
Beta-Amyloid Instigates Dysfunction of Mitochondria in Cardiac Cells

 , , and
, , and 
Abstract
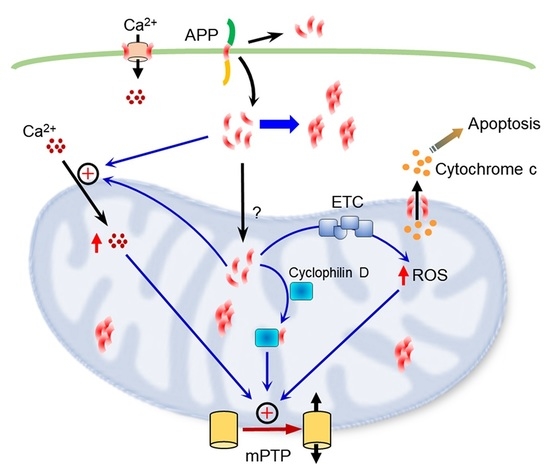
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Isolation of Mitochondria from Rat Hearts
2.3. Mitochondrial Permeability Transition Pore (mPTP) Opening
2.4. Primary Primary Human Coronary Artery Endothelial Cells (HCAEC)
2.5. H9c2 Cardiomyoblasts
2.6. Permeabilization of Cells
2.7. Mitochondrial CRC Assay
2.8. Cellular ROS Assay
2.9. Fluorescence Immunocytochemistry
2.10. Oligomerization and the Treatments of Aβ Peptides
2.11. Analysis of Cell Viability
2.12. Analysis of Cellular ATP, ΔΨm, and mtROS
2.13. Apoptosis Assay
2.14. Statistical Analysis
3. Results
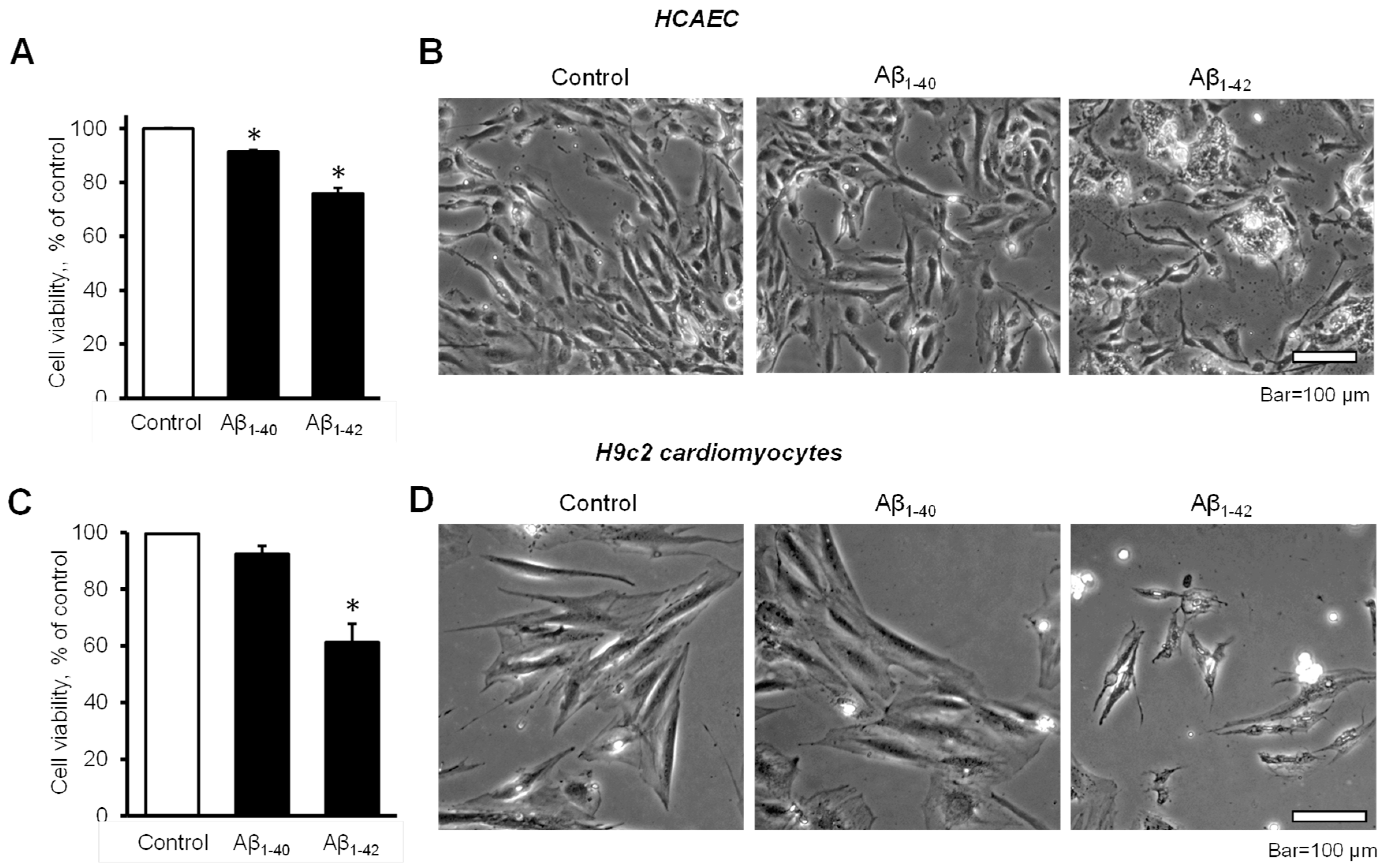
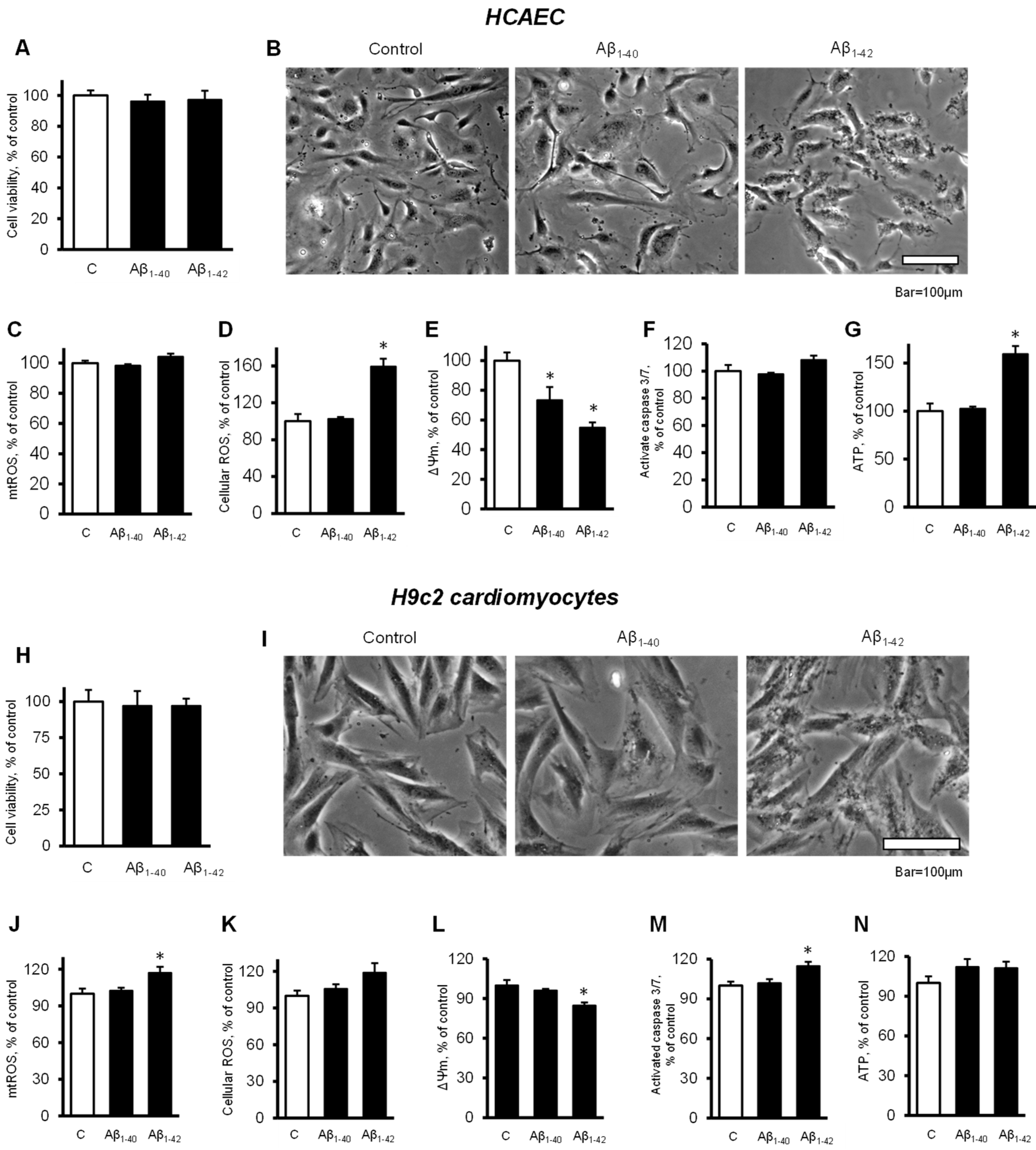
3.1. Aβ Decreased Cell Viability and Impaired Cell Morphology
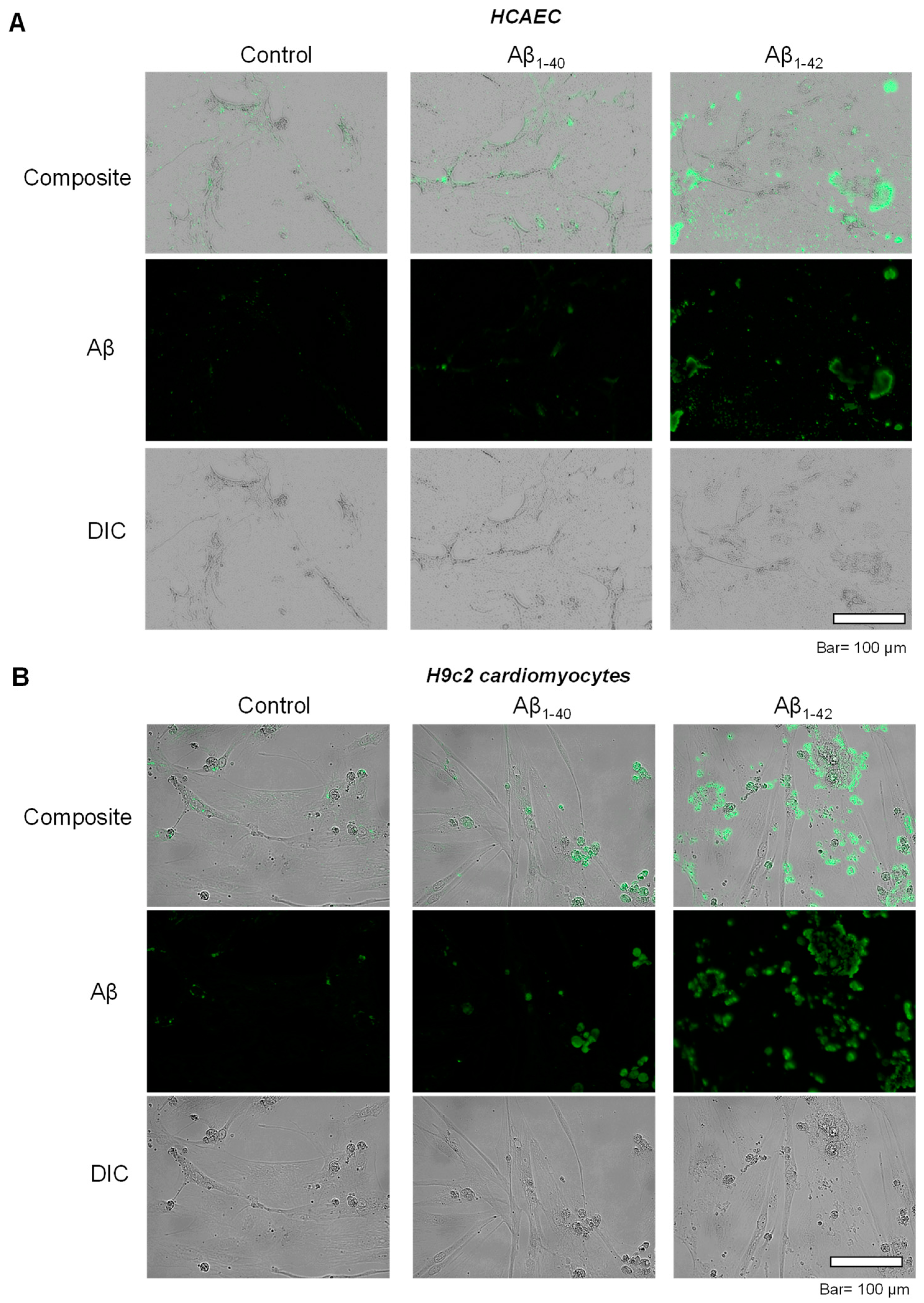
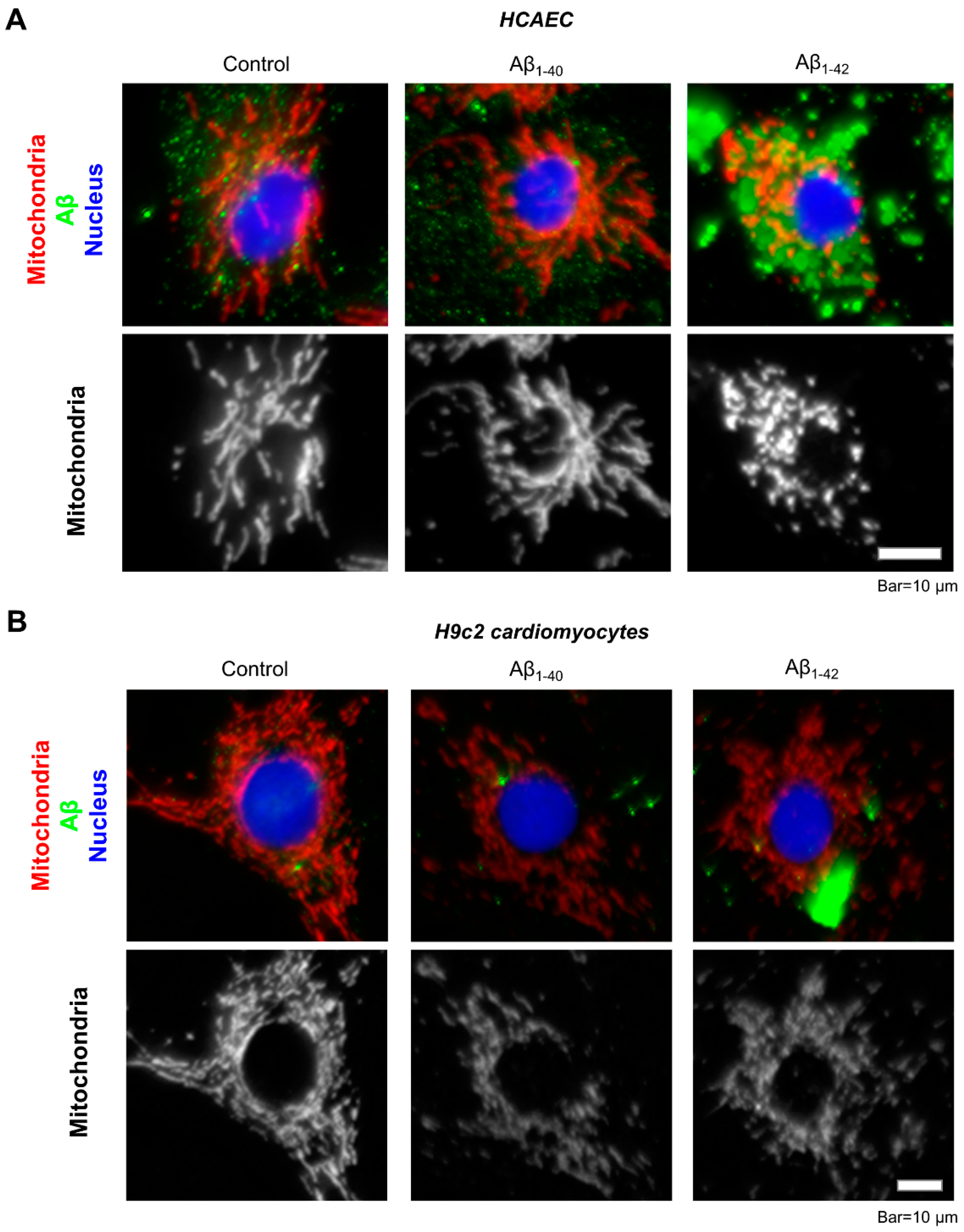
3.2. Aggregated Aβ Accumulated Inside of Cells
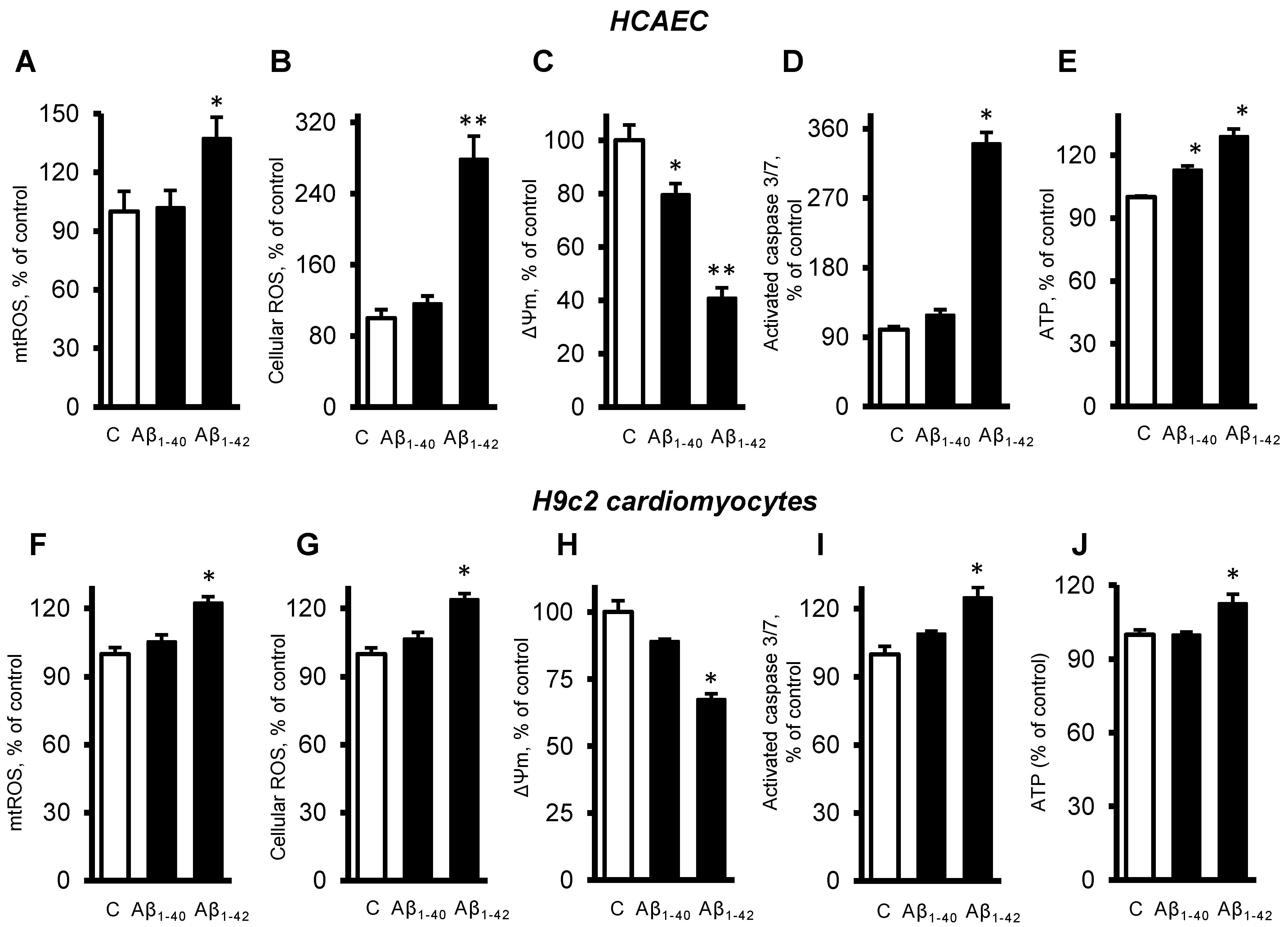
3.3. Aβ Induced Mitochondrial and Cellular Dysfunction
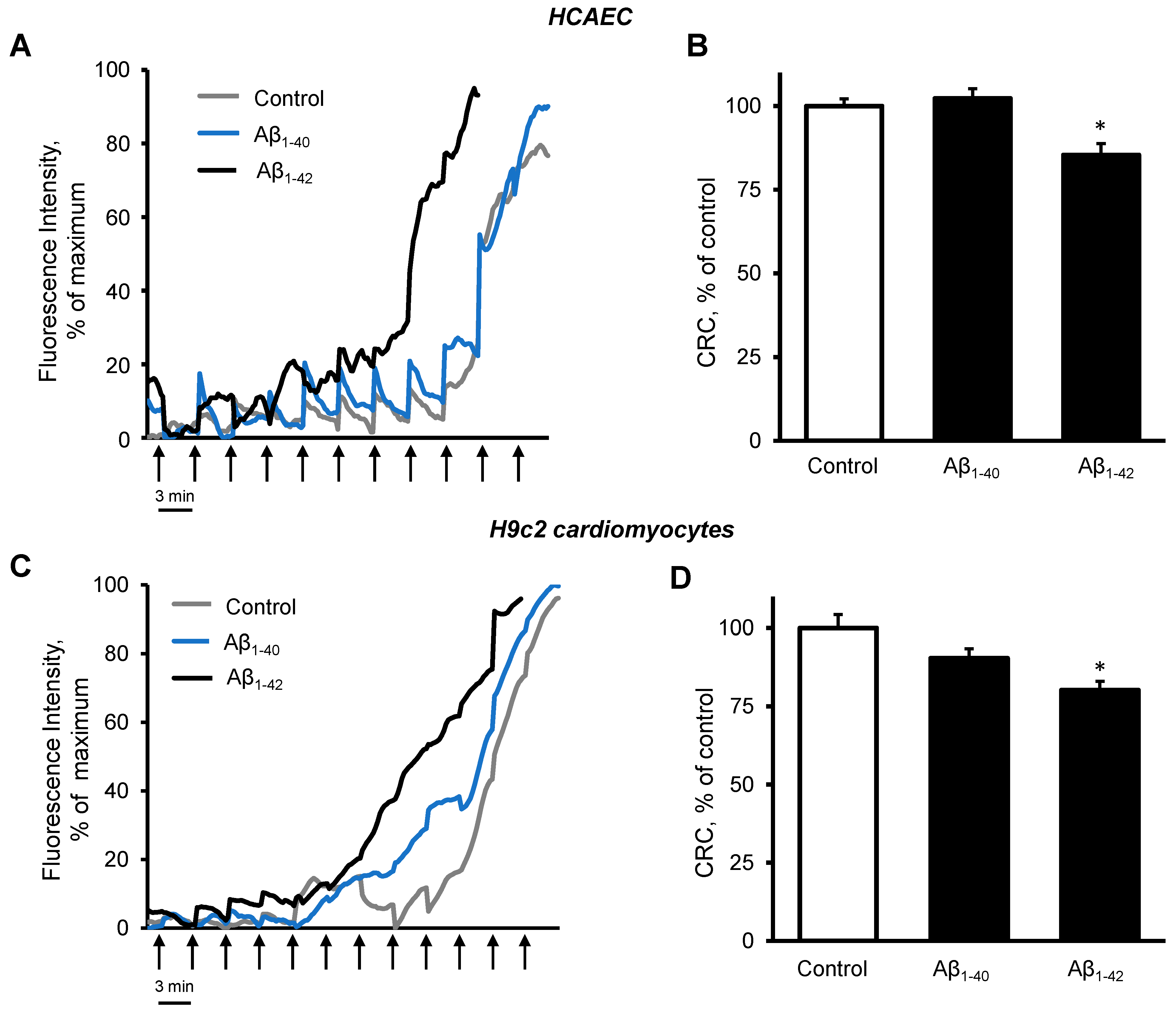
3.4. Cells and Mitochondria Demonstrated Early Response to Aβ
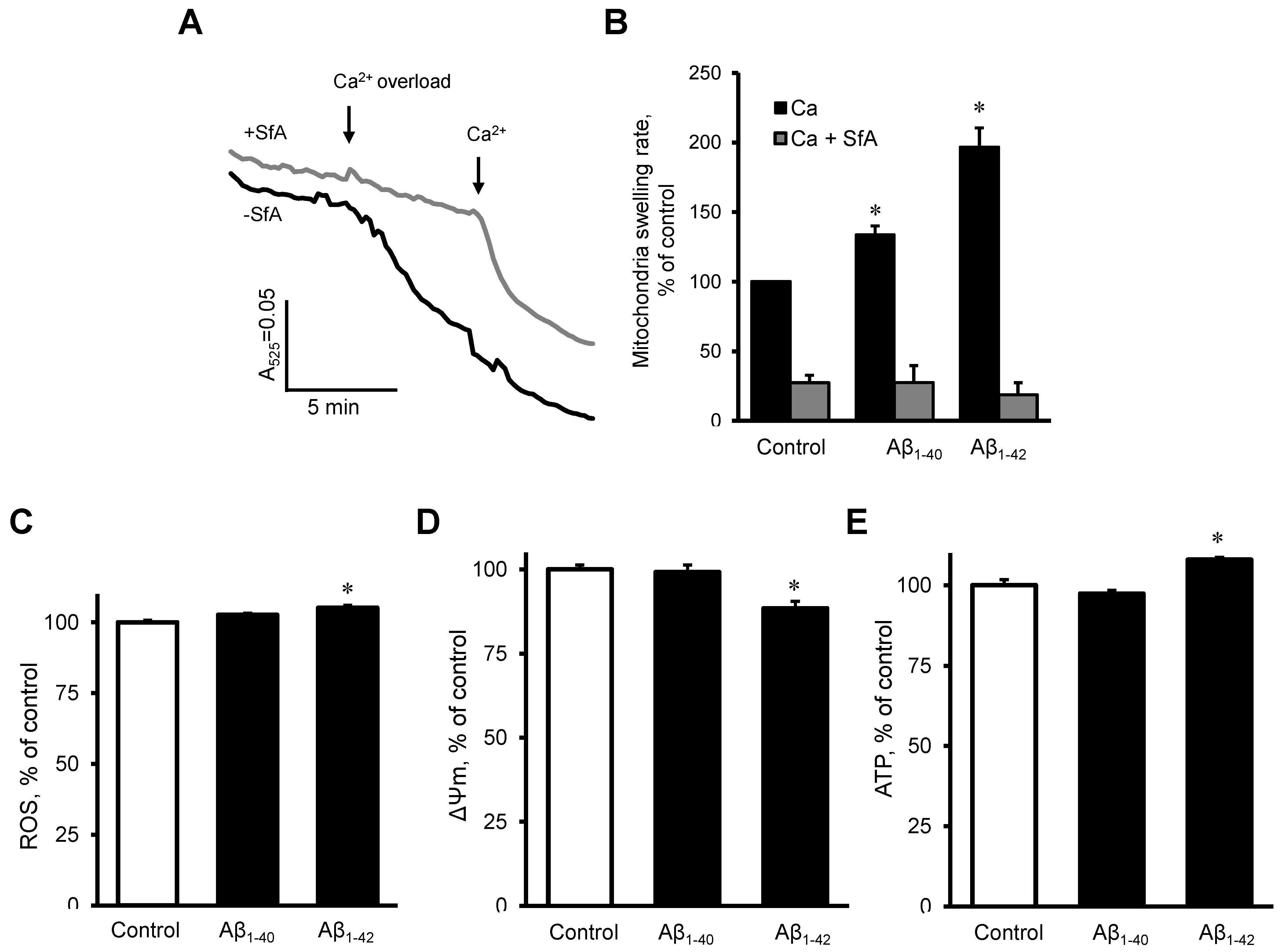
3.5. Aβ induced Dysfunction of Cardiac Mitochondria In Vitro
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Castellani, R.J.; Rolston, R.K.; Smith, M.A. Alzheimer disease. Dis. Mon. 2010, 56, 484–546. [Google Scholar] [CrossRef] [PubMed]
- Tublin, J.M.; Adelstein, J.M.; Del Monte, F.; Combs, C.K.; Wold, L.E. Getting to the Heart of Alzheimer Disease. Circ. Res. 2019, 124, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid. Med. Cell Longev. 2013, 2013, 316523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianni, D.; Li, A.; Tesco, G.; McKay, K.M.; Moore, J.; Raygor, K.; Rota, M.; Gwathmey, J.K.; Dec, G.W.; Aretz, T.; et al. Protein aggregates and novel presenilin gene variants in idiopathic dilated cardiomyopathy. Circulation 2010, 121, 1216–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troncone, L.; Luciani, M.; Coggins, M.; Wilker, E.H.; Ho, C.Y.; Codispoti, K.E.; Frosch, M.P.; Kayed, R.; Del Monte, F. Abeta Amyloid Pathology Affects the Hearts of Patients with Alzheimer’s Disease: Mind the Heart. J. Am. Coll. Cardiol. 2016, 68, 2395–2407. [Google Scholar] [CrossRef]
- Willis, M.S.; Patterson, C. Proteotoxicity and cardiac dysfunction—Alzheimer’s disease of the heart? N. Engl. J. Med. 2013, 368, 455–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunnstrom, H.R.; Englund, E.M. Cause of death in patients with dementia disorders. Eur. J. Neurol. 2009, 16, 488–492. [Google Scholar] [CrossRef]
- Kivipelto, M.; Ngandu, T.; Fratiglioni, L.; Viitanen, M.; Kareholt, I.; Winblad, B.; Helkala, E.L.; Tuomilehto, J.; Soininen, H.; Nissinen, A. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch. Neurol. 2005, 62, 1556–1560. [Google Scholar] [CrossRef] [Green Version]
- Jurkovicova, D.; Goncalvesova, E.; Sedlakova, B.; Hudecova, S.; Fabian, J.; Krizanova, O. Is the ApoE polymorphism associated with dilated cardiomyopathy? Gen. Physiol. Biophys. 2006, 25, 3–10. [Google Scholar]
- Calik, A.N.; Ozcan, K.S.; Yuksel, G.; Gungor, B.; Arugarslan, E.; Varlibas, F.; Ekmekci, A.; Osmonov, D.; Tatlisu, M.A.; Karaca, M.; et al. Altered diastolic function and aortic stiffness in Alzheimer’s disease. Clin. Interv. Aging 2014, 9, 1115–1121. [Google Scholar] [CrossRef] [Green Version]
- Sanna, G.D.; Nusdeo, G.; Piras, M.R.; Forteleoni, A.; Murru, M.R.; Saba, P.S.; Dore, S.; Sotgiu, G.; Parodi, G.; Ganau, A. Cardiac Abnormalities in Alzheimer Disease: Clinical Relevance Beyond Pathophysiological Rationale and Instrumental Findings? JACC Heart Fail 2019, 7, 121–128. [Google Scholar] [CrossRef]
- Jin, W.S.; Bu, X.L.; Wang, Y.R.; Li, L.; Li, W.W.; Liu, Y.H.; Zhu, C.; Yao, X.Q.; Chen, Y.; Gao, C.Y.; et al. Reduced Cardiovascular Functions in Patients with Alzheimer’s Disease. J. Alzheimers Dis. 2017, 58, 919–925. [Google Scholar] [CrossRef]
- Parry, T.L.; Melehani, J.H.; Ranek, M.J.; Willis, M.S. Functional Amyloid Signaling via the Inflammasome, Necrosome, and Signalosome: New Therapeutic Targets in Heart Failure. Front. Cardiovasc. Med. 2015, 2, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.M.; Yam, A.Y.; Wang, X.; Magdangal, E.; Salisbury, C.; Peretz, D.; Zuckermann, R.N.; Connolly, M.D.; Hansson, O.; Minthon, L.; et al. Abeta40 oligomers identified as a potential biomarker for the diagnosis of Alzheimer’s disease. PLoS ONE 2010, 5, e15725. [Google Scholar] [CrossRef]
- Youn, Y.C.; Lee, B.S.; Kim, G.J.; Ryu, J.S.; Lim, K.; Lee, R.; Suh, J.; Park, Y.H.; Pyun, J.M.; Ryu, N.; et al. Blood Amyloid-beta Oligomerization as a Biomarker of Alzheimer’s Disease: A Blinded Validation Study. J. Alzheimers Dis. 2020, 75, 493–499. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteras, N.; Abramov, A.Y. Mitochondrial Calcium Deregulation in the Mechanism of Beta-Amyloid and Tau Pathology. Cells 2020, 9, 2135. [Google Scholar] [CrossRef] [PubMed]
- Parodi-Rullan, R.; Sone, J.Y.; Fossati, S. Endothelial Mitochondrial Dysfunction in Cerebral Amyloid Angiopathy and Alzheimer’s Disease. J. Alzheimers Dis. 2019, 72, 1019–1039. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M.; Wands, J.R. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J. Alzheimers Dis. 2006, 9, 167–181. [Google Scholar] [CrossRef]
- Fossati, S.; Cam, J.; Meyerson, J.; Mezhericher, E.; Romero, I.A.; Couraud, P.O.; Weksler, B.B.; Ghiso, J.; Rostagno, A. Differential activation of mitochondrial apoptotic pathways by vasculotropic amyloid-beta variants in cells composing the cerebral vessel walls. FASEB J. 2010, 24, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Popugaeva, E.; Pchitskaya, E.; Bezprozvanny, I. Dysregulation of neuronal calcium homeostasis in Alzheimer’s disease—A therapeutic opportunity? Biochem. Biophys. Res. Commun. 2017, 483, 998–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, P.R.; Abramov, A.Y. Alpha-synuclein and beta-amyloid—different targets, same players: Calcium, free radicals and mitochondria in the mechanism of neurodegeneration. Biochem. Biophys. Res. Commun. 2017, 483, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- Solesio, M.E.; Peixoto, P.M.; Debure, L.; Madamba, S.M.; de Leon, M.J.; Wisniewski, T.; Pavlov, E.V.; Fossati, S. Carbonic anhydrase inhibition selectively prevents amyloid beta neurovascular mitochondrial toxicity. Aging Cell 2018, 17, e12787. [Google Scholar] [CrossRef] [PubMed]
- Fossati, S.; Giannoni, P.; Solesio, M.E.; Cocklin, S.L.; Cabrera, E.; Ghiso, J.; Rostagno, A. The carbonic anhydrase inhibitor methazolamide prevents amyloid beta-induced mitochondrial dysfunction and caspase activation protecting neuronal and glial cells in vitro and in the mouse brain. Neurobiol. Dis. 2016, 86, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Fossati, S.; Ghiso, J.; Rostagno, A. TRAIL death receptors DR4 and DR5 mediate cerebral microvascular endothelial cell apoptosis induced by oligomeric Alzheimer’s Abeta. Cell Death Dis. 2012, 3, e321. [Google Scholar] [CrossRef] [PubMed]
- Chapa-Dubocq, X.R.; Rodríguez-Graciani, K.M.; Guzmán-Hernández, R.A.; Jang, S.; Brookes, P.S.; Javadov, S. Cardiac Function is not Susceptible to Moderate Disassembly of Mitochondrial Respiratory Supercomplexes. Int. J. Mol. Sci. 2020, 21, 155. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.; Javadov, S. OPA1 regulates respiratory supercomplexes assembly: The role of mitochondrial swelling. Mitochondrion 2020, 51, 30–39. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Javadov, S.; Sickinger, S.; Frotschnig, S.; Grimm, M. H9c2 and HL-1 cells demonstrate distinct features of energy metabolism, mitochondrial function and sensitivity to hypoxia-reoxygenation. Biochim. Biophys. Acta 2015, 1853, 276–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parodi-Rullán, R.; Ghiso, J.; Cabrera, E.; Rostagno, A.; Fossati, S. Alzheimer’s amyloid β heterogeneous species differentially affect brain endothelial cell viability, blood-brain barrier integrity, and angiogenesis. Aging Cell 2020, 19, e13258. [Google Scholar] [CrossRef]
- Stine, W.B., Jr.; Dahlgren, K.N.; Krafft, G.A.; LaDu, M.J. In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J. Biol. Chem. 2003, 278, 11612–11622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, S.; Chapa-Dubocq, X.R.; Tyurina, Y.Y.; St Croix, C.M.; Kapralov, A.A.; Tyurin, V.A.; Bayir, H.; Kagan, V.E.; Javadov, S. Elucidating the contribution of mitochondrial glutathione to ferroptosis in cardiomyocytes. Redox Biol. 2021, 45, 102021. [Google Scholar] [CrossRef]
- Supnet, C.; Bezprozvanny, I. Neuronal calcium signaling, mitochondrial dysfunction, and Alzheimer’s disease. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S487–S498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fossati, S.; Todd, K.; Sotolongo, K.; Ghiso, J.; Rostagno, A. Differential contribution of isoaspartate post-translational modifications to the fibrillization and toxic properties of amyloid-beta and the asparagine 23 Iowa mutation. Biochem. J. 2013. [Google Scholar] [CrossRef] [Green Version]
- Javadov, S.; Karmazyn, M.; Escobales, N. Mitochondrial permeability transition pore opening as a promising therapeutic target in cardiac diseases. J. Pharmacol. Exp. Ther. 2009, 330, 670–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokjohn, T.A.; Van Vickle, G.D.; Maarouf, C.L.; Kalback, W.M.; Hunter, J.M.; Daugs, I.D.; Luehrs, D.C.; Lopez, J.; Brune, D.; Sue, L.I.; et al. Chemical characterization of pro-inflammatory amyloid-beta peptides in human atherosclerotic lesions and platelets. Biochim. Biophys. Acta 2011, 1812, 1508–1514. [Google Scholar] [CrossRef] [Green Version]
- Stamatelopoulos, K.; Sibbing, D.; Rallidis, L.S.; Georgiopoulos, G.; Stakos, D.; Braun, S.; Gatsiou, A.; Sopova, K.; Kotakos, C.; Varounis, C.; et al. Amyloid-beta (1-40) and the risk of death from cardiovascular causes in patients with coronary heart disease. J. Am. Coll. Cardiol. 2015, 65, 904–916. [Google Scholar] [CrossRef] [Green Version]
- Aleardi, A.M.; Benard, G.; Augereau, O.; Malgat, M.; Talbot, J.C.; Mazat, J.P.; Letellier, T.; Dachary-Prigent, J.; Solaini, G.C.; Rossignol, R. Gradual alteration of mitochondrial structure and function by beta-amyloids: Importance of membrane viscosity changes, energy deprivation, reactive oxygen species production, and cytochrome c release. J. Bioenerg. Biomembr. 2005, 37, 207–225. [Google Scholar] [CrossRef]
- Chouraki, V.; Beiser, A.; Younkin, L.; Preis, S.R.; Weinstein, G.; Hansson, O.; Skoog, I.; Lambert, J.C.; Au, R.; Launer, L.; et al. Plasma amyloid-beta and risk of Alzheimer’s disease in the Framingham Heart Study. Alzheimers Dement. 2015, 11, 249–257.e1. [Google Scholar] [CrossRef] [Green Version]
- Despa, F.; Decarli, C. Amylin: What might be its role in Alzheimer’s disease and how could this affect therapy? Expert Rev. Proteomics 2013, 10, 403–405. [Google Scholar] [CrossRef] [Green Version]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parodi-Rullán, R.M.; Javadov, S.; Fossati, S. Dissecting the Crosstalk between Endothelial Mitochondrial Damage, Vascular Inflammation, and Neurodegeneration in Cerebral Amyloid Angiopathy and Alzheimer’s Disease. Cells 2021, 10, 2903. [Google Scholar] [CrossRef] [PubMed]
- Kuchibhotla, K.V.; Goldman, S.T.; Lattarulo, C.R.; Wu, H.Y.; Hyman, B.T.; Bacskai, B.J. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 2008, 59, 214–225. [Google Scholar] [CrossRef] [Green Version]
- Busche, M.A.; Chen, X.; Henning, H.A.; Reichwald, J.; Staufenbiel, M.; Sakmann, B.; Konnerth, A. Critical role of soluble amyloid-beta for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad Sci. USA 2012, 109, 8740–8745. [Google Scholar] [CrossRef] [Green Version]
- Abeti, R.; Abramov, A.Y. Mitochondrial Ca2+ in neurodegenerative disorders. Pharmacol. Res. 2015, 99, 377–381. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Zhang, W.; Rydzewska, M.; Yan, S. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol. Aging 2011, 32, 398–406. [Google Scholar] [CrossRef] [Green Version]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. 2004, 24, 565–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, S.; Chapa-Dubocq, X.R.; Parodi-Rullán, R.M.; Fossati, S.; Javadov, S. Beta-Amyloid Instigates Dysfunction of Mitochondria in Cardiac Cells. Cells 2022, 11, 373. https://doi.org/10.3390/cells11030373
Jang S, Chapa-Dubocq XR, Parodi-Rullán RM, Fossati S, Javadov S. Beta-Amyloid Instigates Dysfunction of Mitochondria in Cardiac Cells. Cells. 2022; 11(3):373. https://doi.org/10.3390/cells11030373
Chicago/Turabian StyleJang, Sehwan, Xavier R. Chapa-Dubocq, Rebecca M. Parodi-Rullán, Silvia Fossati, and Sabzali Javadov. 2022. "Beta-Amyloid Instigates Dysfunction of Mitochondria in Cardiac Cells" Cells 11, no. 3: 373. https://doi.org/10.3390/cells11030373
APA StyleJang, S., Chapa-Dubocq, X. R., Parodi-Rullán, R. M., Fossati, S., & Javadov, S. (2022). Beta-Amyloid Instigates Dysfunction of Mitochondria in Cardiac Cells. Cells, 11(3), 373. https://doi.org/10.3390/cells11030373