
Comparative Analysis of Gene Expression in Fibroblastic Foci in Patients with Idiopathic Pulmonary Fibrosis and Pulmonary Sarcoidosis

 , , , , ,
, , , , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Histopathology
2.2. Laser Capture Microdissection
2.3. Gene Expression Analysis
2.4. Protein Expression Analysis
3. Results
3.1. Clinical Information
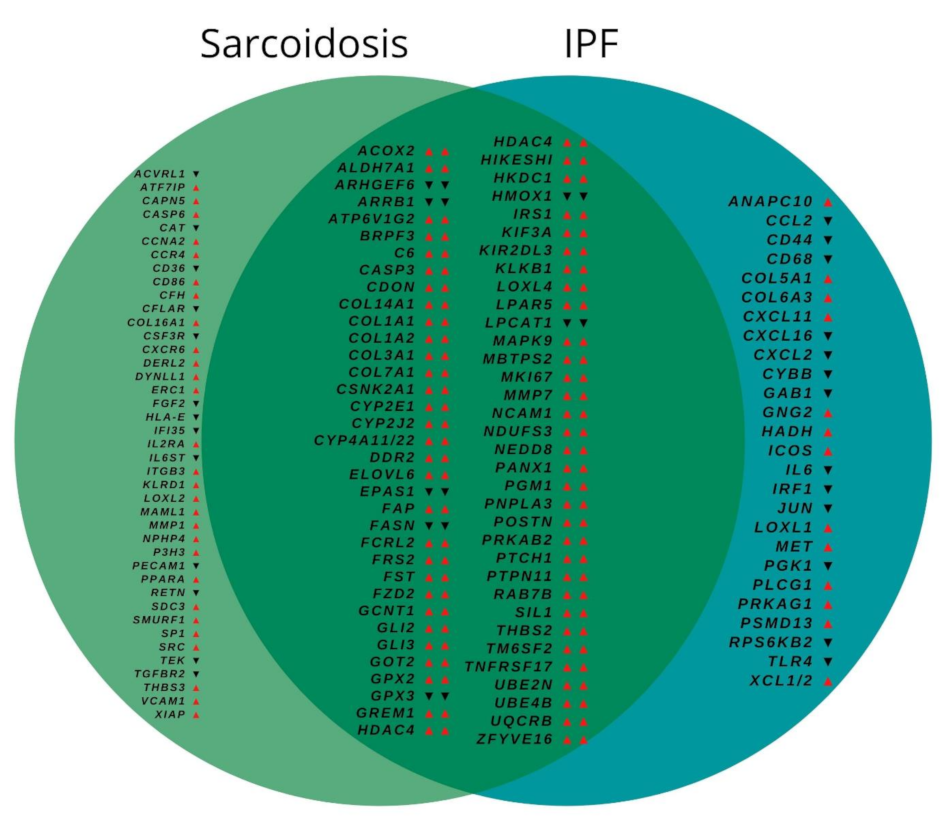
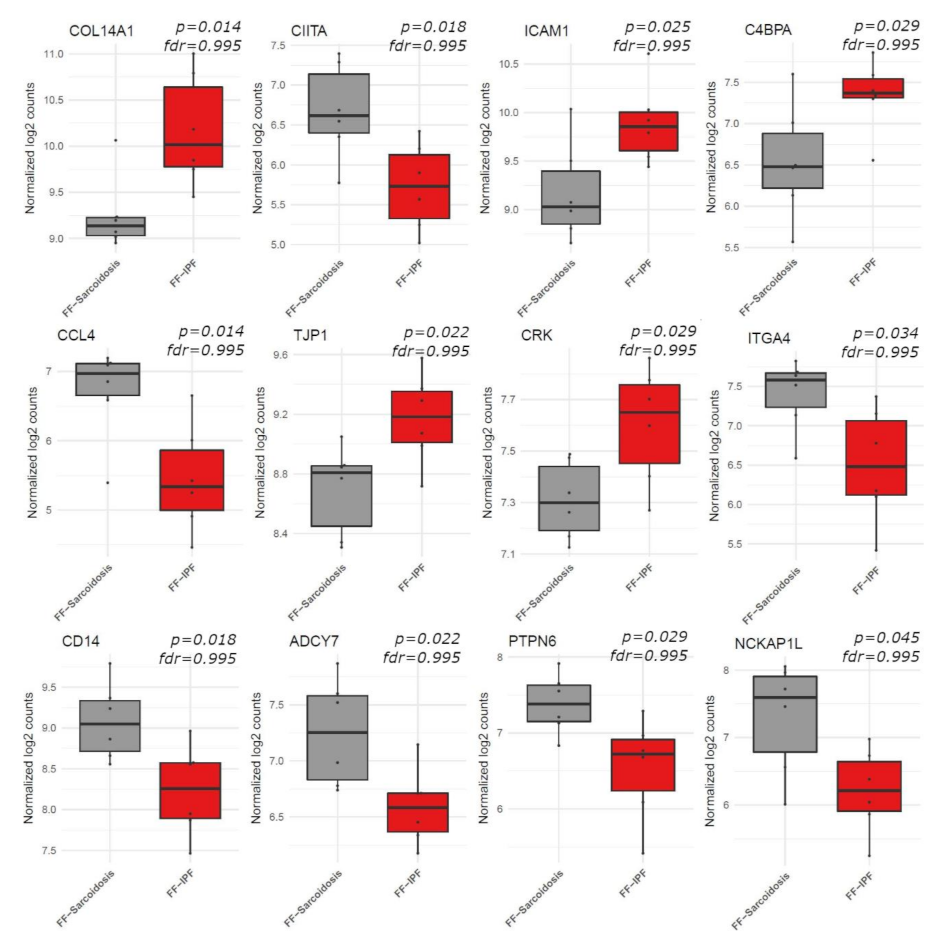
3.2. Gene Expression
3.3. Biological Pathway Analysis
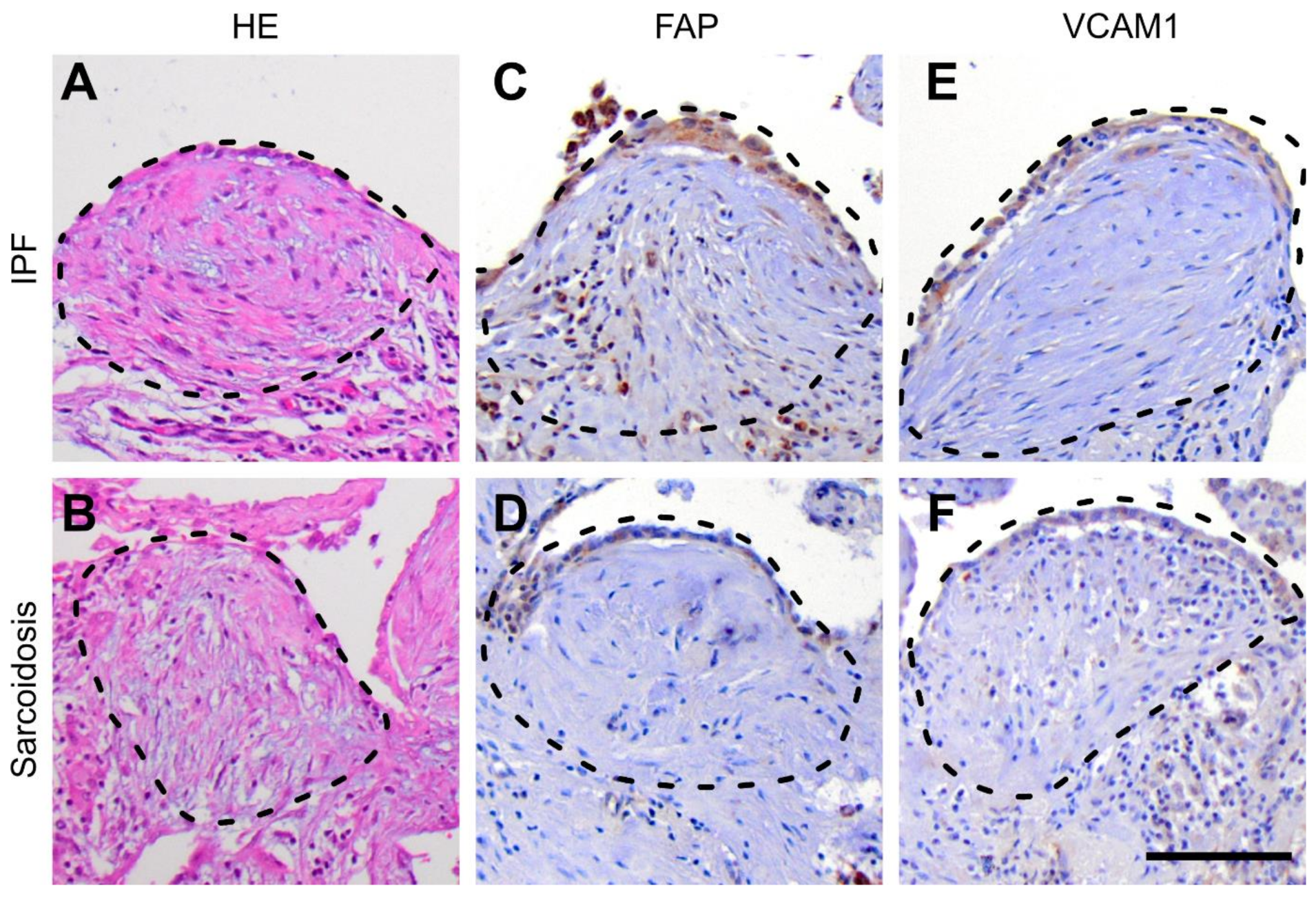
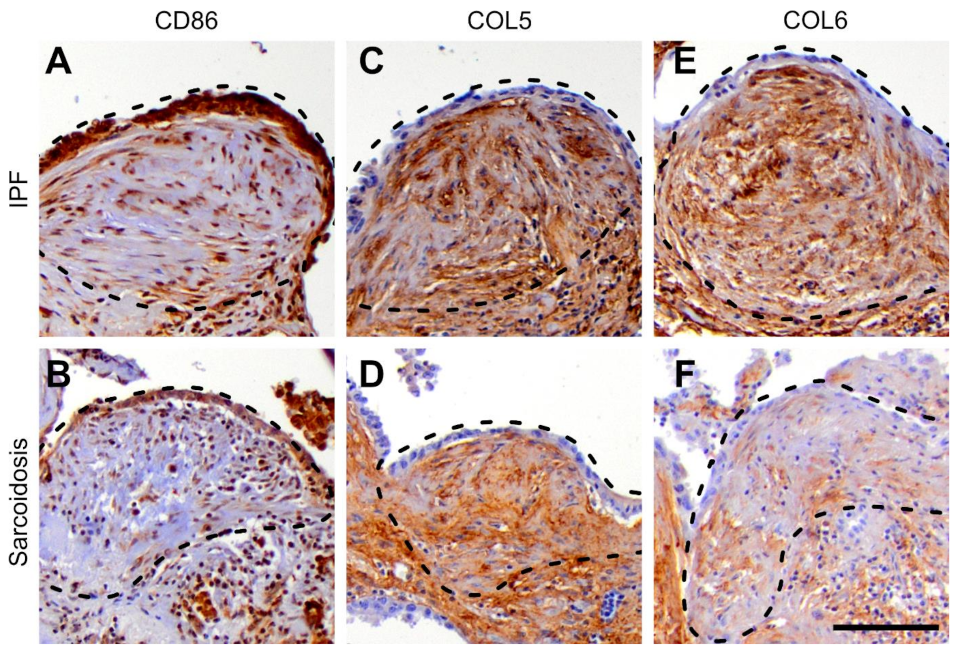
3.4. Protein Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.-F.; Flaherty, K.R.; Lasky, J.A.; et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-based Guidelines for Diagnosis and Management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.A.; Sverzellati, N.; Travis, W.D.; Brown, K.K.; Colby, T.V.; Galvin, J.R.; Goldin, J.G.; Hansell, D.M.; Inoue, Y.; Johkoh, T.; et al. Diagnostic criteria for idiopathic pulmonary fibrosis: A Fleischner Society White Paper. Lancet Respir. Med. 2018, 6, 138–153. [Google Scholar] [CrossRef]
- Harada, T.; Watanabe, K.; Nabeshima, K.; Hamasaki, M.; Iwasaki, H. Prognostic significance of fibroblastic foci in usual interstitial pneumonia and non-specific interstitial pneumonia. Respirology 2013, 18, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chan, K.M.; Schmidt, L.A.; Myers, J.L. Histopathology of Explanted Lungs From Patients With a Diagnosis of Pulmonary Sarcoidosis. Chest 2016, 149, 499–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.R.; Martinez, F.J.; Travis, W.; Lynch, I.J.P. Nonspecific Interstitial Pneumonia (NSIP). Semin. Respir. Crit. Care Med. 2001, 22, 423–434. [Google Scholar] [CrossRef]
- Churg, A.; Bilawich, A.; Wright, J.L. Pathology of Chronic Hypersensitivity Pneumonitis What Is It? What Are the Diagnostic Criteria? Why Do We Care? Arch. Pathol. Lab. Med. 2017, 142, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Baughman, R.P.; Culver, D.A.; Judson, M.A. A Concise Review of Pulmonary Sarcoidosis. Am. J. Respir. Crit. Care Med. 2011, 183, 573–581. [Google Scholar] [CrossRef] [Green Version]
- Rossi, A.; Cavazza, A.; Colby, T.V. Pathology of Sarcoidosis. Clin. Rev. Allergy Immunol. 2015, 49, 36–44. [Google Scholar] [CrossRef]
- Spagnolo, P.; Rossi, G.; Trisolini, R.; Sverzellati, N.; Baughman, R.P.; Wells, A.U. Pulmonary sarcoidosis. Lancet Respir. Med. 2018, 6, 389–402. [Google Scholar] [CrossRef]
- Länger, F.; Stark, H.; Braubach, P.; Ackermann, M.; Hussein, K.; Teiken, K.; Maegel, L.; Kuehnel, M.; Jonigk, D. Schädigungsmuster interstitieller Lungenerkrankungen. Der Pathol. 2018, 39, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Katzenstein, A.L.; Myers, J.L. Idiopathic Pulmonary Fibrosis Clinical Relevance of Pathologic Classification Clinical Features of the Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med. 1998, 157, 1301–1315. [Google Scholar] [CrossRef]
- Guillotin, D.; Taylor, A.R.; Platé, M.; Mercer, P.F.; Edwards, L.M.; Haggart, R.; Miele, G.; McAnulty, R.J.; Maher, T.M.; E Hynds, R.; et al. Transcriptome analysis of IPF fibroblastic foci identifies key pathways involved in fibrogenesis. Thorax 2020, 76, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, Allosteric Signaling Machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Li, S.; Li, W. LOX/LOXL in pulmonary fibrosis: Potential therapeutic targets. J. Drug Target. 2018, 27, 790–796. [Google Scholar] [CrossRef]
- Tjin, G.; White, E.S.; Faiz, A.; Sicard, D.; Tschumperlin, D.J.; Mahar, A.; Kable, E.P.W.; Burgess, J.K. Correction: Lysyl oxidases regulate fibrillar collagen remodelling in idiopathic pulmonary fibrosis. Dis. Models Mech. 2017, 10, 1301–1312. [Google Scholar] [CrossRef] [Green Version]
- Bellaye, P.-S.; Shimbori, C.; Upagupta, C.; Sato, S.; Shi, W.; Gauldie, J.; Ask, K.; Kolb, M. Lysyl Oxidase–Like 1 Protein Deficiency Protects Mice from Adenoviral Transforming Growth Factor-β1–induced Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 461–470. [Google Scholar] [CrossRef]
- Zuo, F.; Kaminski, N.; Eugui, E.; Allard, J.; Yakhini, Z.; Ben-Dor, A.; Lollini, L.; Morris, D.; Kim, Y.; DeLustro, B.; et al. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc. Natl. Acad. Sci. USA 2002, 99, 6292–6297. [Google Scholar] [CrossRef] [Green Version]
- Vuorinen, K.; Myllärniemi, M.; Lammi, L.; Piirilä, P.; Rytilä, P.; Salmenkivi, K.; Kinnula, V.L. Elevated matrilysin levels in bronchoalveolar lavage fluid do not distinguish idiopathic pulmonary fibrosis from other interstitial lung diseases. APMIS 2007, 115, 969–975. [Google Scholar] [CrossRef]
- Leng, D.; Yi, J.; Xiang, M.; Zhao, H.; Zhang, Y. Identification of common signatures in idiopathic pulmonary fibrosis and lung cancer using gene expression modeling. BMC Cancer 2020, 20, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Rosas, I.O.; Richards, T.J.; Konishi, K.; Zhang, Y.; Gibson, K.; Lokshin, A.E.; Lindell, K.O.; Cisneros, J.; MacDonald, S.D.; Pardo, A.; et al. MMP1 and MMP7 as Potential Peripheral Blood Biomarkers in Idiopathic Pulmonary Fibrosis. PLoS Med. 2008, 5, e93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morais, A.; Beltrão, M.; Sokhatska, O.; Costa, D.; Melo, N.; Mota, P.; Marques, A.; Delgado, L. Serum metalloproteinases 1 and 7 in the diagnosis of idiopathic pulmonary fibrosis and other interstitial pneumonias. Respir. Med. 2015, 109, 1063–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.-F.; Jiang, L.; Li, Z.-H.; Kang, J. Change of matrix metalloproteinase-1 and matrix metalloproteinase-7 in serum and bronchoalveolar lavage fluid of patients with idiopathic pulmonary fibrosis and sarcoidosis. Chin. J. Tuberc. Respir. Dis. 2010, 33. [Google Scholar]
- Kelly, M.M.; Leigh, R.; Gilpin, S.E.; Cheng, E.; Martin, G.E.M.; Radford, K.; Cox, G.; Gauldie, J. Cell-specific Gene Expression in Patients with Usual Interstitial Pneumonia. Am. J. Respir. Crit. Care Med. 2006, 174, 557–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isshiki, T.; Matsuyama, H.; Yamaguchi, T.; Morita, T.; Ono, J.; Nunomura, S.; Izuhara, K.; Sakamoto, S.; Homma, S.; Kishi, K. Plasma matrix metalloproteinase 7, CC-chemokine ligand 18, and periostin as markers for pulmonary sarcoidosis. Respir. Investig. 2020, 58, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Stella, G.M.; Gentile, A.; Balderacchi, A.; Meloni, F.; Milan, M.; Benvenuti, S. Ockham’s razor for the MET-driven invasive growth linking idiopathic pulmonary fibrosis and cancer. J. Transl. Med. 2016, 14, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Grimminger, F.; Günther, A.; Vancheri, C. The role of tyrosine kinases in the pathogenesis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1426–1433. [Google Scholar] [CrossRef] [Green Version]
- Richeldi, L.; Du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [Green Version]
- Wollin, L.; Distler, J.H.; Redente, E.F.; Riches, D.W.H.; Stowasser, S.; Schlenker-Herceg, R.; Maher, T.; Kolb, M. Potential of nintedanib in treatment of progressive fibrosing interstitial lung diseases. Eur. Respir. J. 2019, 54, 1900161. [Google Scholar] [CrossRef]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.; Stowasser, S.; Kolb, M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef] [PubMed]
- Lattanzio, R.; Piantelli, M.; Falasca, M. Role of phospholipase C in cell invasion and metastasis. Adv. Biol. Regul. 2013, 53, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tomar, A.; George, S.P.; Khurana, S. Obligatory role for phospholipase C-γ1in villin-induced epithelial cell migration. Am. J. Physiol. Physiol. 2007, 292, C1775–C1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhao, C.; Tian, Y.; Lu, J.; Zhang, G.; Liang, S.; Chen, D.; Liu, X.; Kuang, W.; Zhu, M. Src family kinases and pulmonary fibrosis: A review. Biomed. Pharmacother. 2020, 127, 110183. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Zhu, L.; Eckert, R.L.; Simonson, M.S. TGF-β-regulated collagen type I accumulation: Role of Src-based signals. Am. J. Physiol. Physiol. 2007, 292, C1361–C1369. [Google Scholar] [CrossRef] [Green Version]
- Beyer, C.; Distler, J.H. Tyrosine kinase signaling in fibrotic disorders. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1832, 897–904. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Che, P.; Han, X.; Cai, G.-Q.; Liu, G.; Antony, V.; Luckhardt, T.; Siegal, G.P.; Zhou, Y.; Liu, R.-M.; et al. Therapeutic Targeting of Src Kinase in Myofibroblast Differentiation and Pulmonary Fibrosis. J. Pharmacol. Exp. Ther. 2014, 351, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Jonigk, D.; Rath, B.; Borchert, P.; Braubach, P.; Maegel, L.; Izykowski, N.; Warnecke, G.; Sommer, W.; Kreipe, H.; Blach, R.; et al. Comparative analysis of morphological and molecular motifs in bronchiolitis obliterans and alveolar fibroelastosis after lung and stem cell transplantation. J. Pathol. Clin. Res. 2016, 3, 17–28. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Signaling Pathway | FF-S vs. Controls | FF-IPF vs. Controls | FF-IPF vs. FF-S | |||
|---|---|---|---|---|---|---|
| Regulation | fdr | Regulation | fdr | Regulation | fdr | |
| Cell adhesion 1 | Up | 0.782 | Up | 0.437 | Up | 0.018 |
| ECM structure 1 | Up | 0.165 | Up | 0.063 | Up | 0.018 |
| ECM receptor interaction 1 | Up | 0.623 | Up | 0.437 | Up | 0.018 |
| ECM organization 2 | Up | 0.83 | Up | 0.503 | Up | 0.005 |
| Organization of collagen fibrils within ECM 2 | Up | 0.261 | Up | 0.036 | Up | 0.095 |
| Innate immune system 1 | Down | 0.165 | Down | 0.011 | Down | 0.0001 |
| Type I interferon signaling 1 | Down | 0.165 | Down | 0.031 | Down | 0.002 |
| Type II interferon signaling 1 | Down | 0.343 | Down | 0.206 | Down | 0.009 |
| MHC class I antigen presentation 1 | Down | 0.13 | Down | 0.036 | Down | 0.002 |
| Activation of TNF production 2 | Down | 0.916 | Down | 0.038 | Down | 0.005 |
| Activation of ERK1/ERK2 cascade 2 | Down | 0.14 | Down | 0.036 | Down | 0.884 |
| NLR signaling 2 | Down | 0.276 | Down | 0.036 | Down | 0.241 |
| TLR signaling 1 | Down | 0.926 | Down | 0.202 | Down | 0.001 |
| T-cell receptor signaling 1 | Down | 0.926 | Down | 0.96 | Down | 0.013 |
| Chemokine signaling 1 | Down | 0.957 | Down | 0.769 | Down | 0.018 |
| Macrophages 1 | Down | 0.787 | Down | 0.091 | Down | 0.015 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamp, J.C.; Neubert, L.; Stark, H.; Hinrichs, J.B.; Boekhoff, C.; Seidel, A.D.; Ius, F.; Haverich, A.; Gottlieb, J.; Welte, T.; et al. Comparative Analysis of Gene Expression in Fibroblastic Foci in Patients with Idiopathic Pulmonary Fibrosis and Pulmonary Sarcoidosis. Cells 2022, 11, 664. https://doi.org/10.3390/cells11040664
Kamp JC, Neubert L, Stark H, Hinrichs JB, Boekhoff C, Seidel AD, Ius F, Haverich A, Gottlieb J, Welte T, et al. Comparative Analysis of Gene Expression in Fibroblastic Foci in Patients with Idiopathic Pulmonary Fibrosis and Pulmonary Sarcoidosis. Cells. 2022; 11(4):664. https://doi.org/10.3390/cells11040664
Chicago/Turabian StyleKamp, Jan C., Lavinia Neubert, Helge Stark, Jan B. Hinrichs, Caja Boekhoff, Allison D. Seidel, Fabio Ius, Axel Haverich, Jens Gottlieb, Tobias Welte, and et al. 2022. "Comparative Analysis of Gene Expression in Fibroblastic Foci in Patients with Idiopathic Pulmonary Fibrosis and Pulmonary Sarcoidosis" Cells 11, no. 4: 664. https://doi.org/10.3390/cells11040664
APA StyleKamp, J. C., Neubert, L., Stark, H., Hinrichs, J. B., Boekhoff, C., Seidel, A. D., Ius, F., Haverich, A., Gottlieb, J., Welte, T., Braubach, P., Laenger, F., Hoeper, M. M., Kuehnel, M. P., & Jonigk, D. D. (2022). Comparative Analysis of Gene Expression in Fibroblastic Foci in Patients with Idiopathic Pulmonary Fibrosis and Pulmonary Sarcoidosis. Cells, 11(4), 664. https://doi.org/10.3390/cells11040664