
The Interface of Tumour-Associated Macrophages with Dying Cancer Cells in Immuno-Oncology

 , , , and
, , , and 
Abstract
:1. Introduction
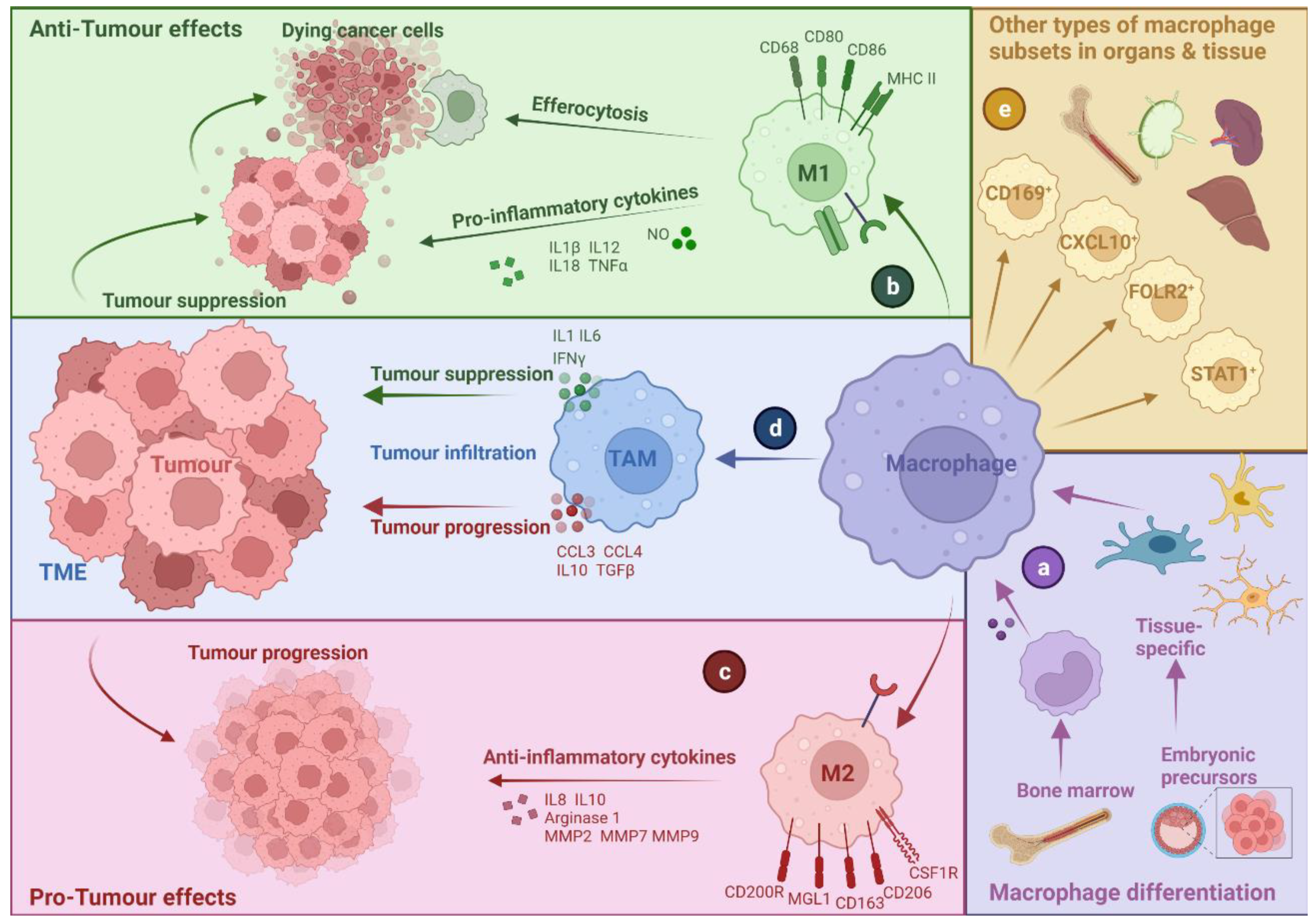
2. Tumour-Associated Macrophages: A Bird’s Eye-View
2.1. TAM Diversity and Classifications
2.2. M1- and M2-like Macrophages: A Narrow Vision for a Wide Spectrum of Phenotypes
2.3. Multi-Faceted Roles of TAMs in Cancer
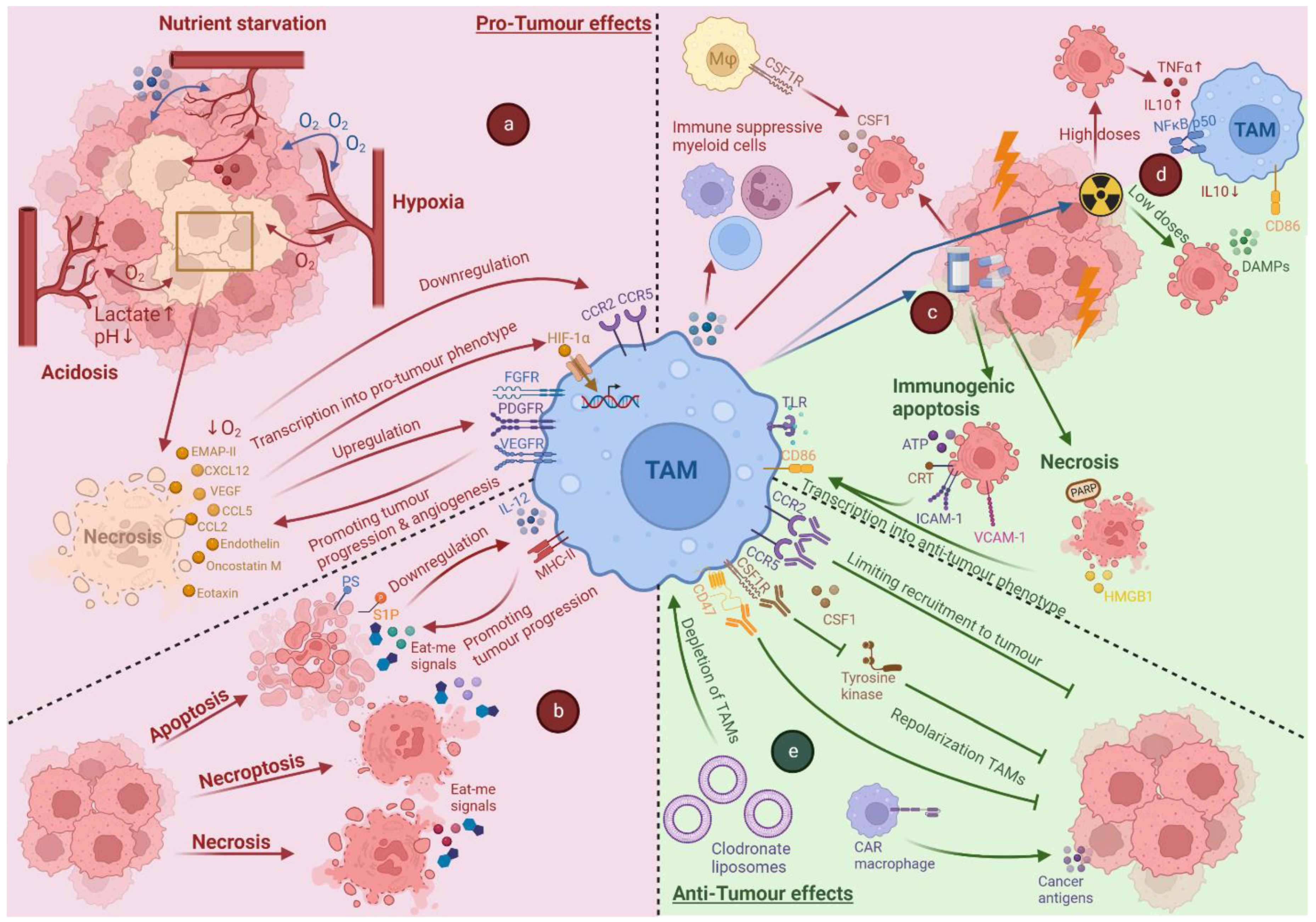
3. Cell Death in the Context of a Tumour Microenvironment
4. Impact of Tumour Associated Stress and Cell Death on TAMs
4.1. Impact of Hypoxia on TAMs
4.2. The Impact of Acidosis and Nutrient Starvation on TAMs
4.3. Cross-Talk between Dying Cancer Cells and TAMs
5. Anti-Cancer Therapy-Induced Cell Death and Its Impact on TAMs
5.1. Impact of Chemotherapy-Induced Cell Death on TAMs
5.2. Impact of Radiotherapy-Induced Cell Death on TAMs
6. Immunotherapy Targeting TAMs
6.1. Limiting the Recruitment of TAMs
6.2. Repolarisation of TAMs
6.3. Depletion of TAMs
6.4. CAR-Macrophages
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef] [PubMed]
- Prenen, H.; Mazzone, M. Tumor-associated macrophages: A short compendium. Cell. Mol. Life Sci. 2019, 76, 1447–1458. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Medetgul-Ernar, K.; Davis, M.M. Standing on the shoulders of mice. Immunity 2022, 55, 1343–1353. [Google Scholar] [CrossRef]
- Hadadi, E.; Deschoemaeker, S.; Venegas, G.V.; Laoui, D. Heterogeneity and function of macrophages in the breast during homeostasis and cancer. Int. Rev. Cell Mol. Biol. 2022, 367, 149–182. [Google Scholar]
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef]
- Moeini, P.; Niedźwiedzka-Rystwej, P. Tumor-Associated Macrophages: Combination of Therapies, the Approach to Improve Cancer Treatment. Int. J. Mol. Sci. 2021, 22, 7239. [Google Scholar] [CrossRef]
- Pittet, M.J.; Michielin, O.; Migliorini, D. Clinical relevance of tumour-associated macrophages. Nat. Rev. Clin. Oncol. 2022, 19, 402–421. [Google Scholar] [CrossRef]
- Evans, R.; Alexander, P. Cooperation of Immune Lymphoid Cells with Macrophages in Tumour Immunity. Nature 1970, 228, 620–622. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geeraerts, X.; Fernández-Garcia, J.; Hartmann, F.J.; de Goede, K.E.; Martens, L.; Elkrim, Y.; Debraekeleer, A.; Stijlemans, B.; Vandekeere, A.; Rinaldi, G.; et al. Macrophages are metabolically heterogeneous within the tumor microenvironment. Cell Rep. 2021, 37, 110171. [Google Scholar] [CrossRef] [PubMed]
- Pombo Antunes, A.R.; Scheyltjens, I.; Lodi, F.; Messiaen, J.; Antoranz, A.; Duerinck, J.; Kancheva, D.; Martens, L.; De Vlaminck, K.; Van Hove, H. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 2021, 24, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Van Hove, H.; Martens, L.; Scheyltjens, I.; De Vlaminck, K.; Pombo Antunes, A.R.; De Prijck, S.; Vandamme, N.; De Schepper, S.; Van Isterdael, G.; Scott, C.L.; et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci. 2019, 22, 1021–1035. [Google Scholar] [CrossRef]
- Chávez-Galán, L.; Olleros, M.L.; Vesin, D.; Garcia, I. Much More than M1 and M2 Macrophages, There are also CD169(+) and TCR(+) Macrophages. Front. Immunol. 2015, 6, 263. [Google Scholar]
- Alborzinia, H.; Can, S.; Holenya, P.; Scholl, C.; Lederer, E.; Kitanovic, I.; Wölfl, S. Real-Time Monitoring of Cisplatin-Induced Cell Death. PLoS ONE 2011, 6, e19714. [Google Scholar] [CrossRef]
- Garrido-Martin, E.M.; Mellows, T.W.; Clarke, J.; Ganesan, A.P.; Wood, O.; Cazaly, A.; Seumois, G.; Chee, S.J.; Alzetani, A.; King, E.V.; et al. M1hot tumor-associated macrophages boost tissue-resident memory T cells infiltration and survival in human lung cancer. J. Immunother. Cancer 2020, 8, e000778. [Google Scholar] [CrossRef]
- Casanova-Acebes, M.; Dalla, E.; Leader, A.M.; LeBerichel, J.; Nikolic, J.; Morales, B.M.; Brown, M.; Chang, C.; Troncoso, L.; Chen, S.T.; et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature 2021, 595, 578–584. [Google Scholar] [CrossRef]
- Aggarwal, N.R.; King, L.S.; D’Alessio, F.R. Diverse macrophage populations mediate acute lung inflammation and resolution. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L709–L725. [Google Scholar] [CrossRef]
- Zhang, N.; Kim, S.H.; Gainullina, A.; Erlich, E.C.; Onufer, E.J.; Kim, J.; Czepielewski, R.S.; Helmink, B.A.; Dominguez, J.R.; Saunders, B.T.; et al. LYVE1+ macrophages of murine peritoneal mesothelium promote omentum-independent ovarian tumor growth. J. Exp. Med. 2021, 218, e20210924. [Google Scholar] [CrossRef]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Mat Zin, A.A.; Ang, K.C.; Ch’ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2019, 9, 1512. [Google Scholar] [CrossRef] [PubMed]
- Larionova, I.; Cherdyntseva, N.; Liu, T.; Patysheva, M.; Rakina, M.; Kzhyshkowska, J. Interaction of tumor-associated macrophages and cancer chemotherapy. OncoImmunology 2019, 8, e1596004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Schioppa, T.; Porta, C.; Allavena, P.; Sica, A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006, 25, 315–322. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P. The interaction of anticancer therapies with tumor-associated macrophages. J. Exp. Med. 2015, 212, 435–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Liu, L.; Che, G.; Yu, N.; Dai, F.; You, Z. The M1 form of tumor-associated macrophages in non-small cell lung cancer is positively associated with survival time. BMC Cancer 2010, 10, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef] [Green Version]
- Krishna, C.; DiNatale, R.G.; Kuo, F.; Srivastava, R.M.; Vuong, L.; Chowell, D.; Gupta, S.; Vanderbilt, C.; Purohit, A.T.; Liu, M.; et al. Single-cell sequencing links multiregional immune landscapes and tissue-resident T cells in ccRCC to tumor topology and therapy efficacy. Cancer Cell 2021, 39, 662–677.e6. [Google Scholar] [CrossRef]
- House, I.G.; Savas, P.; Lai, J.; Chen, A.X.Y.; Oliver, A.J.; Teo, Z.L.; Todd, K.L.; Henderson, M.A.; Giuffrida, L.; Petley, E.V.; et al. Macrophage-Derived CXCL9 and CXCL10 Are Required for Antitumor Immune Responses Following Immune Checkpoint Blockade. Clin. Cancer Res. 2020, 26, 487–504. [Google Scholar] [CrossRef] [Green Version]
- Jaggi, U.; Yang, M.; Matundan, H.H.; Hirose, S.; Shah, P.K.; Sharifi, B.G.; Ghiasi, H. Increased phagocytosis in the presence of enhanced M2-like macrophage responses correlates with increased primary and latent HSV-1 infection. PLoS Pathog. 2020, 16, e1008971. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Elbadawi, M.; Efferth, T. Multiple cell death modalities and their key features (Review). World Acad. Sci. J. 2020, 2, 39–48. [Google Scholar] [CrossRef]
- Poljšak, B.; Milisav, I. Clinical implications of cellular stress responses. Bosn. J. Basic Med. Sci. 2012, 12, 122–126. [Google Scholar] [CrossRef] [Green Version]
- Sprooten, J.; Garg, A.D. Type I interferons and endoplasmic reticulum stress in health and disease. Int. Rev. Cell Mol. Biol. 2020, 350, 63–118. [Google Scholar] [PubMed]
- Sprooten, J.; Agostinis, P.; Garg, A.D. Type I interferons and dendritic cells in cancer immunotherapy. Int. Rev. Cell Mol. Biol. 2019, 348, 217–262. [Google Scholar]
- Jin, S.; DiPaola, R.S.; Mathew, R.; White, E. Metabolic catastrophe as a means to cancer cell death. J. Cell Sci. 2007, 120 Pt 3, 379–383. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Nowis, D.; Golab, J.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. Immunogenic cell death, DAMPs and anticancer therapeutics: An emerging amalgamation. Biochim. Biophys. Acta 2010, 1805, 53–71. [Google Scholar] [CrossRef]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.-T.; Zhou, T.-T.; Liu, B.; Bao, J.-K. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef]
- Bohlson, S.S.; O’Conner, S.D.; Hulsebus, H.J.; Eho, M.-M.; Fraser, D.A. Complement, C1q, and C1q-Related Molecules Regulate Macrophage Polarization. Front. Immunol. 2014, 5, 402. [Google Scholar] [CrossRef] [Green Version]
- Tesniere, A.; Panaretakis, T.; Kepp, O.; Apetoh, L.; Ghiringhelli, F.; Zitvogel, L.; Kroemer, G. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ. 2008, 15, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Dillon, C.P.; Green, D.R. Molecular Cell Biology of Apoptosis and Necroptosis in Cancer. Adv. Exp. Med. Biol. 2016, 930, 1–23. [Google Scholar]
- Ziegler, U.; Groscurth, P. Morphological Features of Cell Death. News Physiol. Sci. 2004, 19, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Sprooten, J.; De Wijngaert, P.; Vanmeerbeerk, I.; Martin, S.; Vangheluwe, P.; Schlenner, S.; Krysko, D.V.; Parys, J.B.; Bultynck, G.; Vandenabeele, P.; et al. Necroptosis in Immuno-Oncology and Cancer Immunotherapy. Cells 2020, 9, 1823. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.; Lu, M.; Petersen, S.; Ashkenazi, A. Apoptosis Initiation Through the Cell-Extrinsic Pathway. Meth. Enzymol. 2014, 544, 99–128. [Google Scholar]
- Fulda, S.; Debatin, K.-M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [Green Version]
- Vande Walle, L.; Lamkanfi, M. Pyroptosis. Curr. Biol. 2016, 26, R568–R572. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Tripathi, C.; Tewari, B.N.; Kanchan, R.K.; Baghel, K.S.; Nautiyal, N.; Shrivastava, R.; Kaur, H.; Bhatt, M.L.B.; Bhadauria, S. Macrophages are recruited to hypoxic tumor areas and acquire a Pro-Angiogenic M2-Polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget 2014, 5, 5350–5368. [Google Scholar] [CrossRef] [Green Version]
- Murdoch, C.; Lewis, C.E. Macrophage migration and gene expression in response to tumor hypoxia. Int. J. Cancer 2005, 117, 701–708. [Google Scholar] [CrossRef]
- Lewis, C.; Murdoch, C. Macrophage Responses to Hypoxia: Implications for Tumor Progression and Anti-Cancer Therapies. Am. J. Pathol. 2005, 167, 627–635. [Google Scholar] [CrossRef]
- Henze, A.-T.; Mazzone, M. The impact of hypoxia on tumor-associated macrophages. J. Clin. Investig. 2016, 126, 3672–3679. [Google Scholar] [CrossRef] [PubMed]
- Vito, A.; El-Sayes, N.; Mossman, K. Hypoxia-Driven Immune Escape in the Tumor Microenvironment. Cells 2020, 9, 992. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Ohno, Y.; Suzuki, N.; Kamei, T.; Koike, K.; Inagawa, H.; Kohchi, C.; Soma, G.-I.; Inoue, M. Correlation of histological localization of tumor-associated macrophages with clinicopathological features in endometrial cancer. Anticancer Res. 2004, 24, 3335–3342. [Google Scholar] [PubMed]
- Leek, R.D.; Lewis, E.C.; Whitehouse, R.; Greenall, M.; Clarke, J.; Harris, A. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996, 56, 4625–4629. [Google Scholar] [PubMed]
- Ferrara, N. Vascular Endothelial Growth Factor: Basic Science and Clinical Progress. Endocr. Rev. 2004, 25, 581–611. [Google Scholar] [CrossRef]
- Matschurat, S.; Knies, U.E.; Person, V.; Fink, L.; Stoelcker, B.; Ebenebe, C.; Behrensdorf, H.A.; Schaper, J.; Clauss, M. Regulation of EMAP II by Hypoxia. Am. J. Pathol. 2003, 162, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Youssef, M.; Heng, Y.M.; Powe, D.G.; Edgson, J.; Ellis, I.; Murray, C. Hypoxia-induced EMAP-II transcription in colorectal cancer. Egypt. J. Immunol. 2010, 17, 121–129. [Google Scholar]
- Korbecki, J.; Kojder, K.; Kapczuk, P.; Kupnicka, P.; Gawrońska-Szklarz, B.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. The Effect of Hypoxia on the Expression of CXC Chemokines and CXC Chemokine Receptors—A Review of Literature. Int. J. Mol. Sci. 2021, 22, 843. [Google Scholar] [CrossRef]
- Grimshaw, M.J. Endothelins and hypoxia-inducible factor in cancer. Endocr. Relat. Cancer 2007, 14, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Grimshaw, M.J.; Wilson, J.L.; Balkwill, F.R. Endothelin-2 is a macrophage chemoattractant: Implications for macrophage distribution in tumors. Eur. J. Immunol. 2002, 32, 2393–2400. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Giannoudis, A.; Lewis, C.E. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 2004, 104, 2224–2234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosco, M.C.; Reffo, G.; Puppo, M.; Varesio, L. Hypoxia inhibits the expression of the CCR5 chemokine receptor in macrophages. Cell. Immunol. 2004, 228, 1–7. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Gabrilovich, D.I. Hypoxia-inducible factors in regulation of immune responses in tumour microenvironment. Immunology 2014, 143, 512–519. [Google Scholar] [CrossRef]
- Laoui, D.; Van Overmeire, E.; Di Conza, G.; Aldeni, C.; Keirsse, J.; Morias, Y.; Movahedi, K.; Houbracken, I.; Schouppe, E.; Elkrim, Y.; et al. Tumor Hypoxia Does Not Drive Differentiation of Tumor-Associated Macrophages but Rather Fine-Tunes the M2-like Macrophage Population. Cancer Res. 2014, 74, 24–30. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Lewis, C.E.; Pollard, J.W. Distinct Role of Macrophages in Different Tumor Microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [Green Version]
- Burke, B.; Giannoudis, A.; Corke, K.P.; Gill, D.; Wells, M.; Ziegler-Heitbrock, L.; Lewis, C.E. Hypoxia-Induced Gene Expression in Human Macrophages: Implications for ischemic tissues and hypoxia-regulated gene therapy. Am. J. Pathol. 2003, 163, 1233–1243. [Google Scholar] [CrossRef]
- Casazza, A.; Laoui, D.; Wenes, M.; Rizzolio, S.; Bassani, N.; Mambretti, M.; Deschoemaeker, S.; Van Ginderachter, J.A.; Tamagnone, L.; Mazzone, M. Impeding Macrophage Entry into Hypoxic Tumor Areas by Sema3A/Nrp1 Signaling Blockade Inhibits Angiogenesis and Restores Antitumor Immunity. Cancer Cell 2013, 24, 695–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Zuo, H.; Xiong, H.; Kolar, M.J.; Chu, Q.; Saghatelian, A.; Siegwart, D.J.; Wan, Y. Gpr132 sensing of lactate mediates tumor–macrophage interplay to promote breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2017, 114, 580–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundø, K.; Trauelsen, M.; Pedersen, S.F.; Schwartz, T.W. Why Warburg Works: Lactate Controls Immune Evasion through GPR81. Cell Metab. 2020, 31, 666–668. [Google Scholar] [CrossRef]
- Wang, J.; Mi, S.; Ding, M.; Li, X.; Yuan, S. Metabolism and polarization regulation of macrophages in the tumor microenvironment. Cancer Lett. 2022, 543, 215766. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-C.; Yan, X.-Y.; Yu, W.-W.; Liang, X.-Q.; Du, X.-Y.; Liu, Z.-C.; Long, J.-P.; Zhao, G.-H.; Liu, H.-B. Lactic acid in macrophage polarization: The significant role in inflammation and cancer. Int. Rev. Immunol. 2022, 41, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, B.; Wang, X.; Guan, G.; Xin, Y.; Sun, Y.D.; Wang, J.H.; Guo, Y.; Zang, Y.J. Macrophage transcriptome modification induced by hypoxia and lactate. Exp. Ther. Med. 2019, 18, 4811–4819. [Google Scholar] [CrossRef]
- Wenes, M.; Shang, M.; Di Matteo, M.; Goveia, J.; Martín-Pérez, R.; Serneels, J.; Prenen, H.; Ghesquière, B.; Carmeliet, P.; Mazzone, M. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab. 2016, 24, 701–715. [Google Scholar] [CrossRef] [Green Version]
- Cappellesso, F.; Orban, M.-P.; Shirgaonkar, N.; Berardi, E.; Serneels, J.; Neveu, M.-A.; di Molfetta, D.; Piccapane, F.; Caroppo, R.; Debellis, L.; et al. Targeting the bicarbonate transporter SLC4A4 overcomes immunosuppression and immunotherapy resistance in pancreatic cancer. Nat. Cancer 2022. [Google Scholar] [CrossRef]
- Rabold, K.; Netea, M.G.; Adema, G.J.; Netea-Maier, R.T. Cellular metabolism of tumor-associated macrophages—Functional impact and consequences. FEBS Lett. 2017, 591, 3022–3041. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Stradmann-Bellinghausen, B.; Yakubov, E.; Savaskan, N.; Régnier-Vigouroux, A. Glioblastoma cells induce differential glutamatergic gene expressions in human tumor-associated microglia/macrophages and monocyte-derived macrophages. Cancer Biol. Ther. 2015, 16, 1205–1213. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, E.M.; Menga, A.; Martín-Pérez, R.; Quinto, A.; Riera-Domingo, C.; De Tullio, G.; Hooper, D.C.; Lamers, W.H.; Ghesquière, B.; McVicar, D.W.; et al. Pharmacologic or Genetic Targeting of Glutamine Synthetase Skews Macrophages toward an M1-like Phenotype and Inhibits Tumor Metastasis. Cell Rep. 2017, 20, 1654–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, M.-H.; Sun, I.-H.; Zhao, L.; Leone, R.D.; Sun, I.M.; Xu, W.; Collins, S.L.; Tam, A.J.; Blosser, R.L.; Patel, C.H.; et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J. Clin. Investig. 2020, 130, 3865–3884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mojsilovic, S.S.; Mojsilovic, S.; Villar, V.H.; Santibanez, J.F. The Metabolic Features of Tumor-Associated Macrophages: Opportunities for Immunotherapy? Anal. Cell Pathol. 2021, 2021, 5523055. [Google Scholar] [CrossRef] [PubMed]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An Interaction between Kynurenine and the Aryl Hydrocarbon Receptor Can Generate Regulatory T Cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 Kinase in T Cells Mediates Proliferative Arrest and Anergy Induction in Response to Indoleamine 2,3-Dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Giannone, G.; Ghisoni, E.; Genta, S.; Scotto, G.; Tuninetti, V.; Turinetto, M.; Valabrega, G. Immuno-Metabolism and Microenvironment in Cancer: Key Players for Immunotherapy. Int. J. Mol. Sci. 2020, 21, 4414. [Google Scholar] [CrossRef]
- Corn, K.C.; Windham, M.A.; Rafat, M. Lipids in the tumor microenvironment: From cancer progression to treatment. Prog. Lipid Res. 2020, 80, 101055. [Google Scholar] [CrossRef]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef] [Green Version]
- Timperi, E.; Gueguen, P.; Molgora, M.; Magagna, I.; Kieffer, Y.; Lopez-Lastra, S.; Sirven, P.; Baudrin, L.G.; Baulande, S.; Nicolas, A.; et al. Lipid-associated macrophages are induced by cancer-associated fibroblasts and mediate immune suppression in breast cancer. Cancer Res. 2022, 82, 3291–3306. [Google Scholar] [CrossRef]
- Masetti, M.; Carriero, R.; Portale, F.; Marelli, G.; Morina, N.; Pandini, M.; Iovino, M.; Partini, B.; Erreni, M.; Ponzetta, A.; et al. Lipid-loaded tumor-associated macrophages sustain tumor growth and invasiveness in prostate cancer. J. Exp. Med. 2022, 219, e20210564. [Google Scholar] [CrossRef]
- Gregory, C.D.; Paterson, M. An apoptosis-driven “onco-regenerative niche”: Roles of tumour-associated macrophages and extracellular vesicles. Philos. Trans. R. Soc. Lond B Biol. Sci. 2018, 373, 1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehne, N.; Mora, J.; Namgaladze, D.; Weigert, A.; Brüne, B. Cancer cell and macrophage cross-talk in the tumor microenvironment. Curr. Opin. Pharmacol. 2017, 35, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Weigert, A.; Tzieply, N.; Von Knethen, A.; Johann, A.M.; Schmidt, H.; Geisslinger, G.; Brüne, B. Tumor Cell Apoptosis Polarizes Macrophages—Role of Sphingosine-1-Phosphate. Mol. Biol. Cell 2007, 18, 3810–3819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voss, J.; Ford, A.C.; Petrova, S.; Melville, L.; Paterson, M.; Pound, J.D.; Holland, P.; Giotti, B.; Freeman, T.; Gregory, C.D. Modulation of macrophage antitumor potential by apoptotic lymphoma cells. Cell Death Differ. 2017, 24, 971–983. [Google Scholar] [CrossRef] [Green Version]
- Gardai, S.J.; Bratton, D.L.; Ogden, C.A.; Henson, P.M. Recognition ligands on apoptotic cells: A perspective. J. Leukoc. Biol. 2006, 79, 896–903. [Google Scholar] [CrossRef]
- Gregory, C.D.; Pound, J.D. Microenvironmental influences of apoptosis in vivo and in vitro. Apoptosis 2010, 15, 1029–1049. [Google Scholar] [CrossRef]
- Fadok, V.A.; Voelker, D.R.; Campbell, P.A.; Cohen, J.J.; Bratton, D.L.; Henson, P.M. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 1992, 148, 2207–2216. [Google Scholar]
- Matsura, T.; Serinkan, B.F.; Jiang, J.; Kagan, E.V. Phosphatidylserine peroxidation/externalization during staurosporine-induced apoptosis in HL-60 cells. FEBS Lett. 2002, 524, 25–30. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Huang, X.; Lynn, K.D.; Thorpe, P.E. Phosphatidylserine-Targeting Antibody Induces M1 Macrophage Polarization and Promotes Myeloid-Derived Suppressor Cell Differentiation. Cancer Immunol. Res. 2013, 1, 256–268. [Google Scholar] [CrossRef] [Green Version]
- Weigert, A.; Mora, J.; Sekar, D.; Syed, S.; Brüne, B. Killing Is Not Enough: How Apoptosis Hijacks Tumor-Associated Macrophages to Promote Cancer Progression. Adv. Exp. Med. Biol. 2016, 930, 205–239. [Google Scholar]
- Cuvillier, O.; Ader, I.; Bouquerel, P.; Brizuela, L.; Gstalder, C.; Malavaud, B. Hypoxia, therapeutic resistance, and sphingosine 1-phosphate. Adv. Cancer Res. 2013, 117, 117–141. [Google Scholar] [PubMed]
- Cuvillier, O.; Ader, I. Hypoxia-inducible factors and sphingosine 1-phosphate signaling. Anti-Cancer Agents Med. Chem. 2011, 11, 854–862. [Google Scholar] [CrossRef]
- Brecht, K.; Weigert, A.; Hu, J.; Popp, R.; Fisslthaler, B.; Korff, T.; Fleming, I.; Geisslinger, G.; Brüne, B. Macrophages programmed by apoptotic cells promote angiogenesis via prostaglandin E2. FASEB J. 2011, 25, 2408–2417. [Google Scholar] [CrossRef]
- Olesch, C.; Sha, W.; Angioni, C.; Sha, L.K.; Açaf, E.; Patrignani, P.; Jakobsson, P.-J.; Radeke, H.H.; Grösch, S.; Geisslinger, G.; et al. MPGES-1-derived PGE2 suppresses CD80 expression on tumor-associated phagocytes to inhibit anti-tumor immune responses in breast cancer. Oncotarget 2015, 6, 10284–10296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 18, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Z.; Yang, Z.; Xie, L.; DeWitt, J.P.; Chen, Y. Cancer therapy in the necroptosis era. Cell Death Differ. 2016, 23, 748–756. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Vanmeerbeek, I.; Sprooten, J.; De Ruysscher, D.; Tejpar, S.; Vandenberghe, P.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; et al. Trial watch: Chemotherapy-induced immunogenic cell death in immuno-oncology. OncoImmunology 2020, 9, 1703449. [Google Scholar] [CrossRef]
- Seebacher, N.A.; Stacy, A.E.; Porter, G.M.; Merlot, A.M. Clinical development of targeted and immune based anti-cancer therapies. J. Exp. Clin. Cancer Res. 2019, 38, 156. [Google Scholar] [CrossRef] [Green Version]
- Shewach, D.S.; Kuchta, R.D. Introduction to Cancer Chemotherapeutics. Chem. Rev. 2009, 109, 2859–2861. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Hogg, G.D.; DeNardo, D.G. Rethinking immune checkpoint blockade: “Beyond the T cell”. J. Immunother. Cancer 2021, 9, e001460. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.B.; Apetoh, L. Radiotherapy and Immunogenic Cell Death. Semin. Radiat. Oncol. 2015, 25, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Z.; Luo, Y. Targeting macrophages in cancer immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 127. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprooten, J.; Coosemans, A.; Garg, A.D. A first-in-class, non-invasive, immunodynamic biomarker approach for precision immuno-oncology. OncoImmunology 2022, 11, 2024692. [Google Scholar] [CrossRef]
- Sprooten, J.; Vankerckhoven, A.; Vanmeerbeek, I.; Borras, D.M.; Berckmans, Y.; Wouters, R.; Laureano, R.S.; Baert, T.; Boon, L.; Landolfo, C.; et al. Peripherally-driven myeloid NFkB and IFN/ISG responses predict malignancy risk, survival, and immunotherapy regime in ovarian cancer. J. Immunother. Cancer 2021, 9, e003609. [Google Scholar] [CrossRef]
- Maharjan, R.; Choi, J.U.; Kweon, S.; Pangeni, R.; Lee, N.K.; Park, S.J.; Chang, K.-Y.; Park, J.W.; Byun, Y. A novel oral metronomic chemotherapy provokes tumor specific immunity resulting in colon cancer eradication in combination with anti-PD-1 therapy. Biomaterials 2022, 281, 121334. [Google Scholar] [CrossRef]
- Michaud, M.; Sukkurwala, A.Q.; Di Sano, F.; Zitvogel, L.; Kepp, O.; Kroemer, G. Synthetic induction of immunogenic cell death by genetic stimulation of endoplasmic reticulum stress. OncoImmunology 2014, 3, e28276. [Google Scholar] [CrossRef]
- Wang, H.-T.; Lee, H.-I.; Guo, J.-H.; Chen, S.-H.; Liao, Z.-K.; Huang, K.-W.; Torng, P.-L. Calreticulin promotes tumor lymphocyte infiltration and enhances the antitumor effects of immunotherapy by up-regulating the endothelial expression of adhesion molecules. Int. J. Cancer 2012, 130, 2892–2902. [Google Scholar] [CrossRef]
- Shakfa, N.; Siemens, D.R.; Koti, M. Revisiting immunogenic cell death to improve treatment response in cancer. Biol. Mech. Adv. Approaches Overcoming Cancer Drug Resist. 2021, 8, 65–90. [Google Scholar]
- Anfray, C.; Ummarino, A.; Andón, F.T.; Allavena, P. Current Strategies to Target Tumor-Associated-Macrophages to Improve Anti-Tumor Immune Responses. Cells 2019, 9, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genard, G.; Lucas, S.; Michiels, C. Reprogramming of Tumor-Associated Macrophages with Anticancer Therapies: Radiotherapy versus Chemo- and Immunotherapies. Front. Immunol. 2017, 8, 828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crittenden, M.R.; Cottam, B.; Savage, T.; Nguyen, C.; Newell, P.; Gough, M.J. Expression of NF-κB p50 in tumor stroma limits the control of tumors by radiation therapy. PLoS ONE 2012, 7, e39295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrandt, G.; Radlingmayr, A.; Rosenthal, S.; Rothe, R.; Jahns, J.; Hindemith, M.; Rödel, F.; Kamprad, F. Low-dose radiotherapy (LD-RT) and the modulation of iNOS expression in adjuvant-induced arthritis in rats. Int. J. Radiat. Biol. 2003, 79, 993–1001. [Google Scholar] [CrossRef]
- Murgaski, A.; Kiss, M.; Van Damme, H.; Kancheva, D.; Vanmeerbeek, I.; Keirsse, J.; Hadadi, E.; Brughmans, J.; Arnouk, S.M.; Hamouda, A.E.I.; et al. Efficacy of CD40 agonists is mediated by distinct cDC subsets and subverted by suppressive macrophages. Cancer Res. 2022, 82, 3785–3801. [Google Scholar] [CrossRef]
- Brom, V.C.; Burger, C.; Wirtz, D.C.; Schildberg, F.A. The Role of Immune Checkpoint Molecules on Macrophages in Cancer, Infection, and Autoimmune Pathologies. Front. Immunol. 2022, 13, 837645. [Google Scholar] [CrossRef]
- Sun, N.-Y.; Chen, Y.-L.; Wu, W.-Y.; Lin, H.-W.; Chiang, Y.-C.; Chang, C.-F.; Tai, Y.-J.; Hsu, H.-C.; Chen, C.-A.; Sun, W.-Z.; et al. Blockade of PD-L1 Enhances Cancer Immunotherapy by Regulating Dendritic Cell Maturation and Macrophage Polarization. Cancers 2019, 11, 1400. [Google Scholar] [CrossRef] [Green Version]
- Koh, M.Y.; Sayegh, N.; Agarwal, N. Seeing the forest for the trees-single-cell atlases link CD8+ T cells and macrophages to disease progression and treatment response in kidney cancer. Cancer Cell 2021, 39, 594–596. [Google Scholar] [CrossRef]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef]
- Chow, A.; Schad, S.; Green, M.D.; Hellmann, M.D.; Allaj, V.; Ceglia, N.; Zago, G.; Shah, N.S.; Sharma, S.K.; Mattar, M.; et al. Tim-4+ cavity-resident macrophages impair anti-tumor CD8+ T cell immunity. Cancer Cell 2021, 39, 973–988.e9. [Google Scholar] [CrossRef]
- Zeng, D.; Ye, Z.; Wu, J.; Zhou, R.; Fan, X.; Wang, G.; Huang, Y.; Wu, J.; Sun, H.; Wang, M.; et al. Macrophage correlates with immunophenotype and predicts anti-PD-L1 response of urothelial cancer. Theranostics 2020, 10, 7002–7014. [Google Scholar] [CrossRef]
- Antoranz, A.; Van Herck, Y.; Bolognesi, M.M.; Lynch, S.M.; Rahman, A.; Gallagher, W.M.; Boecxstaens, V.; Marine, J.-C.; Cattoretti, G.; van den Oord, J.J.; et al. Mapping the Immune Landscape in Metastatic Melanoma Reveals Localized Cell-Cell Interactions That Predict Immunotherapy Response. Cancer Res. 2022, 82, 3275–3290. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, S.; Geeraerts, X.; Bolli, E.; Keirsse, J.; Kiss, M.; Pombo Antunes, A.R.; Van Damme, H.; De Vlaminck, K.; Movahedi, K.; Laoui, D.; et al. Beyond the M-CSF receptor - novel therapeutic targets in tumor-associated macrophages. FEBS J. 2018, 285, 777–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Wang, Y.; Xia, R.; Wei, Y.; Wei, X. Role of the CCL2-CCR2 signalling axis in cancer: Mechanisms and therapeutic targeting. Cell Prolif. 2021, 54, e13115. [Google Scholar] [CrossRef] [PubMed]
- Yasui, H.; Kajiyama, H.; Tamauchi, S.; Suzuki, S.; Peng, Y.; Yoshikawa, N.; Sugiyama, M.; Nakamura, K.; Kikkawa, F. CCL2 secreted from cancer-associated mesothelial cells promotes peritoneal metastasis of ovarian cancer cells through the P38-MAPK pathway. Clin Exp Metastasis 2019, 37, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lu, Y.; Pienta, K.J. Multiple roles of chemokine (CC motif) ligand 2 in promoting prostate cancer growth. J. Natl. Cancer 2010, 102, 522–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brana, I.; Calles, A.; LoRusso, P.M.; Yee, L.K.; Puchalski, T.A.; Seetharam, S.; Zhong, B.; de Boer, C.J.; Tabernero, J.; Calvo, E. Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: An open-label, multicenter phase 1b study. Target Oncol. 2015, 10, 111–123. [Google Scholar] [CrossRef]
- Pienta, K.J.; Machiels, J.-P.; Schrijvers, D.; Alekseev, B.; Shkolnik, M.; Crabb, S.J.; Li, S.; Seetharam, S.; Puchalski, T.A.; Takimoto, C.; et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Investig. New Drugs 2013, 31, 760–768. [Google Scholar] [CrossRef]
- Fei, L.; Ren, X.; Yu, H.; Zhan, Y. Targeting the CCL2/CCR2 axis in cancer immunotherapy: One stone, three birds? Front. Immunol. 2021, 12, 771210. [Google Scholar] [CrossRef]
- Guo, F.; Wang, Y.; Liu, J.; Mok, S.C.; Xue, F.; Zhang, W. CXCL12/CXCR4: A symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene 2016, 35, 816–826. [Google Scholar] [CrossRef]
- Biasci, D.; Smoragiewicz, M.; Connell, C.M.; Wang, Z.; Gao, Y.; Thaventhiran, J.E.D.; Basu, B.; Magiera, L.; Johnson, T.I.; Bax, L.; et al. CXCR4 inhibition in human pancreatic and colorectal cancers induces an integrated immune response. Proc. Natl. Acad. Sci. USA 2020, 117, 28960–28970. [Google Scholar] [CrossRef] [PubMed]
- Petty, A.J.; Owen, D.H.; Yang, Y.; Huang, X. Targeting Tumor-Associated Macrophages in Cancer Immunotherapy. Cancers 2021, 13, 5318. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C. Clinical Development of Colony-Stimulating Factor 1 Receptor (CSF1R) Inhibitors. J. Immunother. Precis. Oncol. 2021, 4, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.-M.; Ries, C.H.; Rüttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef]
- Ao, J.Y.; Zhu, X.D.; Chai, Z.T.; Cai, H.; Zhang, Y.Y. Colony-Stimulating Factor 1 Receptor Blockade Inhibits Tumor Growth by Altering the Polarization of Tumor-Associated Macrophages in Hepatocellular. Mol. Cancer 2017, 16, 1544–1554. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.C.; Gil-Martin, M.; Bauer, T.M.; Naing, A.; Lim, D.W.T. Abstract CT171: Phase I study of BLZ945 alone and with spartalizumab (PDR001) in patients (pts) with advanced solid tumors. Cancer Res. 2020, 80, CT171. [Google Scholar] [CrossRef]
- Gomez-Roca, C.A.; Italiano, A.; Le Tourneau, C.; Cassier, P.A.; Toulmonde, M.; D’Angelo, S.P.; Campone, M.; Weber, K.L.; Loirat, D.; Cannarile, M.A.; et al. Phase I Study of Emactuzumab Single Agent or in Combination with Paclitaxel in Patients with Advanced/Metastatic Solid Tumors Reveals Depletion of Immunosuppressive M2-like Macrophages. Ann. Oncol. 2019, 30, 1381–1392. [Google Scholar] [CrossRef]
- Hensler, M.; Rakova, J.; Kasikova, L.; Lanickova, T.; Pasulka, J.; Holicek, P.; Hraska, M.; Hrnciarova, T.; Kadlecova, P.; Schoenenberger, A.; et al. Peripheral gene signatures reveal distinct cancer patient immunotypes with therapeutic implications for autologous DC-based vaccines. Oncoimmunology 2022, 11, 2101596. [Google Scholar] [CrossRef]
- Kapp, K.; Volz, B.; Oswald, D.; Wittig, B.; Baumann, M.; Schmidt, M. Beneficial modulation of the tumor microenvironment and generation of anti-tumor responses by TLR9 agonist lefitolimod alone and in combination with checkpoint inhibitors. Oncoimmunology 2019, 8, e1659096. [Google Scholar] [CrossRef]
- Sato-Kaneko, F.; Yao, S.; Ahmadi, A.; Zhang, S.S.; Hosoya, T.; Kaneda, M.M.; Varner, J.A.; Pu, M.; Messer, K.S.; Guiducci, C.; et al. Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI Insight 2017, 2, e93397. [Google Scholar] [CrossRef]
- Le Naour, J.; Galluzzi, L.; Zitvogel, L.; Kroemer, G.; Vacchelli, E. Trial watch: TLR3 agonists in cancer therapy. Oncoimmunology 2020, 9, 1771143. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; García-Martínez, E.; Pitter, M.R.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Toll-like receptor agonists in cancer immunotherapy. Oncoimmunology 2018, 7, e1526250. [Google Scholar] [CrossRef] [PubMed]
- Laureano, R.S.; Sprooten, J.; Vanmeerbeerk, I.; Borras, D.M.; Govaerts, J.; Naulaerts, S.; Berneman, Z.N.; Beuselinck, B.; Bol, K.F.; Borst, J.; et al. Trial watch: Dendritic cell (DC)-based immunotherapy for cancer. Oncoimmunology 2022, 11, 2096363. [Google Scholar] [CrossRef] [PubMed]
- Phuengkham, H.; Song, C.; Lim, Y.T. A Designer Scaffold with Immune Nanoconverters for Reverting Immunosuppression and Enhancing Immune Checkpoint Blockade Therapy. Adv. Mater. Weinheim 2019, e1903242. [Google Scholar] [CrossRef]
- Zhang, F.; Parayath, N.N.; Ene, C.I.; Stephan, S.B.; Koehne, A.L.; Coon, M.E.; Holland, E.C.; Stephan, M.T. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat. Commun. 2019, 10, 3974. [Google Scholar] [CrossRef] [Green Version]
- Jia, N.; Wu, H.; Duan, J.; Wei, C.; Wang, K.; Zhang, Y. Polyethyleneimine-coated iron oxide nanoparticles as a vehicle for the delivery of small interfering RNA to macrophages in vitro and in vivo. J. Vis. Exp. 2019. [Google Scholar] [CrossRef] [Green Version]
- Nuhn, L.; Bolli, E.; Massa, S.; Vandenberghe, I.; Movahedi, K.; Devreese, B.; Van Ginderachter, J.A.; De Geest, B.G. Targeting Protumoral Tumor-Associated Macrophages with Nanobody-Functionalized Nanogels through Strain Promoted Azide Alkyne Cycloaddition Ligation. Bioconjug. Chem. 2018, 29, 2394–2405. [Google Scholar] [CrossRef]
- Oronsky, B.; Paulmurugan, R.; Foygel, K.; Scicinski, J.; Knox, S.J.; Peehl, D.; Zhao, H.; Ning, S.; Cabrales, P.; Summers, T.A.; et al. RRx-001: A systemically non-toxic M2-to-M1 macrophage stimulating and prosensitizing agent in Phase II clinical trials. Expert Opin. Investig. Drugs 2017, 26, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Brooke, G.; Holbrook, J.D.; Brown, M.H.; Barclay, A.N. Human lymphocytes interact directly with CD47 through a novel member of the signal regulatory protein (SIRP) family. J. Immunol. 2004, 173, 2562–2570. [Google Scholar] [CrossRef]
- Jalil, A.R.; Andrechak, J.C.; Discher, D.E. Macrophage checkpoint blockade: Results from initial clinical trials, binding analyses, and CD47-SIRPα structure-function. Antib. Ther. 2020, 3, 80–94. [Google Scholar] [CrossRef] [Green Version]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P.; et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Takimoto, C.H.; Feng, D.D.; McKenna, K.; Gip, P.; Liu, J.; Volkmer, J.-P.; Weissman, I.L.; Majeti, R. Therapeutic targeting of the macrophage immune checkpoint CD47 in myeloid malignancies. Front. Oncol. 2019, 9, 1380. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Spreafico, A.; Paterson, A.M.; Peluso, M. Results of a first-in-human phase I study of SRF231, a fully human, high-affinity anti-CD47 antibody. J. Clin. Oncol. 2020, 38, 3064. [Google Scholar] [CrossRef]
- Ansell, S.M.; Maris, M.B.; Lesokhin, A.M.; Chen, R.W. Phase I study of the CD47 blocker TTI-621 in patients with relapsed or refractory hematologic malignancies. Clin. Cancer 2021, 27, 2190–2199. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.M.; DeAngelo, D.J.; Palmer, J.M.; Seet, C.S.; Tallman, M.S.; Wei, X.; Li, Y.F.; Hock, N.; Burgess, M.R.; Hege, K.; et al. A Phase I Study of CC-90002, a Monoclonal Antibody Targeting CD47, in Patients with Relapsed and/or Refractory (R/R) Acute Myeloid Leukemia (AML) and High-Risk Myelodysplastic Syndromes (MDS): Final Results. Blood 2019, 134, 1320. [Google Scholar] [CrossRef]
- Sim, J.; Sockolosky, J.T.; Sangalang, E.; Izquierdo, S.; Pedersen, D.; Harriman, W.; Wibowo, A.S.; Carter, J.; Madan, A.; Doyle, L.; et al. Discovery of high affinity, pan-allelic, and pan-mammalian reactive antibodies against the myeloid checkpoint receptor SIRPα. MAbs 2019, 11, 1036–1052. [Google Scholar] [CrossRef] [Green Version]
- Dowlati, A.; Harvey, R.D.; Carvajal, R.D.; Hamid, O.; Klempner, S.J.; Kauh, J.S.W.; Peterson, D.A.; Yu, D.; Chapman, S.C.; Szpurka, A.M.; et al. LY3022855, an anti-colony stimulating factor-1 receptor (CSF-1R) monoclonal antibody, in patients with advanced solid tumors refractory to standard therapy: Phase 1 dose-escalation trial. Investig. New Drugs 2021. [Google Scholar] [CrossRef]
- Wang, S.; Yang, Y.; Ma, P.; Zha, Y.; Zhang, J.; Lei, A.; Li, N. CAR-macrophage: An extensive immune enhancer to fight cancer. EBioMedicine 2022, 76, 103873. [Google Scholar] [CrossRef]
- Su, S.; Lei, A.; Wang, X.; Lu, H.; Wang, S.; Yang, Y.; Li, N.; Zhang, Y.; Zhang, J. Induced CAR-Macrophages as a Novel Therapeutic Cell Type for Cancer Immune Cell Therapies. Cells 2022, 11, 1652. [Google Scholar] [CrossRef]
- Sly, L.M.; McKay, D.M. Macrophage immunotherapy: Overcoming impediments to realize promise. Trends Immunol. 2022. [Google Scholar] [CrossRef]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tian, L.; Dai, X.; Yu, H.; Wang, J.; Lei, A.; Zhu, M.; Xu, J.; Zhao, W.; Zhu, Y.; et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J Hematol Oncol. 2020, 13, 153. [Google Scholar] [CrossRef] [PubMed]
- Naulaerts, S.; Borras, D.M.; Martinez, A.A.; Messiaen, J.; Van Herck, Y.; Gelens, L.; Venken, T.; Vanmeerbeek, I.; More, S.; Sprooten, J.; et al. Immunogenomic, single-cell and spatial dissection of CD8+T cell exhaustion reveals critical determinants of cancer immunotherapy. bioRxiv 2021. [Google Scholar] [CrossRef]
- Vanmeerbeek, I.; Borras, D.M.; Sprooten, J.; Bechter, O.; Tejpar, S.; Garg, A.D. Early memory differentiation and cell death resistance in T cells predicts melanoma response to sequential anti-CTLA4 and anti-PD1 immunotherapy. Genes Immun. 2021, 22, 108–119. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Mechanism/Target | Cancer Type | Combination Agents | Identifier |
|---|---|---|---|
| CCR2/CCR5 | Pancreatic adenocarcinoma | Radiation, nivolumab, GVAX | NCT03767582 |
| NSCLC, hepatocellular carcinoma | Nivolumab | NCT04123379 | |
| Colorectal cancer, pancreatic cancer | Nivolumab, ipilimumab | NCT04721301 | |
| Colorectal cancer, pancreatic cancer | Nivolumab, chemotherapy | NCT03184870 | |
| CXCL12/CXCR4 | Multiple myeloma | G-CSF | NCT03246529 |
| Pancreatic adenocarcinoma | Cemiplimab | NCT04177810 | |
| Pancreatic adenocarcinoma | Pembrolizumab | NCT02907099 | |
| Pancreatic adenocarcinoma | Cemiplimab, gemcitabine, nab paclitaxel | NCT04543071 | |
| CSF1/CSF1R | Solid tumours | PDR001 | NCT02829723 |
| Sarcoma | Sirolimus | NCT02584647 | |
| Sarcoma, neurofibroma, leukaemia | - | NCT02390752 | |
| Melanoma | Vemurafenib, cobimetinib | NCT03101254 | |
| Breast cancer | Nivolumab, paclitaxel, carboplatin | NCT04331067 | |
| Leukaemia | - | NCT03922100 | |
| CD47/SIRPα | Myelodysplastic syndrome | Azacitidine | NCT04485065 |
| Hodgkin Lymphoma | Pembrolizumab | NCT04788043 | |
| B-cell lymphoma | Venetoclax, obinutuzumab | NCT04599634 | |
| Solid tumours | - | NCT04900519 | |
| Myelodysplastic Syndrome | Azacitidine | NCT04900350 | |
| Solid tumours | BI 754091 | NCT03990233 | |
| Solid tumours, lymphoma | - | NCT04306224 | |
| CAR macrophages | Solid tumours | - | NCT04660929 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanmeerbeek, I.; Govaerts, J.; Laureano, R.S.; Sprooten, J.; Naulaerts, S.; Borras, D.M.; Laoui, D.; Mazzone, M.; Van Ginderachter, J.A.; Garg, A.D. The Interface of Tumour-Associated Macrophages with Dying Cancer Cells in Immuno-Oncology. Cells 2022, 11, 3890. https://doi.org/10.3390/cells11233890
Vanmeerbeek I, Govaerts J, Laureano RS, Sprooten J, Naulaerts S, Borras DM, Laoui D, Mazzone M, Van Ginderachter JA, Garg AD. The Interface of Tumour-Associated Macrophages with Dying Cancer Cells in Immuno-Oncology. Cells. 2022; 11(23):3890. https://doi.org/10.3390/cells11233890
Chicago/Turabian StyleVanmeerbeek, Isaure, Jannes Govaerts, Raquel S. Laureano, Jenny Sprooten, Stefan Naulaerts, Daniel M. Borras, Damya Laoui, Massimiliano Mazzone, Jo A. Van Ginderachter, and Abhishek D. Garg. 2022. "The Interface of Tumour-Associated Macrophages with Dying Cancer Cells in Immuno-Oncology" Cells 11, no. 23: 3890. https://doi.org/10.3390/cells11233890
APA StyleVanmeerbeek, I., Govaerts, J., Laureano, R. S., Sprooten, J., Naulaerts, S., Borras, D. M., Laoui, D., Mazzone, M., Van Ginderachter, J. A., & Garg, A. D. (2022). The Interface of Tumour-Associated Macrophages with Dying Cancer Cells in Immuno-Oncology. Cells, 11(23), 3890. https://doi.org/10.3390/cells11233890