
Isoform-Specific Role of GSK-3 in High Fat Diet Induced Obesity and Glucose Intolerance

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of Global Conditional GSK-3α and GSK-3β Knockout Mouse Model
2.2. Mice
2.3. Diet
2.4. Body Composition
2.5. Oral Glucose Tolerance Test (GTT)
2.6. Insulin Tolerance Test (ITT)
2.7. Statistical Analysis
3. Results
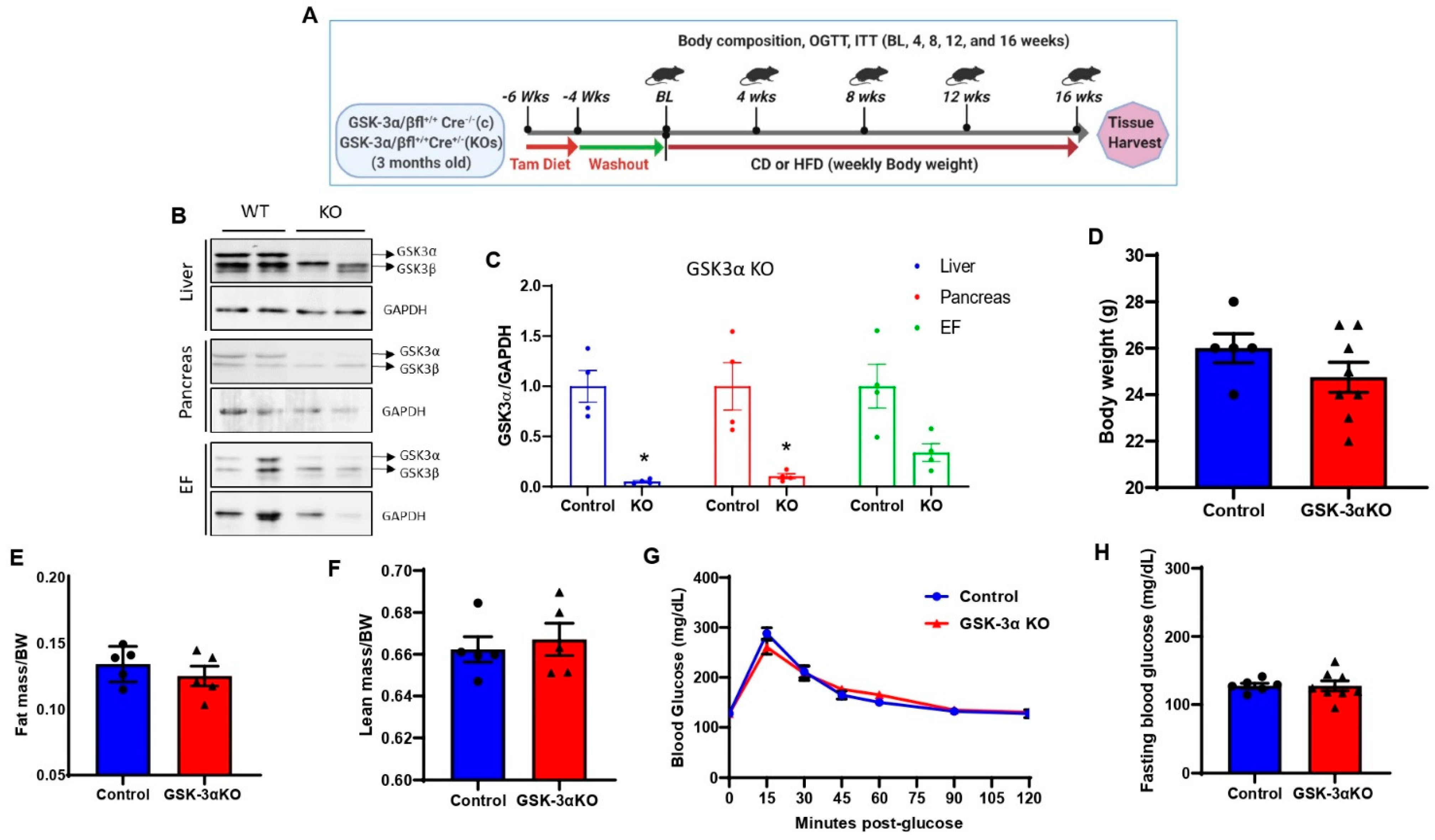
3.1. Creation and Characterization of Global Conditional GSK-3α Knock-Out (KO) Mice
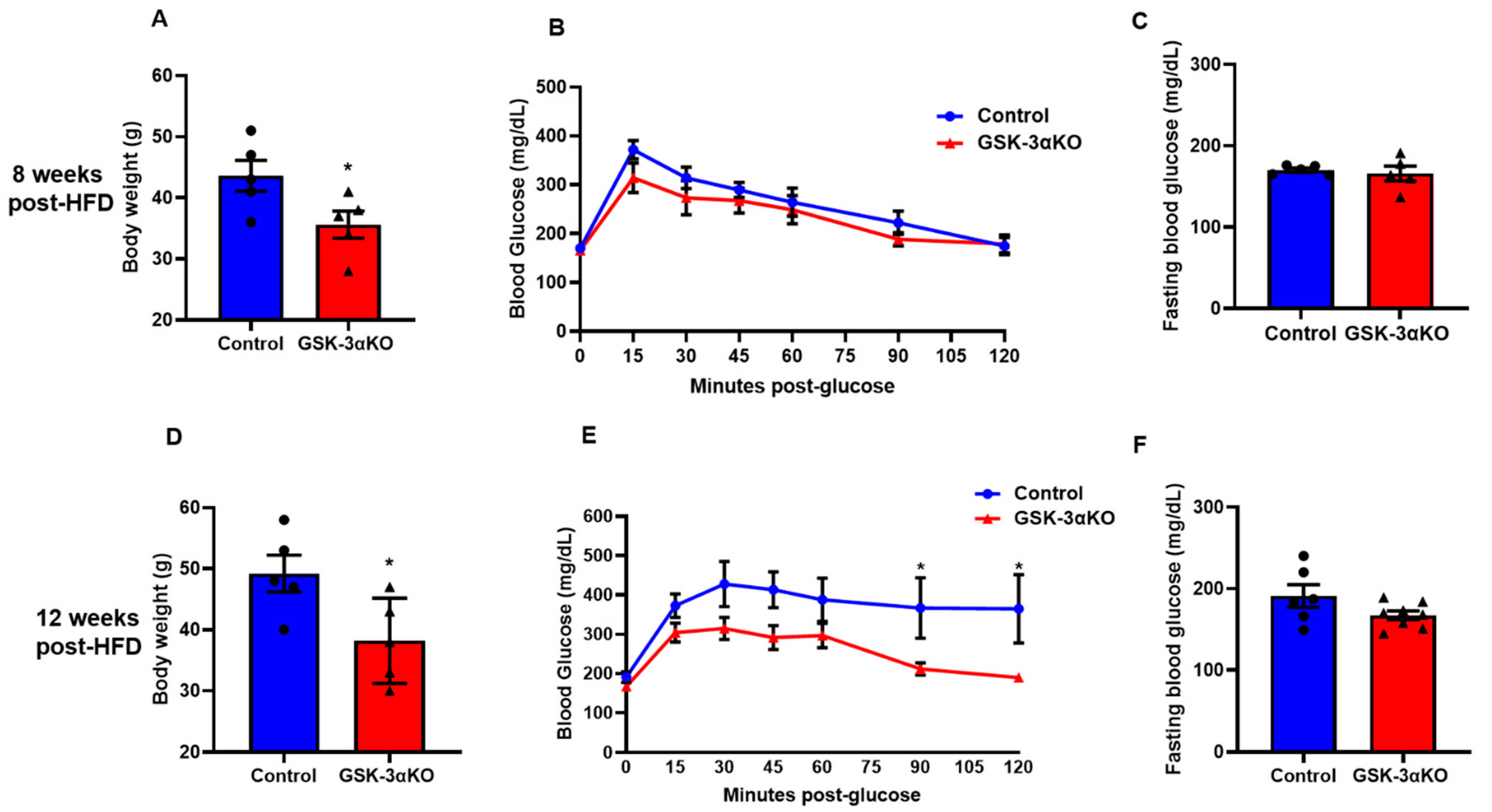
3.2. Conditional Global GSK-3α Deletion Protects from HFD-Induced Obesity but Plays a Minimal Role in Glucose Clearance
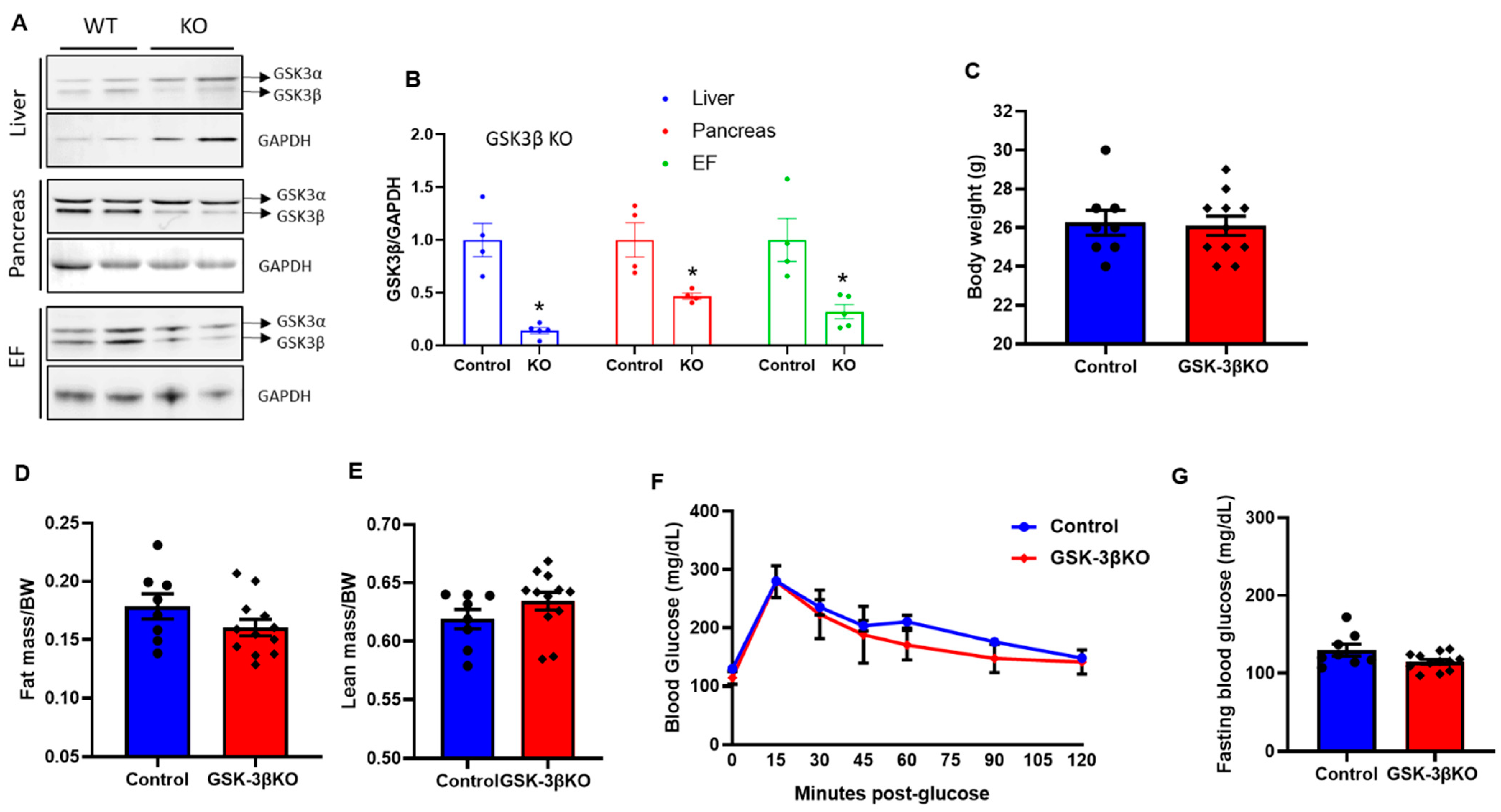
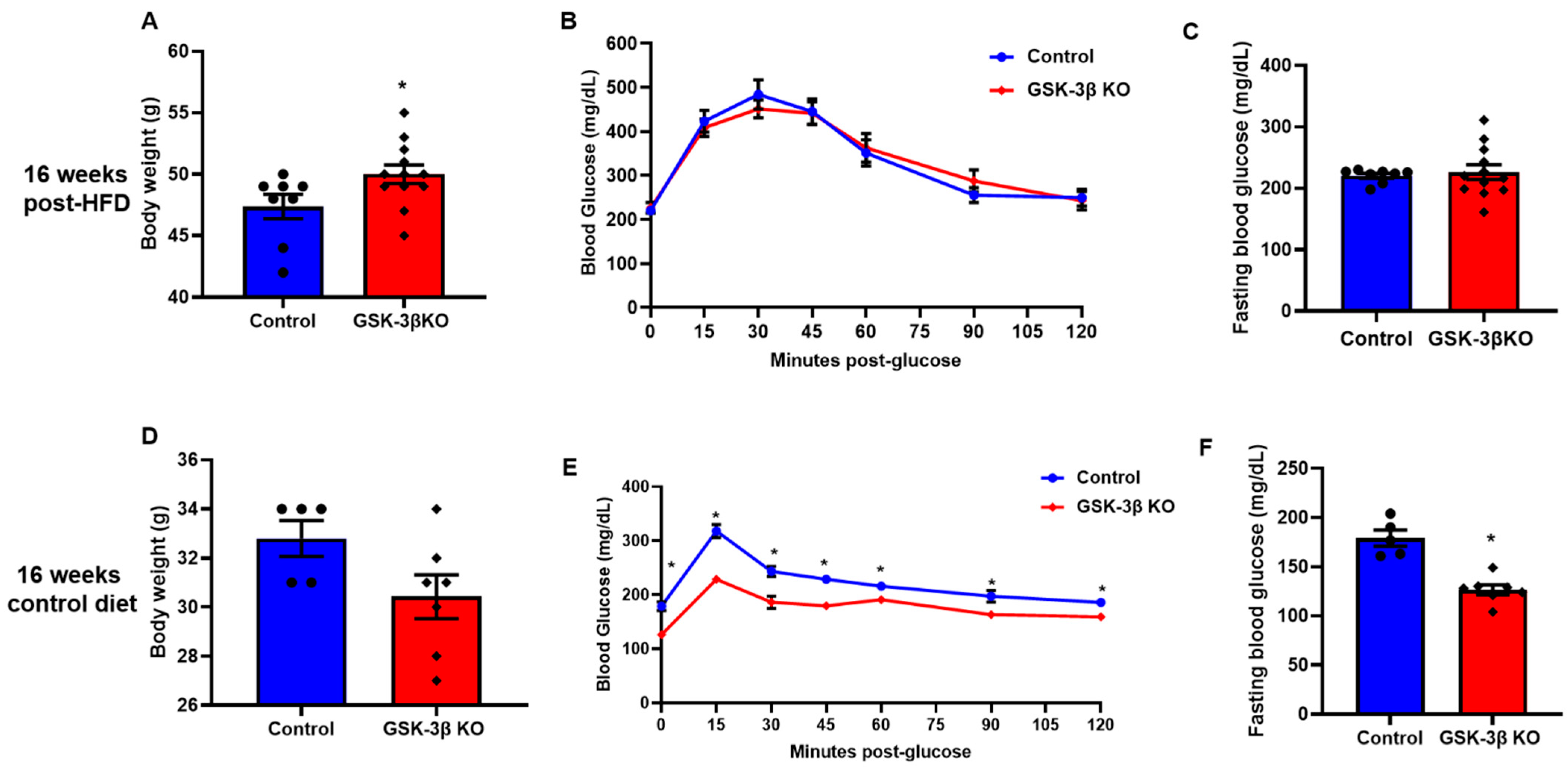
3.3. Baseline Characteristics of Conditional GSK-3β KOs
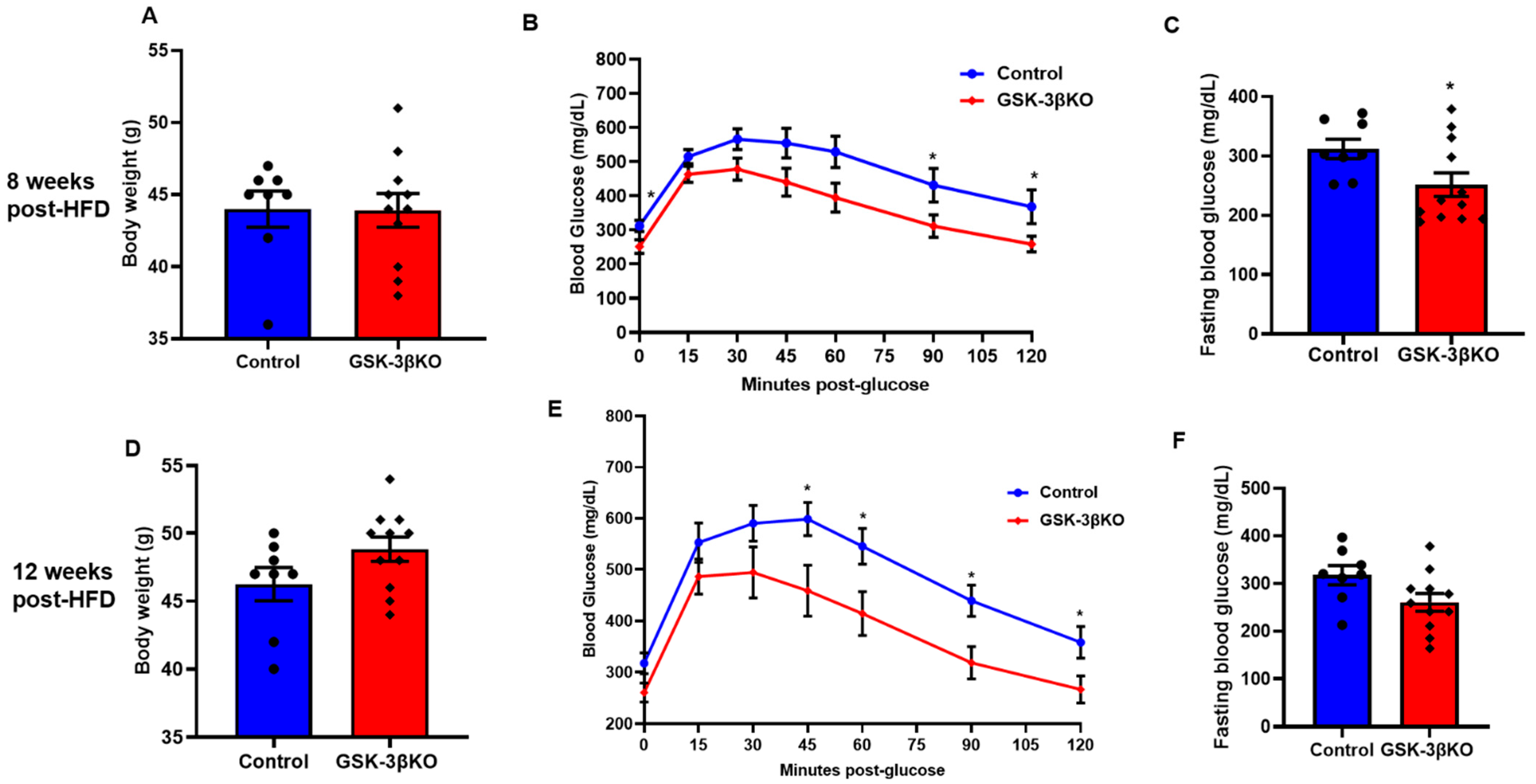
3.4. Conditional Global GSK-3β Deletion Protects from HFD-Induced Glucose Intolerance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Malik, V.S.; Willett, W.C.; Hu, F.B. Global obesity: Trends, risk factors and policy implications. Nat. Rev. Endocrinol. 2013, 9, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Tinajero, M.G.; Malik, V.S. An Update on the Epidemiology of Type 2 Diabetes: A Global Perspective. Endocrinol. Metab. Clin. N. Am. 2021, 50, 337–355. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare, M.; Soric, M.; Bovet, P.; Miranda, J.J.; Bhutta, Z.; Stevens, G.A.; Laxmaiah, A.; Kengne, A.P.; Bentham, J. The epidemiological burden of obesity in childhood: A worldwide epidemic requiring urgent action. BMC Med. 2019, 17, 212. [Google Scholar] [CrossRef] [Green Version]
- Bluher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Eldar-Finkelman, H.; Schreyer, S.A.; Shinohara, M.M.; LeBoeuf, R.C.; Krebs, E.G. Increased glycogen synthase kinase-3 activity in diabetes- and obesity-prone C57BL/6J mice. Diabetes 1999, 48, 1662–1666. [Google Scholar] [CrossRef] [PubMed]
- Kaidanovich, O.; Eldar-Finkelman, H. The role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Expert Opin. Ther. Targets 2002, 6, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, E.J.; Dokken, B.B. Role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Curr. Drug Targets 2006, 7, 1435–1441. [Google Scholar] [CrossRef]
- MacAulay, K.; Woodgett, J.R. Targeting glycogen synthase kinase-3 (GSK-3) in the treatment of Type 2 diabetes. Expert Opin. Ther. Targets 2008, 12, 1265–1274. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Gupte, M.; Umbarkar, P.; Singh, A.P.; Sui, J.Y.; Force, T.; Lal, H. Entanglement of GSK-3beta, beta-catenin and TGF-beta1 signaling network to regulate myocardial fibrosis. J. Mol. Cell. Cardiol. 2017, 110, 109–120. [Google Scholar] [CrossRef]
- Lal, H.; Ahmad, F.; Woodgett, J.; Force, T. The GSK-3 family as therapeutic target for myocardial diseases. Circ. Res. 2015, 116, 138–149. [Google Scholar] [CrossRef]
- Patel, P.; Woodgett, J.R. Glycogen Synthase Kinase 3: A Kinase for All Pathways? Curr. Top Dev. Biol. 2017, 123, 277–302. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Pan, W. GSK3: A multifaceted kinase in Wnt signaling. Trends Biochem. Sci. 2010, 35, 161–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Lal, H.; Chen, X.; Shang, X.; Song, J.; Li, Y.; Kerkela, R.; Doble, B.W.; MacAulay, K.; DeCaul, M.; et al. GSK-3alpha directly regulates beta-adrenergic signaling and the response of the heart to hemodynamic stress in mice. J. Clin. Investig. 2010, 120, 2280–2291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lal, H.; Zhou, J.; Ahmad, F.; Zaka, R.; Vagnozzi, R.J.; Decaul, M.; Woodgett, J.; Gao, E.; Force, T. Glycogen synthase kinase-3alpha limits ischemic injury, cardiac rupture, post-myocardial infarction remodeling and death. Circulation 2012, 125, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Kerkela, R.; Kockeritz, L.; Macaulay, K.; Zhou, J.; Doble, B.W.; Beahm, C.; Greytak, S.; Woulfe, K.; Trivedi, C.M.; Woodgett, J.R.; et al. Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J. Clin. Investig. 2008, 118, 3609–3618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Freeman, T.A.; Ahmad, F.; Shang, X.; Mangano, E.; Gao, E.; Farber, J.; Wang, Y.; Ma, X.L.; Woodgett, J.; et al. GSK-3alpha is a central regulator of age-related pathologies in mice. J. Clin. Investig. 2013, 123, 1821–1832. [Google Scholar] [CrossRef] [PubMed]
- Frame, S.; Zheleva, D. Targeting glycogen synthase kinase-3 in insulin signalling. Expert Opin. Ther. Targets 2006, 10, 429–444. [Google Scholar] [CrossRef]
- Liberman, Z.; Eldar-Finkelman, H. Serine 332 phosphorylation of insulin receptor substrate-1 by glycogen synthase kinase-3 attenuates insulin signaling. J. Biol. Chem. 2005, 280, 4422–4428. [Google Scholar] [CrossRef] [Green Version]
- Leng, S.; Zhang, W.; Zheng, Y.; Liberman, Z.; Rhodes, C.J.; Eldar-Finkelman, H.; Sun, X.J. Glycogen synthase kinase 3 beta mediates high glucose-induced ubiquitination and proteasome degradation of insulin receptor substrate 1. J. Endocrinol. 2010, 206, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Dokken, B.B.; Sloniger, J.A.; Henriksen, E.J. Acute selective glycogen synthase kinase-3 inhibition enhances insulin signaling in prediabetic insulin-resistant rat skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E1188–E1194. [Google Scholar] [CrossRef]
- Gupte, M.; Umbarkar, P.; Singh, A.P.; Zhang, Q.; Tousif, S.; Lal, H. Deletion of Cardiomyocyte Glycogen Synthase Kinase-3 Beta (GSK-3beta) Improves Systemic Glucose Tolerance with Maintained Heart Function in Established Obesity. Cells 2020, 9, 1120. [Google Scholar] [CrossRef]
- Kim, K.M.; Lee, K.S.; Lee, G.Y.; Jin, H.; Durrance, E.S.; Park, H.S.; Choi, S.H.; Park, K.S.; Kim, Y.B.; Jang, H.C.; et al. Anti-diabetic efficacy of KICG1338, a novel glycogen synthase kinase-3beta inhibitor, and its molecular characterization in animal models of type 2 diabetes and insulin resistance. Mol. Cell. Endocrinol. 2015, 409, 1–10. [Google Scholar] [CrossRef]
- Liu, Y.; Tanabe, K.; Baronnier, D.; Patel, S.; Woodgett, J.; Cras-Meneur, C.; Permutt, M.A. Conditional ablation of Gsk-3beta in islet beta cells results in expanded mass and resistance to fat feeding-induced diabetes in mice. Diabetologia 2010, 53, 2600–2610. [Google Scholar] [CrossRef] [Green Version]
- Ullah, A.; Ali, N.; Ahmad, S.; Rahman, S.U.; Alghamdi, S.; Bannunah, A.M.; Ali, R.; Aman, A.; Khan, J.; Hussain, H.; et al. Glycogen synthase kinase-3 (GSK-3) a magic enzyme: It’s role in diabetes mellitus and glucose homeostasis, interactions with fluroquionlones: A mini-review. Braz. J. Biol. 2021, 10, e250179. [Google Scholar] [CrossRef] [PubMed]
- Srivani, G.; Sharvirala, R.; Veerareddy, P.R.; Pal, D.; Kiran, G. GSK-3 Inhibitors as New Leads to Treat Type-II Diabetes. Curr. Drug Targets 2021, 22, 1555–1567. [Google Scholar] [CrossRef]
- Nikoulina, S.E.; Ciaraldi, T.P.; Mudaliar, S.; Mohideen, P.; Carter, L.; Henry, R.R. Potential role of glycogen synthase kinase-3 in skeletal muscle insulin resistance of type 2 diabetes. Diabetes 2000, 49, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Henriksen, E.J. Dysregulation of glycogen synthase kinase-3 in skeletal muscle and the etiology of insulin resistance and type 2 diabetes. Curr. Diabetes Rev. 2010, 6, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Hao, C.M.; Redha, R.; Wasserman, D.H.; McGuinness, O.P.; Breyer, M.D. Glycogen synthase kinase 3 inhibition improves insulin-stimulated glucose metabolism but not hypertension in high-fat-fed C57BL/6J mice. Diabetologia 2007, 50, 452–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ring, D.B.; Johnson, K.W.; Henriksen, E.J.; Nuss, J.M.; Goff, D.; Kinnick, T.R.; Ma, S.T.; Reeder, J.W.; Samuels, I.; Slabiak, T.; et al. Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes 2003, 52, 588–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Force, T.; Woodgett, J.R. Unique and overlapping functions of GSK-3 isoforms in cell differentiation and proliferation and cardiovascular development. J. Biol. Chem. 2009, 284, 9643–9647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, P.; Sciarretta, S.; Galeotti, J.; Volpe, M.; Sadoshima, J. Differential roles of GSK-3beta during myocardial ischemia and ischemia/reperfusion. Circ. Res. 2011, 109, 502–511. [Google Scholar] [CrossRef]
- Cho, J.; Rameshwar, P.; Sadoshima, J. Distinct roles of glycogen synthase kinase (GSK)-3alpha and GSK-3beta in mediating cardiomyocyte differentiation in murine bone marrow-derived mesenchymal stem cells. J. Biol. Chem. 2009, 284, 36647–36658. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, T.; Zhai, P.; Maejima, Y.; Hong, C.; Gao, S.; Tian, B.; Goto, K.; Takagi, H.; Tamamori-Adachi, M.; Kitajima, S.; et al. Distinct roles of GSK-3alpha and GSK-3beta phosphorylation in the heart under pressure overload. Proc. Natl. Acad. Sci. USA 2008, 105, 20900–20905. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Ahmad, F.; Parikh, S.; Hoffman, N.E.; Rajan, S.; Verma, V.K.; Song, J.; Yuan, A.; Shanmughapriya, S.; Guo, Y.; et al. Loss of Adult Cardiac Myocyte GSK-3 Leads to Mitotic Catastrophe Resulting in Fatal Dilated Cardiomyopathy. Circ. Res. 2016, 118, 1208–1222. [Google Scholar] [CrossRef]
- MacAulay, K.; Doble, B.W.; Patel, S.; Hansotia, T.; Sinclair, E.M.; Drucker, D.J.; Nagy, A.; Woodgett, J.R. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007, 6, 329–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, F.; Lal, H.; Zhou, J.; Vagnozzi, R.J.; Yu, J.E.; Shang, X.; Woodgett, J.R.; Gao, E.; Force, T. Cardiomyocyte-specific deletion of Gsk3alpha mitigates post-myocardial infarction remodeling, contractile dysfunction, and heart failure. J. Am. Coll. Cardiol. 2014, 64, 696–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.; Doble, B.W.; MacAulay, K.; Sinclair, E.M.; Drucker, D.J.; Woodgett, J.R. Tissue-specific role of glycogen synthase kinase 3beta in glucose homeostasis and insulin action. Mol. Cell. Biol. 2008, 28, 6314–6328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Macaulay, K.; Woodgett, J.R. Tissue-specific analysis of glycogen synthase kinase-3alpha (GSK-3alpha) in glucose metabolism: Effect of strain variation. PLoS ONE 2011, 6, e15845. [Google Scholar] [CrossRef] [Green Version]
- McCamphill, P.K.; Stoppel, L.J.; Senter, R.K.; Lewis, M.C.; Heynen, A.J.; Stoppel, D.C.; Sridhar, V.; Collins, K.A.; Shi, X.; Pan, J.Q.; et al. Selective inhibition of glycogen synthase kinase 3alpha corrects pathophysiology in a mouse model of fragile X syndrome. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Wagner, F.F.; Benajiba, L.; Campbell, A.J.; Weiwer, M.; Sacher, J.R.; Gale, J.P.; Ross, L.; Puissant, A.; Alexe, G.; Conway, A.; et al. Exploiting an Asp-Glu “switch” in glycogen synthase kinase 3 to design paralog-selective inhibitors for use in acute myeloid leukemia. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.; Taylor, B.; Jin, Q.; Nguyen-Tran, V.; Meeusen, S.; Zhang, Y.Q.; Kamireddy, A.; Swafford, A.; Powers, A.F.; Walker, J.; et al. Inhibition of DYRK1A and GSK3B induces human beta-cell proliferation. Nat. Commun. 2015, 6, 8372. [Google Scholar] [CrossRef]
- Stein, J.; Milewski, W.M.; Hara, M.; Steiner, D.F.; Dey, A. GSK-3 inactivation or depletion promotes beta-cell replication via down regulation of the CDK inhibitor, p27 (Kip1). Islets 2011, 3, 21–34. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Tanabe, K.; Bernal-Mizrachi, E.; Permutt, M.A. Mice with beta cell overexpression of glycogen synthase kinase-3beta have reduced beta cell mass and proliferation. Diabetologia 2008, 51, 623–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mussmann, R.; Geese, M.; Harder, F.; Kegel, S.; Andag, U.; Lomow, A.; Burk, U.; Onichtchouk, D.; Dohrmann, C.; Austen, M. Inhibition of GSK3 promotes replication and survival of pancreatic beta cells. J. Biol. Chem. 2007, 282, 12030–12037. [Google Scholar] [CrossRef] [Green Version]
- Praharaj, S.K. Metformin for Lithium-induced Weight Gain: A Case Report. Clin. Psychopharmacol. Neurosci. 2016, 14, 101–103. [Google Scholar] [CrossRef] [Green Version]
- Atmaca, M.; Kuloglu, M.; Tezcan, E.; Ustundag, B. Weight gain and serum leptin levels in patients on lithium treatment. Neuropsychobiology 2002, 46, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Reekie, J.; Hosking, S.P.; Prakash, C.; Kao, K.T.; Juonala, M.; Sabin, M.A. The effect of antidepressants and antipsychotics on weight gain in children and adolescents. Obes. Rev. 2015, 16, 566–580. [Google Scholar] [CrossRef]
- Sfera, A.; Osorio, C.; Inderias, L.A.; Parker, V.; Price, A.I.; Cummings, M. The Obesity-Impulsivity Axis: Potential Metabolic Interventions in Chronic Psychiatric Patients. Front. Psychiatry 2017, 8, 20. [Google Scholar] [CrossRef] [Green Version]
- Haupt, D.W.; Newcomer, J.W. Abnormalities in glucose regulation associated with mental illness and treatment. J. Psychosom. Res. 2002, 53, 925–933. [Google Scholar] [CrossRef]
- de Groot, T.; Damen, L.; Kosse, L.; Alsady, M.; Doty, R.; Baumgarten, R.; Sheehan, S.; van der Vlag, J.; Korstanje, R.; Deen, P.M.T. Lithium reduces blood glucose levels, but aggravates albuminuria in BTBR-ob/ob mice. PLoS ONE 2017, 12, e0189485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Anshul, F.; Malhotra, D.K.; Jaume, J.; Dworkin, L.D.; Gong, R. Microdose Lithium Protects against Pancreatic Islet Destruction and Renal Impairment in Streptozotocin-Elicited Diabetes. Antioxidants 2021, 10, 138. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, P.; Amdisen, A.; Schou, M. Clinically significant side effects of lithium treatment. A survey of 237 patients in long-term treatment. Acta Psychiatr. Scand. 1980, 62, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Chengappa, K.N.; Chalasani, L.; Brar, J.S.; Parepally, H.; Houck, P.; Levine, J. Changes in body weight and body mass index among psychiatric patients receiving lithium, valproate, or topiramate: An open-label, nonrandomized chart review. Clin. Ther. 2002, 24, 1576–1584. [Google Scholar] [CrossRef]
- Peselow, E.D.; Dunner, D.L.; Fieve, R.R.; Lautin, A. Lithium carbonate and weight gain. J. Affect. Disord. 1980, 2, 303–310. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupte, M.; Tousif, S.; Lemon, J.J.; Toro Cora, A.; Umbarkar, P.; Lal, H. Isoform-Specific Role of GSK-3 in High Fat Diet Induced Obesity and Glucose Intolerance. Cells 2022, 11, 559. https://doi.org/10.3390/cells11030559
Gupte M, Tousif S, Lemon JJ, Toro Cora A, Umbarkar P, Lal H. Isoform-Specific Role of GSK-3 in High Fat Diet Induced Obesity and Glucose Intolerance. Cells. 2022; 11(3):559. https://doi.org/10.3390/cells11030559
Chicago/Turabian StyleGupte, Manisha, Sultan Tousif, Jacob J. Lemon, Angelica Toro Cora, Prachi Umbarkar, and Hind Lal. 2022. "Isoform-Specific Role of GSK-3 in High Fat Diet Induced Obesity and Glucose Intolerance" Cells 11, no. 3: 559. https://doi.org/10.3390/cells11030559
APA StyleGupte, M., Tousif, S., Lemon, J. J., Toro Cora, A., Umbarkar, P., & Lal, H. (2022). Isoform-Specific Role of GSK-3 in High Fat Diet Induced Obesity and Glucose Intolerance. Cells, 11(3), 559. https://doi.org/10.3390/cells11030559