
The Translational Bridge between Inflammation and Hepatocarcinogenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Acute Liver Failure
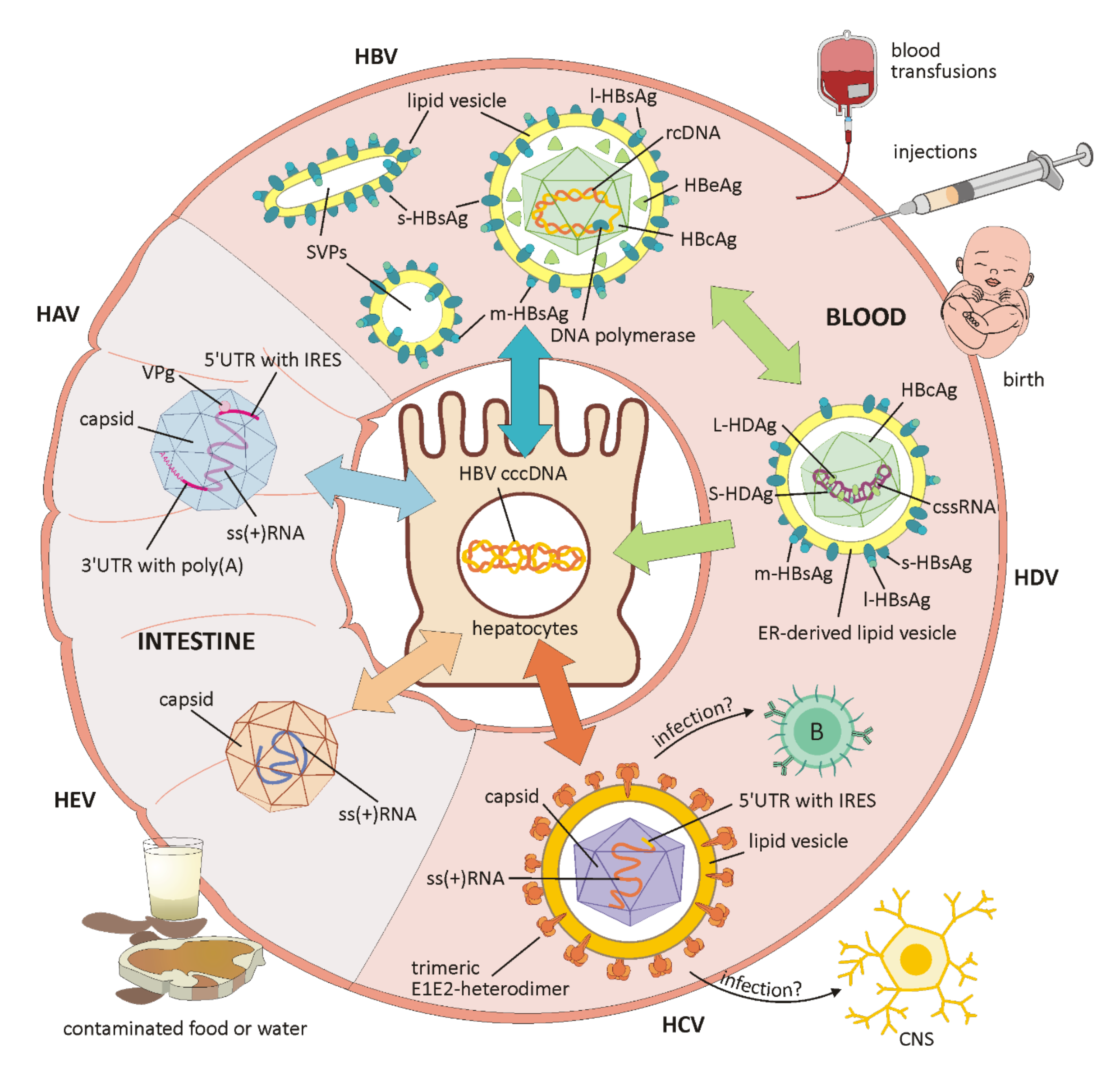
3. Viral Hepatitis
3.1. Hepatitis A (HAV)
3.2. Hepatitis B (HBV)
3.3. Hepatitis C (HCV)
3.4. Hepatitis D (HDV)
3.5. Hepatitis E (HEV)
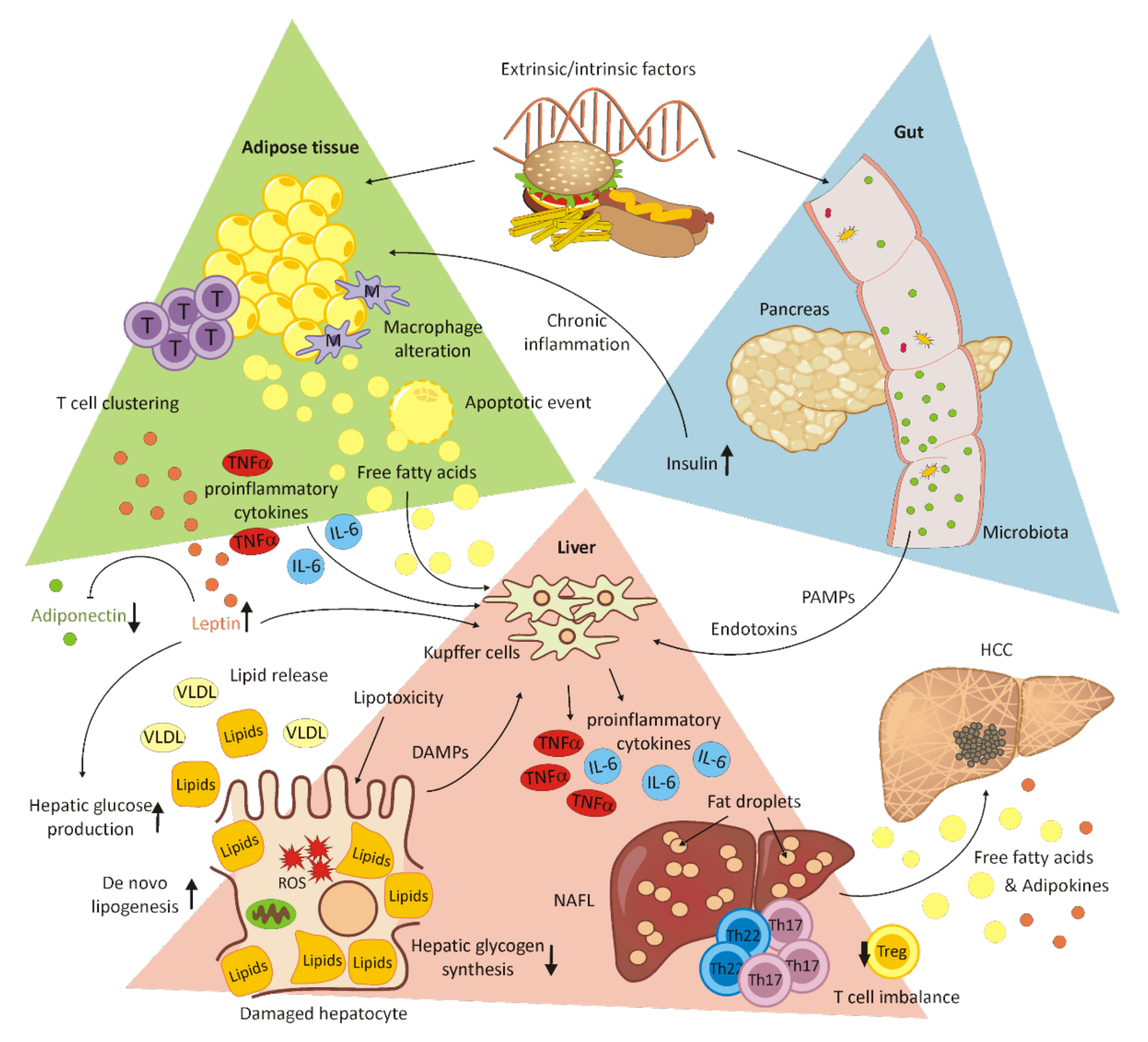
4. Alcohol-Induced Liver Disease
5. Non-Alcoholic Fatty Liver Disease
6. Autoimmune Hepatitis
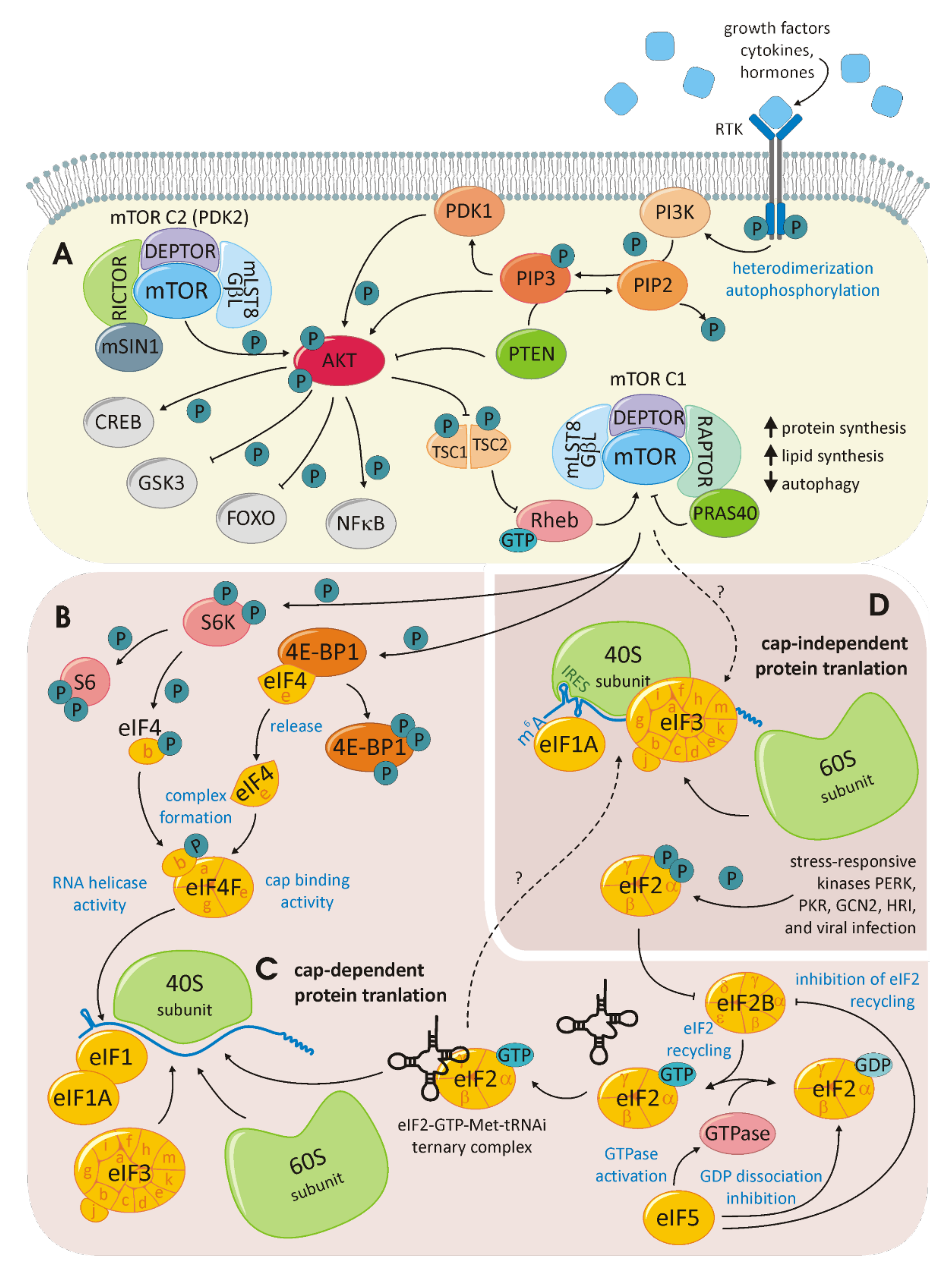
7. HCC/mTOR/eIFs
7.1. mTOR Signaling
7.2. HCC Therapy
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Wang, K. Autophagy and apoptosis in liver injury. Cell Cycle 2015, 14, 1631–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stravitz, R.T.; Lee, W.M. Acute liver failure. Lancet 2019, 394, 869–881. [Google Scholar] [CrossRef]
- Bangash, M.N.; Patel, J.; Parekh, D. COVID-19 and the liver: Little cause for concern. Lancet Gastroenterol. Hepatol. 2020, 5, 529–530. [Google Scholar] [CrossRef] [Green Version]
- Germani, G.; Theocharidou, E.; Adam, R.; Karam, V.; Wendon, J.; O’Grady, J.; Burra, P.; Senzolo, M.; Mirza, D.; Castaing, D.; et al. Liver transplantation for acute liver failure in Europe: Outcomes over 20 years from the ELTR database. J. Hepatol. 2012, 57, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Mysore, K.R.; Leung, D.H. Hepatitis B and C. Clin. Liver Dis. 2018, 22, 703–722. [Google Scholar] [CrossRef]
- Patterson, J.; Hussey, H.S.; Silal, S.; Goddard, L.; Setshedi, M.; Spearman, W.; Hussey, G.D.; Kagina, B.M.; Muloiwa, R. Systematic review of the global epidemiology of viral-induced acute liver failure. BMJ Open 2020, 10, e037473. [Google Scholar] [CrossRef]
- Trautwein, C.K.A. Liver Immunology: Principles and Practice; Springer Science+Business Media: New York, NY, USA, 2014. [Google Scholar]
- Katarey, D.; Verma, S. Drug-induced liver injury. Clin. Med. 2016, 16, s104–s109. [Google Scholar] [CrossRef]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef] [Green Version]
- Lavanchy, D. Chronic viral hepatitis as a public health issue in the world. Best Pract. Res. Clin. Gastroenterol. 2008, 22, 991–1008. [Google Scholar] [CrossRef]
- Cobb, B.R.; Valsamakis, A. Chronic Hepatitis B, C, and D. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Sarpel, D.; Baichoo, E.; Dieterich, D.T. Chronic hepatitis B and C infection in the United States: A review of current guidelines, disease burden and cost effectiveness of screening. Expert Rev. Anti-Infect. Ther. 2016, 14, 511–521. [Google Scholar] [CrossRef]
- Thomas, D.L. Global Elimination of Chronic Hepatitis. N. Engl. J. Med. 2019, 380, 2041–2050. [Google Scholar] [CrossRef] [PubMed]
- Herrscher, C.; Roingeard, P.; Blanchard, E. Hepatitis B Virus Entry into Cells. Cells 2020, 9, 1486. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.Y.; Chen, G.J.; Lee, Y.L.; Huang, Y.C.; Cheng, A.; Sun, H.Y.; Chang, S.Y.; Liu, C.E.; Hung, C.C. Hepatitis A virus infection and hepatitis A vaccination in human immunodeficiency virus-positive patients: A review. World J. Gastroenterol. 2017, 23, 3589–3606. [Google Scholar] [CrossRef] [PubMed]
- Randazzo, W.; Sánchez, G. Hepatitis A infections from food. J. Appl. Microbiol. 2020, 129, 1120–1132. [Google Scholar] [CrossRef]
- Lemon, S.M.; Ott, J.J.; Van Damme, P.; Shouval, D. Type A viral hepatitis: A summary and update on the molecular virology, epidemiology, pathogenesis and prevention. J. Hepatol. 2018, 68, 167–184. [Google Scholar] [CrossRef] [Green Version]
- Revill, P.A.; Tu, T.; Netter, H.J.; Yuen, L.K.W.; Locarnini, S.A.; Littlejohn, M. The evolution and clinical impact of hepatitis B virus genome diversity. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 618–634. [Google Scholar] [CrossRef]
- Brunetto, M.R.; Colombatto, P.; Bonino, F. Personalized therapy in chronic viral hepatitis. Mol. Aspects Med. 2008, 29, 103–111. [Google Scholar] [CrossRef]
- Fletcher, N.F.; McKeating, J.A. Hepatitis C virus and the brain. J. Viral. Hepat. 2012, 19, 301–306. [Google Scholar] [CrossRef]
- Gonzalez, S.A.; Keeffe, E.B. Chronic viral hepatitis: Epidemiology, molecular biology, and antiviral therapy. Front. Biosci. 2011, 16, 225–250. [Google Scholar] [CrossRef] [PubMed]
- Sevvana, M.; Keck, Z.; Foung, S.K.; Kuhn, R.J. Structural perspectives on HCV humoral immune evasion mechanisms. Curr. Opin. Virol. 2021, 49, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Forton, D.M.; Karayiannis, P.; Mahmud, N.; Taylor-Robinson, S.D.; Thomas, H.C. Identification of unique hepatitis C virus quasispecies in the central nervous system and comparative analysis of internal translational efficiency of brain, liver, and serum variants. J. Virol. 2004, 78, 5170–5183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laursen, T.L.; Sandahl, T.D.; Kazankov, K.; George, J.; Grønbæk, H. Liver-related effects of chronic hepatitis C antiviral treatment. World J. Gastroenterol. 2020, 26, 2931–2947. [Google Scholar] [CrossRef]
- Zeng, H.; Li, L.; Hou, Z.; Zhang, Y.; Tang, Z.; Liu, S. Direct-acting Antiviral in the Treatment of Chronic Hepatitis C: Bonuses and Challenges. Int. J. Med. Sci. 2020, 17, 892–902. [Google Scholar] [CrossRef] [Green Version]
- Foster, G.R. Quality of life considerations for patients with chronic hepatitis C. J. Viral Hepat. 2009, 16, 605–611. [Google Scholar] [CrossRef]
- Stöhr, S.; Costa, R.; Sandmann, L.; Westhaus, S.; Pfaender, S.; Anggakusuma; Dazert, E.; Meuleman, P.; Vondran, F.W.R.; Manns, M.P.; et al. Host cell mTORC1 is required for HCV RNA replication. Gut 2016, 65, 2017–2028. [Google Scholar] [CrossRef] [Green Version]
- Kroczynska, B.; Rafidi, R.L.; Majchrzak-Kita, B.; Kosciuczuk, E.M.; Blyth, G.T.; Jemielity, J.; Warminska, Z.; Saleiro, D.; Mehrotra, S.; Arslan, A.D.; et al. Interferon γ (IFNγ) Signaling via Mechanistic Target of Rapamycin Complex 2 (mTORC2) and Regulatory Effects in the Generation of Type II Interferon Biological Responses. J. Biol. Chem. 2016, 291, 2389–2396. [Google Scholar] [CrossRef] [Green Version]
- Metz, P.; Reuter, A.; Bender, S.; Bartenschlager, R. Interferon-stimulated genes and their role in controlling hepatitis C virus. J. Hepatol. 2013, 59, 1331–1341. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Park, S.M.; Park, J.H.; Keum, S.J.; Jang, S.K. eIF2A mediates translation of hepatitis C viral mRNA under stress conditions. Embo J. 2011, 30, 2454–2464. [Google Scholar] [CrossRef] [Green Version]
- Hui, D.J.; Bhasker, C.R.; Merrick, W.C.; Sen, G.C. Viral stress-inducible protein p56 inhibits translation by blocking the interaction of eIF3 with the ternary complex eIF2.GTP.Met-tRNAi. J. Biol. Chem. 2003, 278, 39477–39482. [Google Scholar] [CrossRef] [Green Version]
- Terenzi, F.; Hui, D.J.; Merrick, W.C.; Sen, G.C. Distinct induction patterns and functions of two closely related interferon-inducible human genes, ISG54 and ISG56. J. Biol. Chem. 2006, 281, 34064–34071. [Google Scholar] [CrossRef] [Green Version]
- Mentha, N.; Clément, S.; Negro, F.; Alfaiate, D. A review on hepatitis D: From virology to new therapies. J. Adv. Res. 2019, 17, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Brancaccio, G.; Gaeta, G.B. Treatment of chronic hepatitis due to hepatitis B and hepatitis delta virus coinfection. Int. J. Antimicrob. Agents 2019, 54, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Urban, S. Interplay between Hepatitis D Virus and the Interferon Response. Viruses 2020, 12, 1334. [Google Scholar] [CrossRef] [PubMed]
- Suskind, D.L.; Rosenthal, P. Chronic viral hepatitis. Adolesc. Med. Clin. 2004, 15, 145–158. [Google Scholar] [CrossRef]
- Fujiwara, S.; Yokokawa, Y.; Morino, K.; Hayasaka, K.; Kawabata, M.; Shimizu, T. Chronic hepatitis E: A review of the literature. J. Viral Hepat. 2014, 21, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Aslan, A.T.; Balaban, H.Y. Hepatitis E virus: Epidemiology, diagnosis, clinical manifestations, and treatment. World J. Gastroenterol. 2020, 26, 5543–5560. [Google Scholar] [CrossRef]
- Pingale, K.D.; Kanade, G.D.; Karpe, Y.A. Hepatitis E virus polymerase binds to IFIT1 to protect the viral RNA from IFIT1-mediated translation inhibition. J. Gen. Virol. 2019, 100, 471–483. [Google Scholar] [CrossRef]
- Yin, X.; Feng, Z. Hepatitis E Virus Entry. Viruses 2019, 11, 883. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Cederbaum, A.I. Cytochrome P450s and Alcoholic Liver Disease. Curr. Pharm. Des. 2018, 24, 1502–1517. [Google Scholar] [CrossRef]
- Marroni, C.A.; Fleck, A.M., Jr.; Fernandes, S.A.; Galant, L.H.; Mucenic, M.; de Mattos Meine, M.H.; Mariante-Neto, G.; de Mello Brandão, A.B. Liver transplantation and alcoholic liver disease: History, controversies, and considerations. World J. Gastroenterol. 2018, 24, 2785–2805. [Google Scholar] [CrossRef]
- Stickel, F.; Datz, C.; Hampe, J.; Bataller, R. Pathophysiology and Management of Alcoholic Liver Disease: Update 2016. Gut Liver 2017, 11, 173–188. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S. Alcohol, liver disease and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 235–246. [Google Scholar] [CrossRef]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Primers 2018, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Scott, E.; Anstee, Q.M. Genetics of alcoholic liver disease and non-alcoholic steatohepatitis. Clin. Med. 2018, 18, s54–s59. [Google Scholar] [CrossRef]
- Kourkoumpetis, T.; Sood, G. Pathogenesis of Alcoholic Liver Disease. Clin. Liver Dis. 2019, 23, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Philips, C.A.; Pande, A.; Shasthry, S.M.; Jamwal, K.D.; Khillan, V.; Chandel, S.S.; Kumar, G.; Sharma, M.K.; Maiwall, R.; Jindal, A.; et al. Healthy Donor Fecal Microbiota Transplantation in Steroid-Ineligible Severe Alcoholic Hepatitis: A Pilot Study. Clin. Gastroenterol. Hepatol. 2017, 15, 600–602. [Google Scholar] [CrossRef]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Khoshbin, K.; Camilleri, M. Effects of dietary components on intestinal permeability in health and disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G589–G608. [Google Scholar] [CrossRef]
- Szabo, G. Gut-liver axis in alcoholic liver disease. Gastroenterology 2015, 148, 30–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaza-Díaz, J.; Solís-Urra, P.; Rodríguez-Rodríguez, F.; Olivares-Arancibia, J.; Navarro-Oliveros, M.; Abadía-Molina, F.; Álvarez-Mercado, A.I. The Gut Barrier, Intestinal Microbiota, and Liver Disease: Molecular Mechanisms and Strategies to Manage. Int. J. Mol. Sci. 2020, 21, 8351. [Google Scholar] [CrossRef] [PubMed]
- Buch, S.; Stickel, F.; Trépo, E.; Way, M.; Herrmann, A.; Nischalke, H.D.; Brosch, M.; Rosendahl, J.; Berg, T.; Ridinger, M.; et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat. Genet. 2015, 47, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Lackner, C.; Spindelboeck, W.; Haybaeck, J.; Douschan, P.; Rainer, F.; Terracciano, L.; Haas, J.; Berghold, A.; Bataller, R.; Stauber, R.E. Histological parameters and alcohol abstinence determine long-term prognosis in patients with alcoholic liver disease. J. Hepatol. 2017, 66, 610–618. [Google Scholar] [CrossRef]
- Lackner, C.; Tiniakos, D. Fibrosis and alcohol-related liver disease. J. Hepatol. 2019, 70, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Parola, M.; Pinzani, M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Asp. Med. 2019, 65, 37–55. [Google Scholar] [CrossRef]
- Villanueva, A.; Portela, A.; Sayols, S.; Battiston, C.; Hoshida, Y.; Mendez-Gonzalez, J.; Imbeaud, S.; Letouze, E.; Hernandez-Gea, V.; Cornella, H.; et al. DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Hepatology 2015, 61, 1945–1956. [Google Scholar] [CrossRef] [Green Version]
- Tomic, D.; Kemp, W.W.; Roberts, S.K. Nonalcoholic fatty liver disease: Current concepts, epidemiology and management strategies. Eur. J. Gastroenterol. Hepatol. 2018, 30, 1103–1115. [Google Scholar] [CrossRef]
- Mishra, A.; Younossi, Z.M. Epidemiology and Natural History of Non-alcoholic Fatty Liver Disease. J. Clin. Exp. Hepatol. 2012, 2, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Sayiner, M.; Koenig, A.; Henry, L.; Younossi, Z.M. Epidemiology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis in the United States and the Rest of the World. Clin. Liver Dis. 2016, 20, 205–214. [Google Scholar] [CrossRef]
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715–732. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef] [PubMed]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Negro, F.; Hallaji, S.; Younossi, Y.; Lam, B.; Srishord, M. Nonalcoholic Fatty Liver Disease in Lean Individuals in the United States. Medicine 2012, 91, 319–327. [Google Scholar] [CrossRef]
- Stepanova, M.; Rafiq, N.; Younossi, Z.M. Components of metabolic syndrome are independent predictors of mortality in patients with chronic liver disease: A population-based study. Gut 2010, 59, 1410–1415. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Stepanova, M.; Afendy, M.; Fang, Y.; Younossi, Y.; Mir, H.; Srishord, M. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin. Gastroenterol. Hepatol. 2011, 9, 524–530.e521. [Google Scholar] [CrossRef]
- Amarapurkar, D.; Kamani, P.; Patel, N.; Gupte, P.; Kumar, P.; Agal, S.; Baijal, R.; Lala, S.; Chaudhary, D.; Deshpande, A. Prevalence of non-alcoholic fatty liver disease: Population based study. Ann. Hepatol. 2007, 6, 161–163. [Google Scholar] [CrossRef]
- Bedogni, G.; Miglioli, L.; Masutti, F.; Tiribelli, C.; Marchesini, G.; Bellentani, S. Prevalence of and risk factors for nonalcoholic fatty liver disease: The Dionysos nutrition and liver study. Hepatology 2005, 42, 44–52. [Google Scholar] [CrossRef]
- Kojima, S.-I.; Watanabe, N.; Numata, M.; Ogawa, T.; Matsuzaki, S. Increase in the prevalence of fatty liver in Japan over the past 12 years: Analysis of clinical background. J. Gastroenterol. 2003, 38, 954–961. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Behling, C.; Newbury, R.; Deutsch, R.; Nievergelt, C.; Schork, N.J.; Lavine, J.E. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology 2005, 42, 641–649. [Google Scholar] [CrossRef]
- Pappachan, J.M.; Babu, S.; Krishnan, B.; Ravindran, N.C. Non-alcoholic Fatty Liver Disease: A Clinical Update. J. Clin. Transl. Hepatol. 2017, 5, 384–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, L.A.; Angulo, P.; Lindor, K.D. Nonalcoholic fatty liver disease. CMAJ 2005, 172, 899–905. [Google Scholar] [CrossRef] [Green Version]
- Maurice, J.; Manousou, P. Non-alcoholic fatty liver disease. Clin. Med. 2018, 18, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.F.; Pories, W.J.; Caro, J.F. Liver pathology in diabetes mellitus and morbid obesity. Clinical, pathological, and biochemical considerations. Pathol. Ann. 1989, 24 Pt 1, 275–302. [Google Scholar]
- Wanless, I.R.; Lentz, J.S. Fatty liver hepatitis (steatohepatitis) and obesity: An autopsy study with analysis of risk factors. Hepatology 1990, 12, 1106–1110. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Janney, C.G.; Di Bisceglie, A.M.; Neuschwander-Tetri, B.A.; Bacon, B.R. Nonalcoholic steatohepatitis: A proposal for grading and staging the histological lesions. Am. J. Gastroenterol. 1999, 94, 2467–2474. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Otero, Y.F.; Stafford, J.M.; McGuinness, O.P. Pathway-selective insulin resistance and metabolic disease: The importance of nutrient flux. J. Biol. Chem. 2014, 289, 20462–20469. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Chen, J.; Huang, J.; Li, Z.; Gong, Y.; Zou, B.; Liu, X.; Ding, L.; Li, P.; Zhu, Z.; et al. HIF-2α upregulation mediated by hypoxia promotes NAFLD-HCC progression by activating lipid synthesis via the PI3K-AKT-mTOR pathway. Aging 2019, 11, 10839–10860. [Google Scholar] [CrossRef]
- Marengo, A.; Rosso, C.; Bugianesi, E. Liver Cancer: Connections with Obesity, Fatty Liver, and Cirrhosis. Annu. Rev. Med. 2016, 67, 103–117. [Google Scholar] [CrossRef]
- Van Herck, M.A.; Weyler, J.; Kwanten, W.J.; Dirinck, E.L.; De Winter, B.Y.; Francque, S.M.; Vonghia, L. The Differential Roles of T Cells in Non-alcoholic Fatty Liver Disease and Obesity. Front. Immunol. 2019, 10, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutti, S.; Albano, E. Adaptive immunity: An emerging player in the progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Czaja, A.J. Autoimmune hepatitis. Part A: Pathogenesis. Expert Rev. Gastroenterol. Hepatol. 2007, 1, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Komori, A. Recent updates on the management of autoimmune hepatitis. Clin. Mol. Hepatol. 2021, 27, 58–69. [Google Scholar] [CrossRef]
- Krawitt, E.L. Autoimmune hepatitis. N. Engl. J. Med. 2006, 354, 54–66. [Google Scholar] [CrossRef] [Green Version]
- Manns, M.P.; Czaja, A.J.; Gorham, J.D.; Krawitt, E.L.; Mieli-Vergani, G.; Vergani, D.; Vierling, J.M. Diagnosis and management of autoimmune hepatitis. Hepatology 2010, 51, 2193–2213. [Google Scholar] [CrossRef]
- Sakaguchi, S. Regulatory T cells: Key controllers of immunologic self-tolerance. Cell 2000, 101, 455–458. [Google Scholar] [CrossRef] [Green Version]
- Mieli-Vergani, G.; Vergani, D.; Czaja, A.J.; Manns, M.P.; Krawitt, E.L.; Vierling, J.M.; Lohse, A.W.; Montano-Loza, A.J. Autoimmune hepatitis. Nat. Rev. Dis. Primers 2018, 4, 18017. [Google Scholar] [CrossRef]
- Lee, G.R. The Balance of Th17 versus Treg Cells in Autoimmunity. Int. J. Mol. Sci. 2018, 19, 730. [Google Scholar] [CrossRef] [Green Version]
- Romagnani, S. T-cell subsets (Th1 versus Th2). Ann. Allergy Asthma. Immunol. 2000, 85, 9–18. [Google Scholar] [CrossRef]
- Webb, G.J.; Hirschfield, G.M.; Krawitt, E.L.; Gershwin, M.E. Cellular and Molecular Mechanisms of Autoimmune Hepatitis. Annu. Rev. Pathol. 2018, 13, 247–292. [Google Scholar] [CrossRef] [PubMed]
- Vergani, D.; Alvarez, F.; Bianchi, F.B.; Cançado, E.L.; Mackay, I.R.; Manns, M.P.; Nishioka, M.; Penner, E. Liver autoimmune serology: A consensus statement from the committee for autoimmune serology of the International Autoimmune Hepatitis Group. J. Hepatol. 2004, 41, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Gawrieh, S. Autoimmune Hepatitis in the Elderly: Diagnosis and Pharmacologic Management. Drugs Aging 2018, 35, 589–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covelli, C.; Sacchi, D.; Sarcognato, S.; Cazzagon, N.; Grillo, F.; Baciorri, F.; Fanni, D.; Cacciatore, M.; Maffeis, V.; Guido, M. Pathology of autoimmune hepatitis. Pathologica 2021, 113, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Ohira, H.; Abe, K.; Zeniya, M.; Abe, M.; Arinaga-Hino, T.; Torimura, T.; Yoshizawa, K.; Takaki, A.; Kang, J.H.; et al. Differences in autoimmune hepatitis based on inflammation localization. Med. Mol. Morphol. 2021, 54, 8–13. [Google Scholar] [CrossRef]
- Gurung, A.; Assis, D.N.; McCarty, T.R.; Mitchell, K.A.; Boyer, J.L.; Jain, D. Histologic features of autoimmune hepatitis: A critical appraisal. Hum. Pathol. 2018, 82, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.W.; Lo, R.C.; Chan, L.K.; Ng, I.O. Molecular Pathogenesis of Hepatocellular Carcinoma. Liver Cancer 2016, 5, 290–302. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Ramon, Y.C.S.; Castellvi, J.; Hümmer, S.; Peg, V.; Pelletier, J.; Sonenberg, N. Beyond molecular tumor heterogeneity: Protein synthesis takes control. Oncogene 2018, 37, 2490–2501. [Google Scholar] [CrossRef] [Green Version]
- Ferrín, G.; Guerrero, M.; Amado, V.; Rodríguez-Perálvarez, M.; De la Mata, M. Activation of mTOR Signaling Pathway in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, E.J.; Wankell, M.; Palamuthusingam, P.; McFarlane, C.; Hebbard, L. Targeting the PI3K/Akt/mTOR Pathway in Hepatocellular Carcinoma. Biomedicines 2021, 9, 1639. [Google Scholar] [CrossRef] [PubMed]
- Rebouissou, S.; Nault, J.C. Advances in molecular classification and precision oncology in hepatocellular carcinoma. J. Hepatol. 2020, 72, 215–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sia, D.; Villanueva, A. Signaling pathways in hepatocellular carcinoma. Oncology 2011, 81 (Suppl. 1), 18–23. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Hall, M.N. Regulation of mTORC2 Signaling. Genes 2020, 11, 1045. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Peterson, T.R.; Sabatini, D.M. eIF3: A connecTOR of S6K1 to the translation preinitiation complex. Mol. Cell 2005, 20, 655–657. [Google Scholar] [CrossRef]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef] [Green Version]
- Sriram, A.; Bohlen, J.; Teleman, A.A. Translation acrobatics: How cancer cells exploit alternate modes of translational initiation. EMBO Rep. 2018, 19, e45947. [Google Scholar] [CrossRef]
- Walters, B.; Thompson, S.R. Cap-Independent Translational Control of Carcinogenesis. Front. Oncol. 2016, 6, 128. [Google Scholar] [CrossRef] [Green Version]
- Coots, R.A.; Liu, X.M.; Mao, Y.; Dong, L.; Zhou, J.; Wan, J.; Zhang, X.; Qian, S.B. m(6)A Facilitates eIF4F-Independent mRNA Translation. Mol. Cell 2017, 68, 504–514.e507. [Google Scholar] [CrossRef] [PubMed]
- Hronová, V.; Mohammad, M.P.; Wagner, S.; Pánek, J.; Gunišová, S.; Zeman, J.; Poncová, K.; Valášek, L.S. Does eIF3 promote reinitiation after translation of short upstream ORFs also in mammalian cells? RNA Biol. 2017, 14, 1660–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, R.J.; Hellen, C.U.; Pestova, T.V. Termination and post-termination events in eukaryotic translation. Adv. Protein Chem. Struct. Biol. 2012, 86, 45–93. [Google Scholar] [CrossRef] [PubMed]
- Sahin, F.; Kannangai, R.; Adegbola, O.; Wang, J.; Su, G.; Torbenson, M. mTOR and P70 S6 kinase expression in primary liver neoplasms. Clin. Cancer Res. 2004, 10, 8421–8425. [Google Scholar] [CrossRef] [Green Version]
- Bracic Tomazic, S.; Schatz, C.; Haybaeck, J. Translational Regulation in Hepatocellular Carcinogenesis. Drug Des. Dev. Ther. 2021, 15, 4359–4369. [Google Scholar] [CrossRef]
- Ding, Z.; Dong, Z.; Chen, Z.; Hong, J.; Yan, L.; Li, H.; Yao, S.; Yan, Y.; Yang, Y.; Yang, C.; et al. Viral Status and Efficacy of Immunotherapy in Hepatocellular Carcinoma: A Systematic Review With Meta-Analysis. Front. Immunol. 2021, 12, 733530. [Google Scholar] [CrossRef]
- Zhang, L.; Ding, J.; Li, H.Y.; Wang, Z.H.; Wu, J. Immunotherapy for advanced hepatocellular carcinoma, where are we? Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188441. [Google Scholar] [CrossRef]
- Hao, P.; Yu, J.; Ward, R.; Liu, Y.; Hao, Q.; An, S.; Xu, T. Eukaryotic translation initiation factors as promising targets in cancer therapy. Cell Commun. Signal. 2020, 18, 175. [Google Scholar] [CrossRef]
- Scheurer, J.; Reisser, T.; Leithäuser, F.; Messmann, J.J.; Holzmann, K.; Debatin, K.M.; Strauss, G. Rapamycin-based graft-versus-host disease prophylaxis increases the immunosuppressivity of myeloid-derived suppressor cells without affecting T cells and anti-tumor cytotoxicity. Clin. Exp. Immunol. 2020, 202, 407–422. [Google Scholar] [CrossRef]
- Zarin, D.A.; Tse, T.; Williams, R.J.; Califf, R.M.; Ide, N.C. The ClinicalTrials.gov results database—Update and key issues. N. Engl. J. Med. 2011, 364, 852–860. [Google Scholar] [CrossRef] [Green Version]
- Sehgal, S.N. Sirolimus: Its discovery, biological properties, and mechanism of action. Transp. Proc. 2003, 35, S7–S14. [Google Scholar] [CrossRef]
- Chresta, C.M.; Davies, B.R.; Hickson, I.; Harding, T.; Cosulich, S.; Critchlow, S.E.; Vincent, J.P.; Ellston, R.; Jones, D.; Sini, P.; et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010, 70, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Badawi, M.; Kim, J.; Dauki, A.; Sutaria, D.; Motiwala, T.; Reyes, R.; Wani, N.; Kolli, S.; Jiang, J.; Coss, C.C.; et al. CD44 positive and sorafenib insensitive hepatocellular carcinomas respond to the ATP-competitive mTOR inhibitor INK128. Oncotarget 2018, 9, 26032–26045. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gufler, S.; Seeboeck, R.; Schatz, C.; Haybaeck, J. The Translational Bridge between Inflammation and Hepatocarcinogenesis. Cells 2022, 11, 533. https://doi.org/10.3390/cells11030533
Gufler S, Seeboeck R, Schatz C, Haybaeck J. The Translational Bridge between Inflammation and Hepatocarcinogenesis. Cells. 2022; 11(3):533. https://doi.org/10.3390/cells11030533
Chicago/Turabian StyleGufler, Sabine, Rita Seeboeck, Christoph Schatz, and Johannes Haybaeck. 2022. "The Translational Bridge between Inflammation and Hepatocarcinogenesis" Cells 11, no. 3: 533. https://doi.org/10.3390/cells11030533
APA StyleGufler, S., Seeboeck, R., Schatz, C., & Haybaeck, J. (2022). The Translational Bridge between Inflammation and Hepatocarcinogenesis. Cells, 11(3), 533. https://doi.org/10.3390/cells11030533