlos, K.; Jones, E.Y. Structure of the Wnt signaling enhancer LYPD 6 and its interactions with the Wnt coreceptor LRP 6. FEBS Lett. 2018, 592, 3152–3162. [Google Scholar] [CrossRef]
- Clevers, H.; Loh, K.M.; Nusse, R. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 2014, 346, 1248012. [Google Scholar] [CrossRef] [PubMed]
- Shang, F.; Taylor, A. Oxidative stress and recovery from oxidative stress are associated with altered ubiquitin conjugating and proteolytic activities in bovine lens epithelial cells. Biochem. J. 1995, 307, 297–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singleton, B.K.; Torres-Arzayus, M.I.; Rottinghaus, S.T.; Taccioli, G.E.; Jeggo, P.A. The C terminus of Ku80 activates the DNA-dependent protein kinase catalytic subunit. Mol. Cell. Biol. 1999, 19, 3267–3277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Okada, H.; Yamauchi, M.; Oka, Y.; Kodama, S.; Watanabe, M. Qualitative and quantitative analysis of phosphorylated ATM foci induced by low-dose ionizing radiation. Radiat. Res. 2006, 165, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Short, S.C.; Bourne, S.; Martindale, C.; Woodcock, M.; Jackson, S.P. DNA damage responses at low radiation doses. Radiat. Res. 2005, 164, 292–302. [Google Scholar] [CrossRef]
- Boei, J.J.W.A.; Vermeulen, S.; Skubakova, M.M.; Meijers, M.; Loenen, W.A.M.; Wolterbeek, R.; Mullenders, L.H.F.; Vrieling, H.; Giphart-Gassler, M. No threshold for the induction of chromosomal damage at clinically relevant low doses of X rays. Radiat. Res. 2012, 177, 602–613. [Google Scholar] [CrossRef]
- Manning, G.; Taylor, K.; Finnon, P.; Lemon, J.A.; Boreham, D.R.; Badie, C. Quantifying murine bone marrow and blood radiation dose response following 18F-FDG PET with DNA damage biomarkers. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2014, 770, 29–36. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Jones, M.J.K.; Yin, Y.; Crist, S.B.; Colnaghi, L.; Sims, R.J.; Rothenberg, E.; Jallepalli, P.V.; Huang, T.T. ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol. Cell 2015, 58, 323–338. [Google Scholar] [CrossRef] [Green Version]
- Leimbacher, P.-A.; Jones, S.E.; Shorrocks, A.-M.K.; de Marco Zompit, M.; Day, M.; Blaauwendraad, J.; Bundschuh, D.; Bonham, S.; Fischer, R.; Fink, D.; et al. MDC1 interacts with TOPBP1 to maintain chromosomal stability during mitosis. Mol. Cell 2019, 74, 571–583.e8. [Google Scholar] [CrossRef] [Green Version]
- Lomax, M.E.; Folkes, L.K.; O’Neill, P. Biological consequences of radiation-induced DNA damage: Relevance to radiotherapy. Clin. Oncol. 2013, 25, 578–585. [Google Scholar] [CrossRef] [Green Version]
- Harper, J.V.; Anderson, J.A.; O’Neill, P. Radiation induced DNA DSBs: Contribution from stalled replication forks? DNA Repair 2010, 9, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Chao, C.; Saito, S.; Mazur, S.J.; Murphy, M.E.; Appella, E.; Xu, Y. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat. Cell Biol. 2005, 7, 165–171. [Google Scholar] [CrossRef]
- Kavanagh, B.D.; Dent, P.; Schmidt-Ullrich, R.K.; Chen, P.; Mikkelsen, R.B. Calcium-dependent stimulation of mitogen-activated protein kinase activity in A431 cells by low doses of ionizing radiation. Radiat. Res. 1998, 149, 579. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Blenis, J.; Yuan, J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc. Natl. Acad. Sci. USA 2008, 105, 6584–6589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purvis, J.E.; Karhohs, K.W.; Mock, C.; Batchelor, E.; Loewer, A.; Lahav, G. p53 dynamics control cell fate. Science 2012, 336, 1440–1444. [Google Scholar] [CrossRef] [Green Version]
- Paek, A.L.; Liu, J.C.; Loewer, A.; Forrester, W.C.; Lahav, G. Cell-to-cell variation in p53 dynamics leads to fractional killing. Cell 2016, 165, 631–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Jiang, T.; Wang, H.; Tao, S.; Lau, A.; Fang, D.; Zhang, D. Does Nrf2 contribute to p53-mediated control of cell survival and death? Antioxid. Redox Signal. 2012, 17, 1670–1675. [Google Scholar] [CrossRef] [Green Version]
- Faraonio, R.; Vergara, P.; Marzo, D.; Pierantoni, M.; Napolitano, M.; Russo, T.; Cimino, F. p53 Suppresses the Nrf2-dependent Transcription of Antioxidant Response Genes. J. Biol. Chem. 2006, 281, 39776–39784. [Google Scholar] [CrossRef] [Green Version]
- Paek, A.L.; Liu, J.C.; Loewer, A.; Forrester, W.C.; Lahav, G. MYC controls human pluripotent stem cell fate decisions through regulation of metabolic flux. Cell Stem Cell 2017, 21, 502–516.e9. [Google Scholar]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR signalling in growth, metabolism, and disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Benassi, B.; Fanciulli, M.; Fiorentino, F.; Porrello, A.; Chiorino, G.; Loda, M.; Zupi, G.; Biroccio, A. c-Myc phosphorylation is required for cellular response to oxidative stress. Mol. Cell 2006, 21, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H. Involvement of Wnt signaling in primary cilia assembly and disassembly. FEBS J. 2020, 287, 5027–5038. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Xia, W.; Liu, J.-C.; Yang, J.-Y.; Lee, D.-F.; Xia, J.; Bartholomeusz, G.; Li, Y.; Pan, Y.; Li, Z.; et al. Erk associates with and primes GSK-3β for its inactivation resulting in upregulation of β-catenin. Mol. Cell 2005, 19, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Leach, J.K.; Van Tuyle, G.; Lin, P.-S.; Schmidt-Ullrich, R.; Mikkelsen, R.B. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res. 2001, 61, 3894–3901. [Google Scholar] [PubMed]
- Azzam, E.I.; Jay-Gerin, J.P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Kam, W.W.-Y.; Banati, R.B. Effects of ionizing radiation on mitochondria. Free. Radic. Biol. Med. 2013, 65, 607–619. [Google Scholar] [CrossRef]
- Kim, G.J.; Chandrasekaran, K.; Morgan, W.F. Mitochondrial dysfunction, persistently elevated levels of reactive oxygen species and radiation-induced genomic instability: A review. Mutagenesis 2006, 21, 361–367. [Google Scholar] [CrossRef] [Green Version]
- Spitz, D.R.; Azzam, E.I.; Li, J.J.; Gius, D. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: A unifying concept in stress response biology. Cancer Metastasis Rev. 2004, 23, 311–322. [Google Scholar] [CrossRef]
- Lo, S.C.; Hannink, M. PGAM5, a Bcl-XL-interacting protein, is a novel substrate for the redox-regulated Keap1-dependent ubiquitin ligase complex. J. Biol. Chem. 2006, 281, 37893–37903. [Google Scholar] [CrossRef] [Green Version]
- Holmström, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The multifaceted role of Nrf2 in mitochondrial function. Curr. Opin. Toxicol. 2016, 2, 80–91. [Google Scholar] [CrossRef] [Green Version]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; DeNicola, G.M. The complex interplay between antioxidants and ROS in cancer. Trends Cell Biol. 2020, 30, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Oji, A.; Noda, T.; Fujihara, Y.; Miyata, H.; Kim, Y.J.; Muto, M.; Nozawa, K.; Matsumura, T.; Isotani, A.; Ikawa, M. CRISPR/Cas9 mediated genome editing in ES cells and its application for chimeric analysis in mice. Sci. Rep. 2016, 6, 31666. [Google Scholar] [CrossRef] [PubMed]
- Renaud, J.-B.; Boix, C.; Charpentier, M.; De Cian, A.; Cochennec, J.; Duvernois-Berthet, E.; Perrouault, L.; Tesson, L.; Edouard, J.; Thinard, R.; et al. Improved genome editing efficiency and flexibility using modified oligonucleotides with TALEN and CRISPR-Cas9 nucleases. Cell Rep. 2016, 14, 2263–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Yang, D.; Xu, J.; Zhu, T.; Chen, Y.E.; Zhang, J. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat. Commun. 2016, 7, 10548. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Scavuzzo, M.A.; Chmielowiec, J.; Sharp, R.; Bajic, A.; Borowiak, M. Enrichment of G2/M cell cycle phase in human pluripotent stem cells enhances HDR-mediated gene repair with customizable endonucleases. Sci. Rep. 2016, 6, 21264. [Google Scholar] [CrossRef] [Green Version]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kühn, R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sampadi, B.; Vermeulen, S.; Mišovic, B.; Boei, J.J.; Batth, T.S.; Chang, J.-G.; Paulsen, M.T.; Magnuson, B.; Schimmel, J.; Kool, H.; et al. Divergent Molecular and Cellular Responses to Low and High-Dose Ionizing Radiation. Cells 2022, 11, 3794. https://doi.org/10.3390/cells11233794
Sampadi B, Vermeulen S, Mišovic B, Boei JJ, Batth TS, Chang J-G, Paulsen MT, Magnuson B, Schimmel J, Kool H, et al. Divergent Molecular and Cellular Responses to Low and High-Dose Ionizing Radiation. Cells. 2022; 11(23):3794. https://doi.org/10.3390/cells11233794
Chicago/Turabian StyleSampadi, Bharath, Sylvia Vermeulen, Branislav Mišovic, Jan J. Boei, Tanveer S. Batth, Jer-Gung Chang, Michelle T. Paulsen, Brian Magnuson, Joost Schimmel, Hanneke Kool, and et al. 2022. "Divergent Molecular and Cellular Responses to Low and High-Dose Ionizing Radiation" Cells 11, no. 23: 3794. https://doi.org/10.3390/cells11233794
APA StyleSampadi, B., Vermeulen, S., Mišovic, B., Boei, J. J., Batth, T. S., Chang, J. -G., Paulsen, M. T., Magnuson, B., Schimmel, J., Kool, H., Olie, C. S., Everts, B., Vertegaal, A. C. O., Olsen, J. V., Ljungman, M., Jeggo, P. A., Mullenders, L. H. F., & Vrieling, H. (2022). Divergent Molecular and Cellular Responses to Low and High-Dose Ionizing Radiation. Cells, 11(23), 3794. https://doi.org/10.3390/cells11233794