
Altered Expression of Zonula occludens-1 Affects Cardiac Na+ Channels and Increases Susceptibility to Ventricular Arrhythmias

 , , , , ,
, , , , ,
Abstract
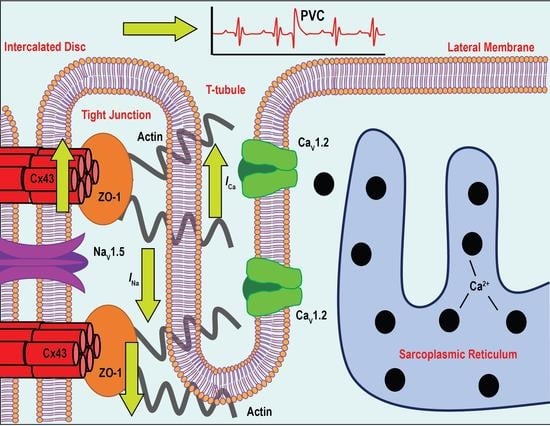
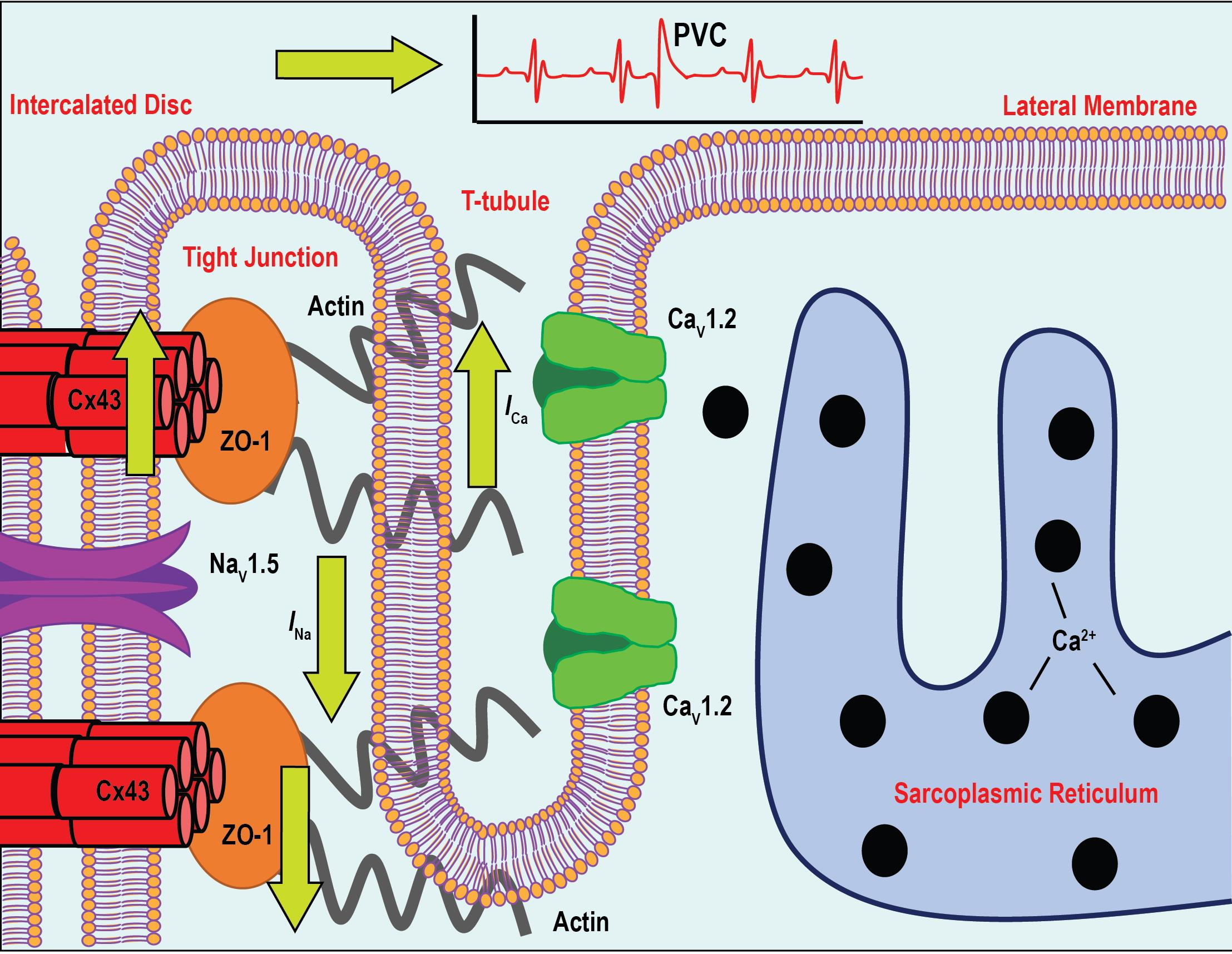
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Procurement of Human Heart Samples
2.2. mRNA Expression Analysis
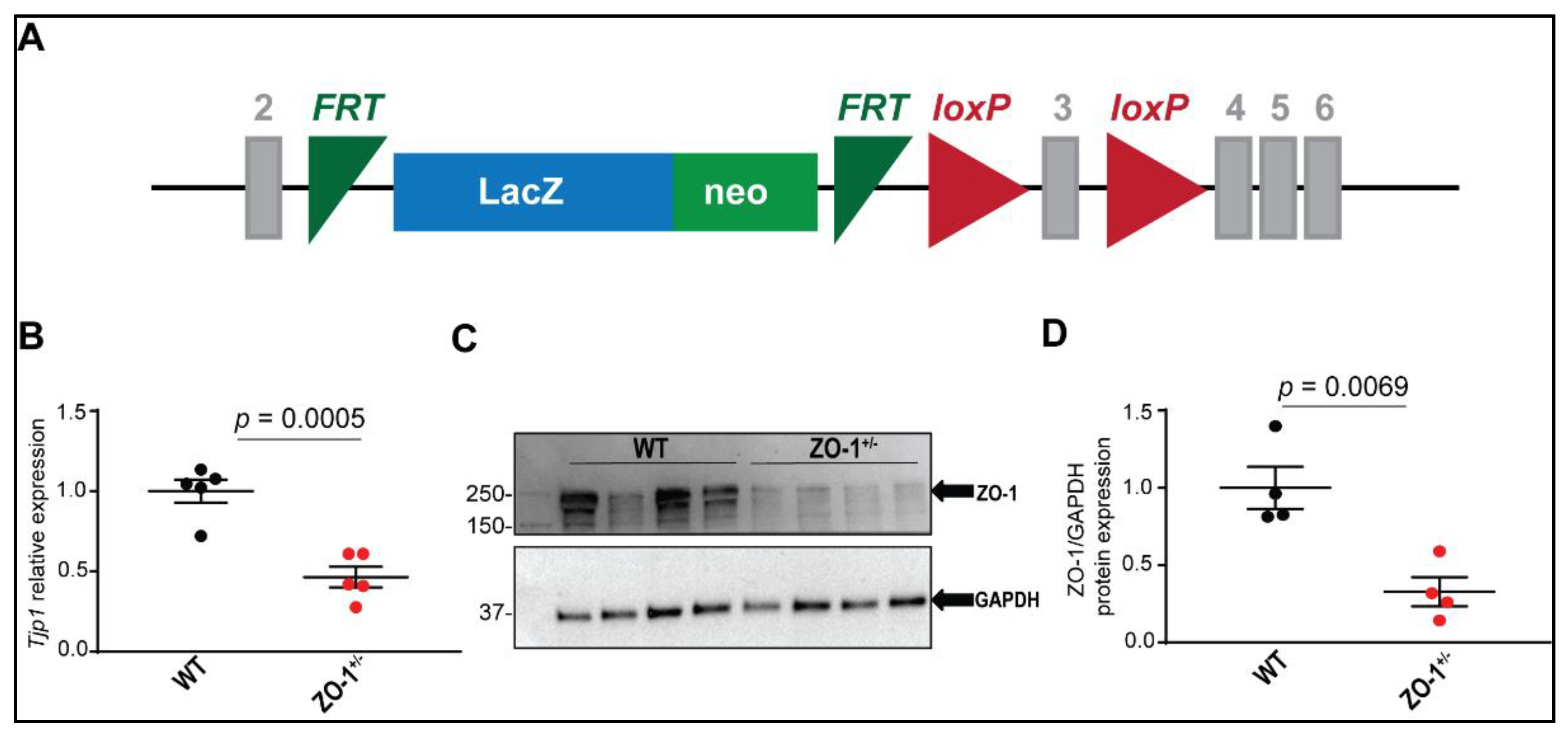
2.3. Mouse Model
2.4. Quantitative RT-PCR
2.5. Tissue Preparation for Immunoblotting
2.6. Immunofluorescence
2.7. Immunoblotting
2.8. Electrocardiograms and Telemetry
2.9. Echocardiograms
2.10. Electrophysiology
2.11. Transmission Electron Microscopy
2.12. Statistics
3. Results
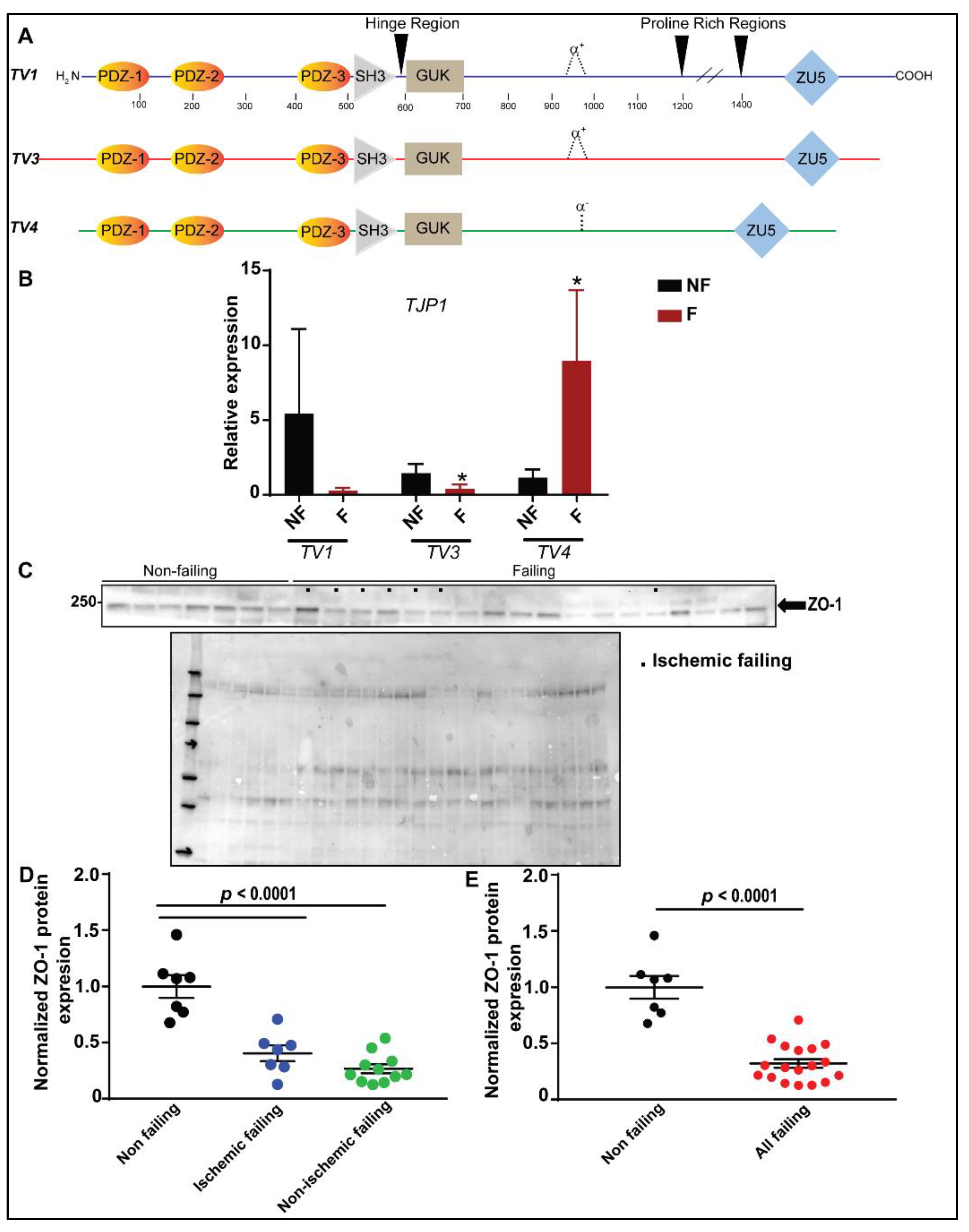
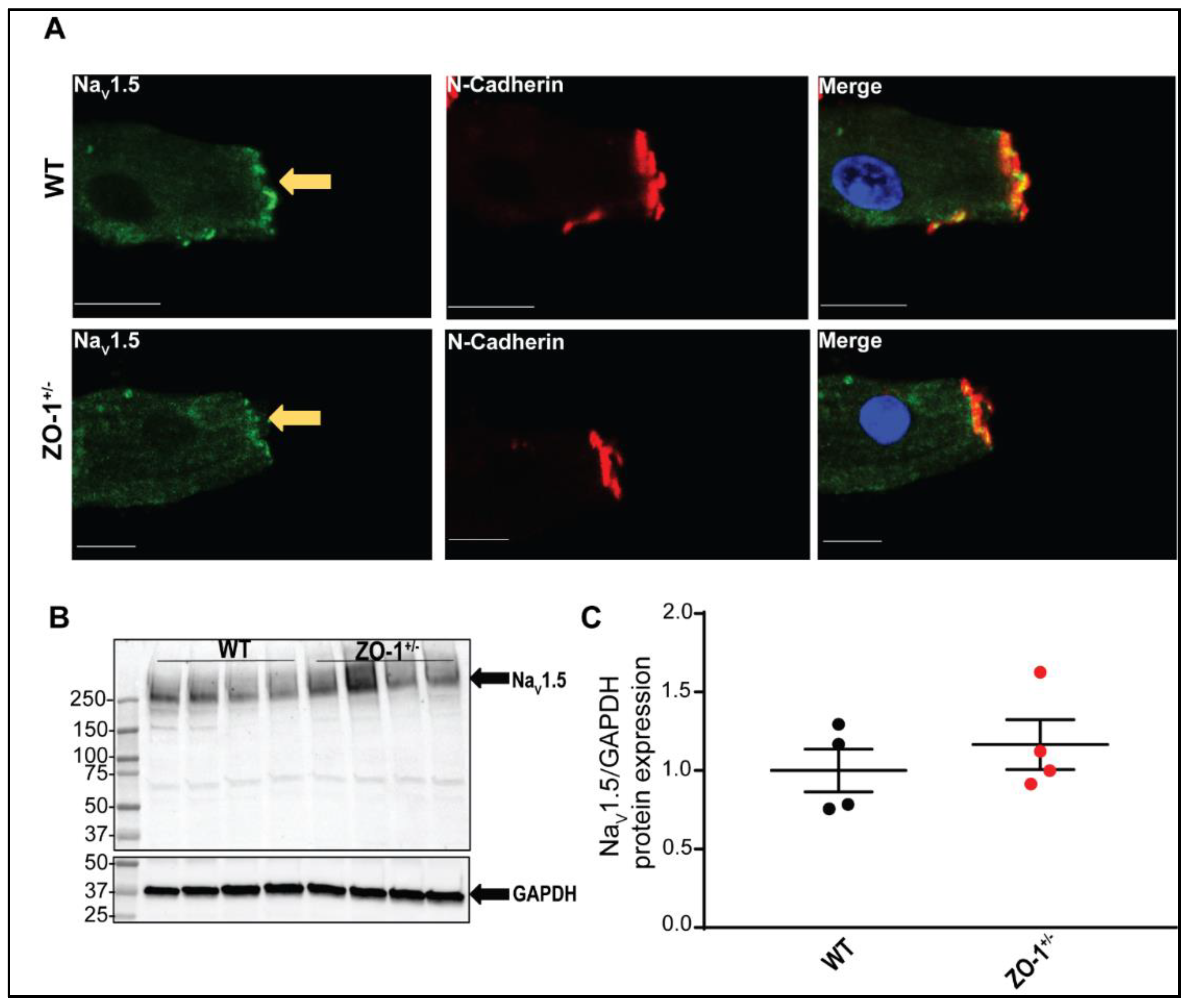
3.1. Identification and Expression-Profile of Different ZO-1 Isoforms in Human Heart
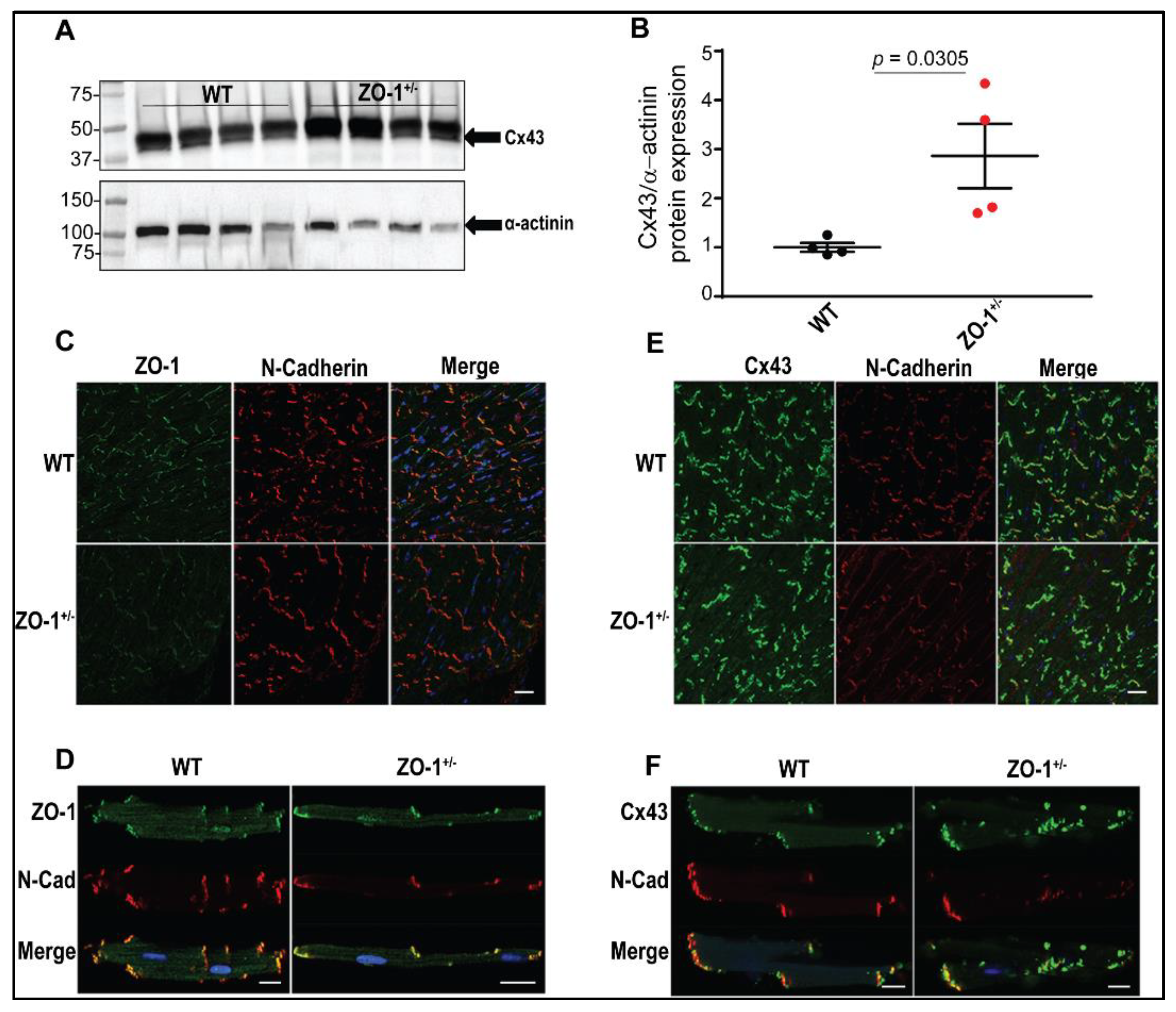
3.2. Reduced ZO-1 Expression Results in Altered Connexin-43 Protein Expression and Localization
3.3. Haploinsufficiency of ZO-1 in Mice Does Not Affect Cardiac Function
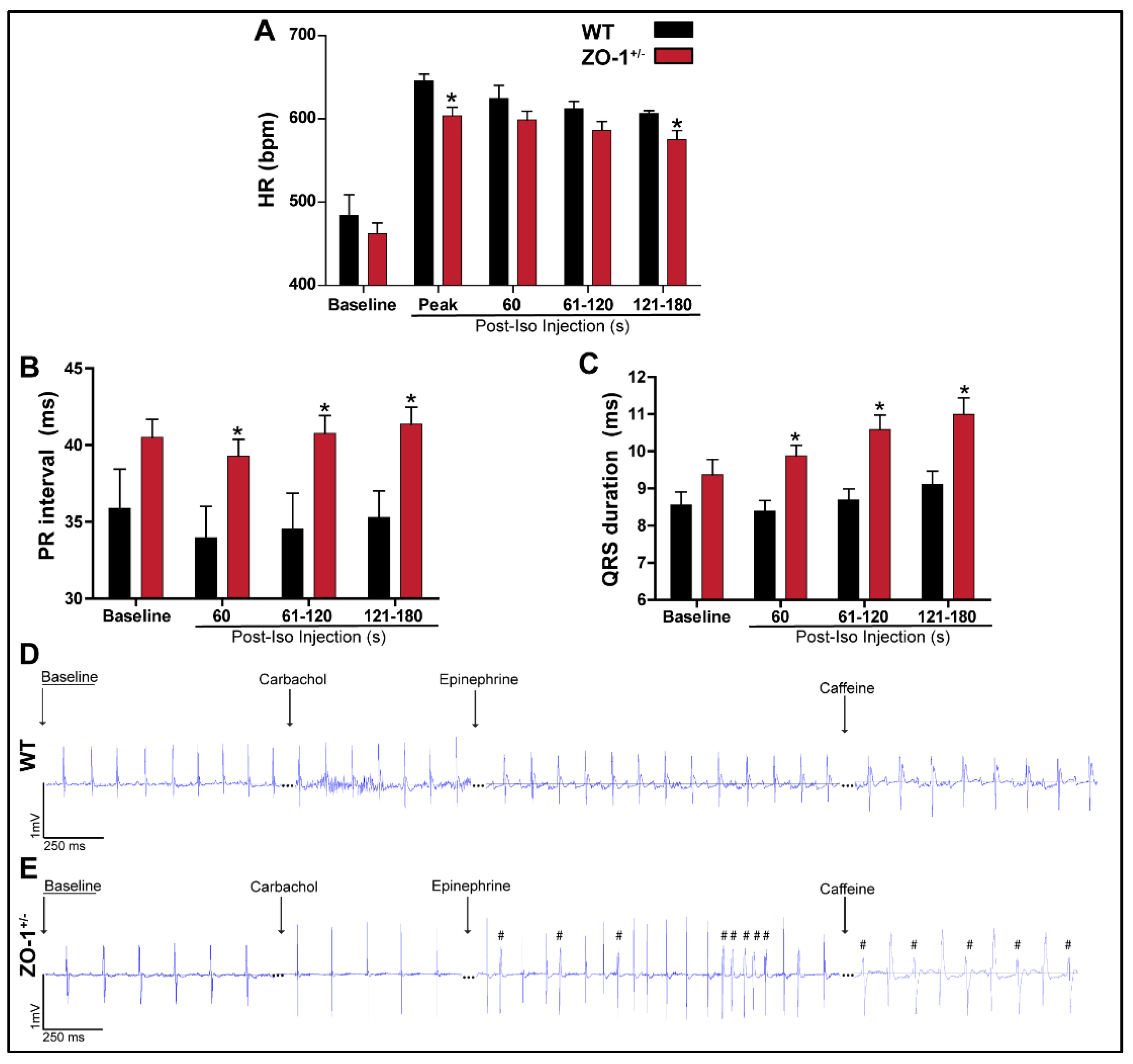
3.4. Adrenergic Stimulation of ZO-1+/− Adult Mice Induces ECG Abnormalities and Arrhythmia
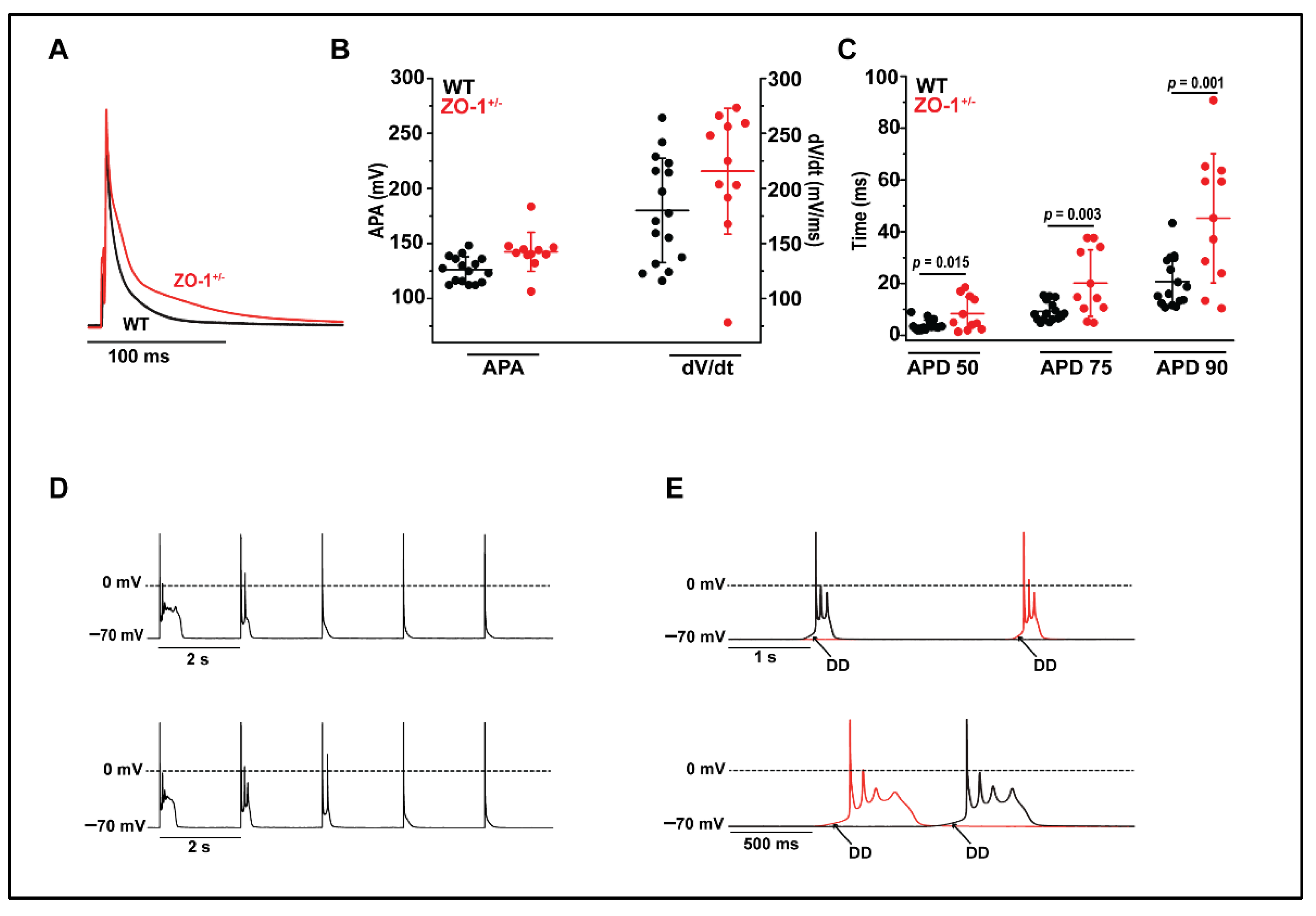
3.5. ZO-1+/− Myocytes Display Prolonged Action Potential Duration and Spontaneous Diastolic Depolarization
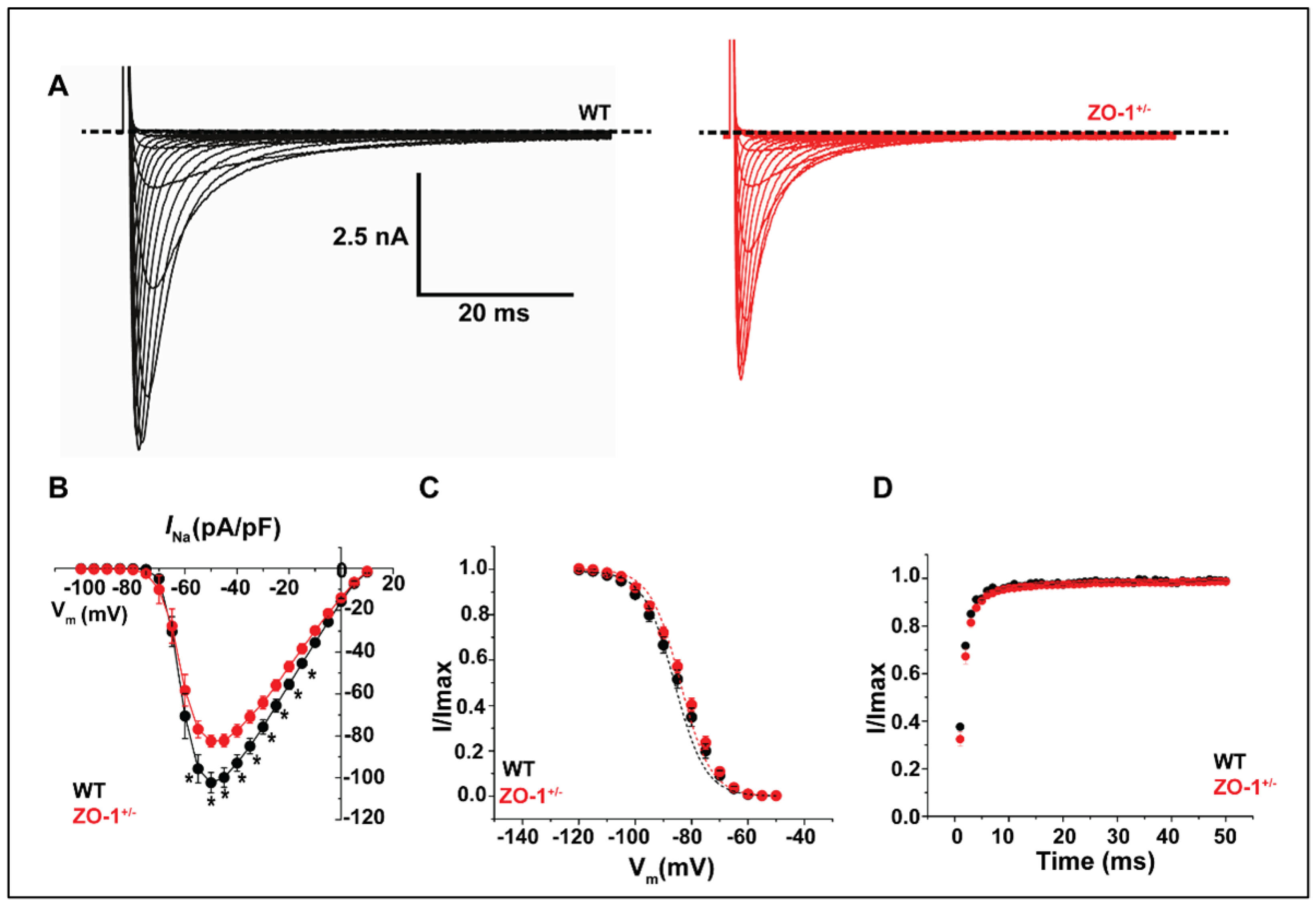
3.6. ZO-1 Haploinsufficiency Attenuates INa Current Density
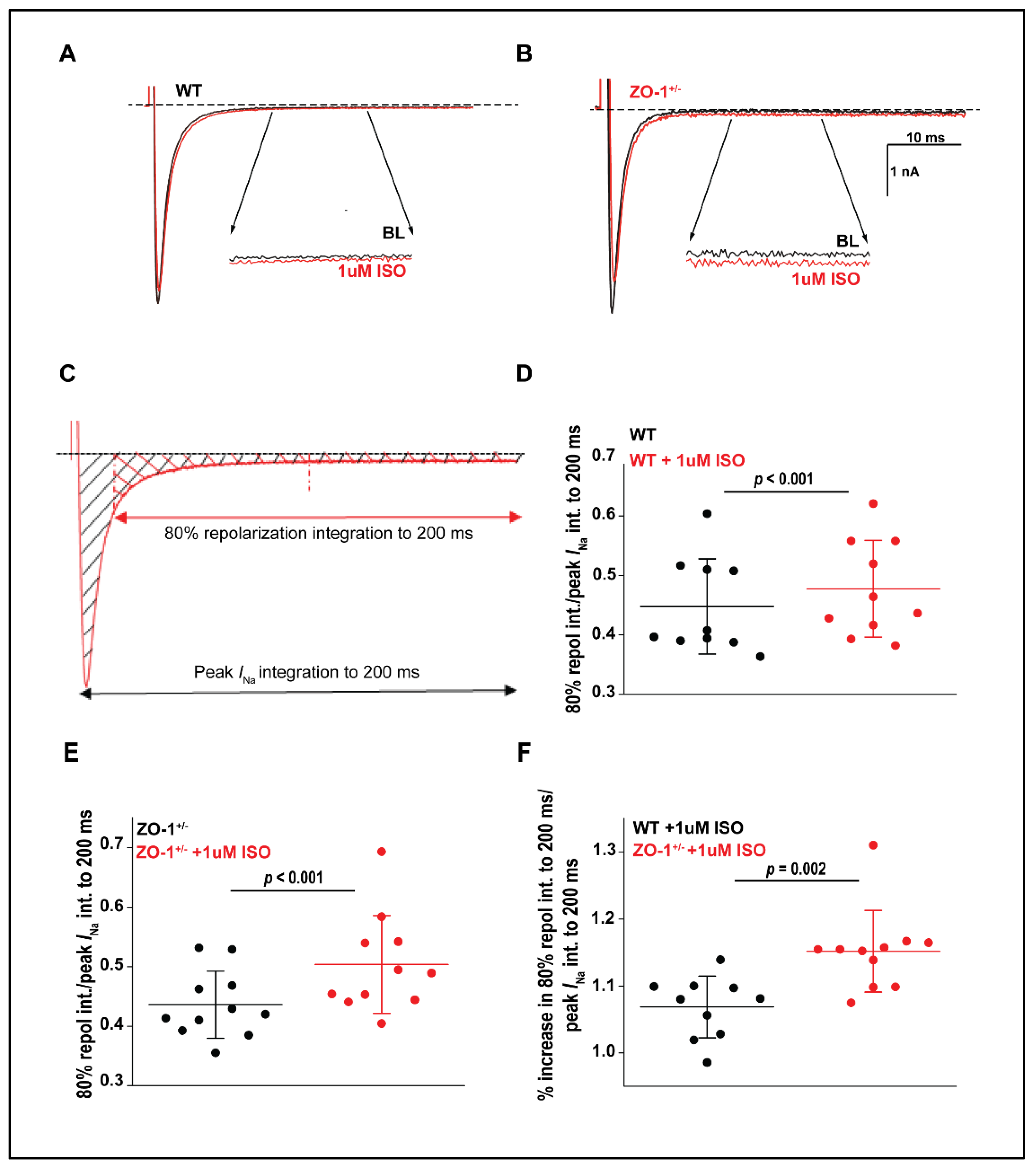
3.7. Late Sodium Current in ZO-1 Deficient Mice Displays an Enhanced Sensitivity to Adrenergic Stimulation
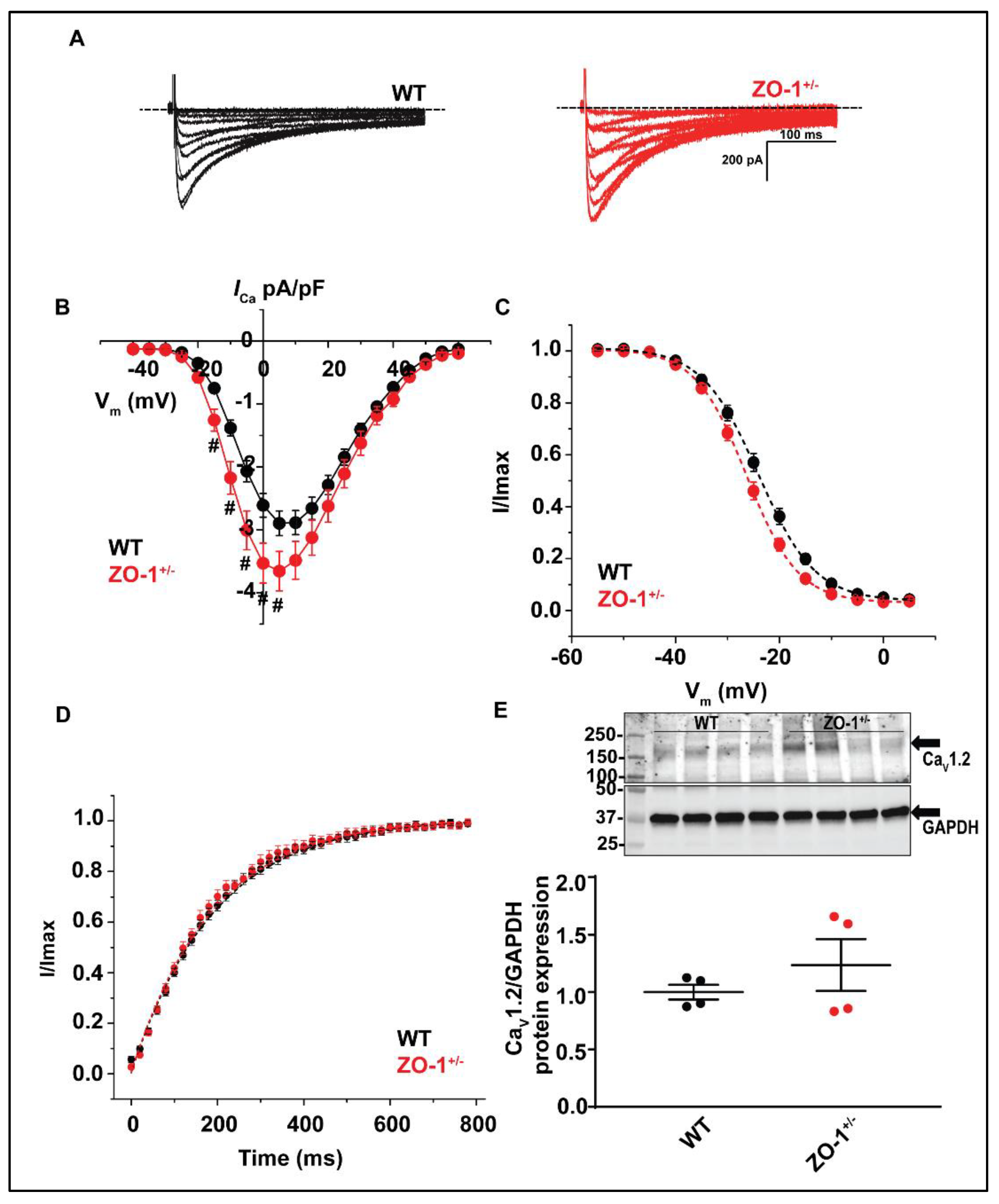
3.8. ZO-1 Haploinsufficiency Displays an Increase in ICa Current Density
4. Discussion
5. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Laing, J.G.; Saffitz, J.E.; Steinberg, T.H.; Yamada, K.A. Diminished zonula occludens-1 expression in the failing human heart. Cardiovasc. Pathol. 2007, 16, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Rhett, J.M.; Ongstad, E.L.; Jourdan, J.; Gourdie, R.G. Cx43 Associates with Na(v)1.5 in the Cardiomyocyte Perinexus. J. Membr. Biol. 2012, 245, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herve, J.C.; Derangeon, M.; Sarrouilhe, D.; Bourmeyster, N. Influence of the scaffolding protein Zonula Occludens (ZOs) on membrane channels. Bba-Biomembr. 2014, 1838, 595–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, C.A.; Lampe, P.D. Injury-triggered Akt phosphorylation of Cx43: A ZO-1-driven molecular switch that regulates gap junction size. J. Cell Sci. 2014, 127, 455–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zemljic-Harpf, A.E.; Godoy, J.C.; Platoshyn, O.; Asfaw, E.K.; Busija, A.R.; Domenighetti, A.A.; Ross, R.S. Vinculin directly binds zonula occludens-1 and is essential for stabilizing connexin-43-containing gap junctions in cardiac myocytes. J. Cell Sci. 2014, 127, 1104–1116. [Google Scholar] [CrossRef] [Green Version]
- Toyofuku, T.; Yabuki, M.; Otsu, K.; Kuzuya, T.; Hori, M.; Tada, M. Direct association of the gap junction protein connexin-43 with ZO-1 in cardiac myocytes. J. Biol. Chem. 1998, 273, 12725–12731. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.Y.; Fong, M.Y.; Min, Y.F.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.F.; Chin, A.R.; et al. Cancer-Secreted miR-105 Destroys Vascular Endothelial Barriers to Promote Metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Y.G.; Peng, H.; Mastej, V.; Chen, W.G. MicroRNA Regulation of Endothelial Junction Proteins and Clinical Consequence. Mediat Inflamm 2016, 2016, 5078627. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Yuan, J.; Nazertehrani, S.; Ni, Z.M.; Liu, S.M. Chronic Kidney Disease Causes Disruption of Gastric and Small Intestinal Epithelial Tight Junction. Am. J. Nephrol. 2013, 38, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Mazzon, E.; Cuzzocrea, S. Role of TNF-alpha in lung tight junction alteration in mouse model of acute lung inflammation. Resp. Res. 2007, 8, 75. [Google Scholar] [CrossRef] [Green Version]
- Kostin, S. Zonula occludens-1 and connexin 43 expression in the failing human heart. J. Cell Mol. Med. 2007, 11, 892–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce, A.F.; Rothery, S.; Dupont, E.; Severs, N.J. Gap junction remodelling in human heart failure is associated with increased interaction of connexin43 with ZO-1. Cardiovasc. Res. 2008, 77, 757–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bortoli, M.; Postma, A.V.; Poloni, G.; Calore, M.; Minervini, G.; Mazzotti, E.; Rigato, I.; Ebert, M.; Lorenzon, A.; Vazza, G.; et al. Whole-Exome Sequencing Identifies Pathogenic Variants in TJP1 Gene Associated With Arrhythmogenic Cardiomyopathy. Circ.-Genom. Precis. Me. 2018, 11, e002123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, W.L.; Nadadur, R.D.; Brennan, J.A.; Smith, H.L.; Shen, K.M.; Gadek, M.; Laforest, B.; Wang, M.Y.; Gemel, J.; Li, Y.; et al. ZO-1 Regulates Intercalated Disc Composition and Atrioventricular Node Conduction. Circ. Res. 2020, 127, E28–E43. [Google Scholar] [CrossRef]
- Zhang, J.; Vincent, K.P.; Peter, A.K.; Klos, M.; Cheng, H.; Huang, S.M.; Towne, J.K.; Ferng, D.; Gu, Y.; Dalton, N.D.; et al. Cardiomyocyte Expression of ZO-1 Is Essential for Normal Atrioventricular Conduction but Does Not Alter Ventricular Function. Circ. Res. 2020, 127, 284–297. [Google Scholar] [CrossRef]
- Katsuno, T.; Umeda, K.; Matsui, T.; Hata, M.; Tamura, A.; Itoh, M.; Takeuchi, K.; Fujimori, T.; Nabeshima, Y.; Noda, T.; et al. Deficiency of zonula occludens-1 causes embryonic lethal phenotype associated with defected yolk sac angiogenesis and apoptosis of embryonic cells. Mol. Biol. Cell 2008, 19, 2465–2475. [Google Scholar] [CrossRef] [Green Version]
- Cavus, O.; Williams, J.; Musa, H.; El Refaey, M.; Gratz, D.; Shaheen, R.; Schwieterman, N.A.; Koenig, S.; Antwi-Boasiako, S.; Young, L.J.; et al. Giant ankyrin-G regulates cardiac function. J. Biol. Chem. 2021, 296, 100507. [Google Scholar] [CrossRef]
- Koval, O.M.; Snyder, J.S.; Wolf, R.M.; Pavlovicz, R.E.; Glynn, P.; Curran, J.; Leymaster, N.D.; Dun, W.; Wright, P.J.; Cardona, N.; et al. Ca2+/Calmodulin-Dependent Protein Kinase II-Based Regulation of Voltage-Gated Na+ Channel in Cardiac Disease. Circulation 2012, 126, 2084–2094. [Google Scholar] [CrossRef] [Green Version]
- Makara, M.A.; Curran, J.; Little, S.C.; Musa, H.; Polina, I.; Smith, S.A.; Wright, P.J.; Unudurthi, S.D.; Snyder, J.; Bennett, V.; et al. Ankyrin-G Coordinates Intercalated Disc Signaling Platform to Regulate Cardiac Excitability In Vivo. Circ. Res. 2014, 115, 929–938. [Google Scholar] [CrossRef] [Green Version]
- Lubbers, E.R.; Murphy, N.P.; Musa, H.; Huang, C.Y.M.; Gupta, R.; Price, M.V.; Han, M.; Daoud, G.E.; Gratz, D.; El Refaey, M.; et al. Defining new mechanistic roles for alpha II spectrin in cardiac function. J. Biol. Chem. 2019, 294, 15557. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, M.A.; Ziman, A.P.; Strong, J.; Zhang, Y.H.; Hartford, A.K.; Ward, C.W.; Randall, W.R.; Kontrogianni-Konstantopoulos, A.; Bloch, R.J. Integrity of the network sarcoplasmic reticulum in skeletal muscle requires small ankyrin 1. J. Cell Sci. 2011, 124, 3619–3630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevenson, B.R.; Siliciano, J.D.; Mooseker, M.S.; Goodenough, D.A. Identification of ZO-1: A high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J. Cell Biol. 1986, 103, 755–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willott, E.; Balda, M.S.; Heintzelman, M.; Jameson, B.; Anderson, J.M. Localization and differential expression of two isoforms of the tight junction protein ZO-1. Am. J. Physiol. 1992, 262, C1119–C1124. [Google Scholar] [CrossRef] [PubMed]
- Balda, M.S.; Anderson, J.M. Two classes of tight junctions are revealed by ZO-1 isoforms. Am. J. Physiol. 1993, 264, C918–C924. [Google Scholar] [CrossRef]
- Rhett, J.M.; Jourdan, J.; Gourdie, R.G. Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol. Biol. Cell 2011, 22, 1516–1528. [Google Scholar] [CrossRef]
- Palatinus, J.A.; O’Quinn, M.P.; Barker, R.J.; Harris, B.S.; Jourdan, J.; Gourdie, R.G. ZO-1 determines adherens and gap junction localization at intercalated disks. Am. J. Physiol-Heart C 2011, 300, H583–H594. [Google Scholar] [CrossRef] [Green Version]
- Abriel, H. Roles and regulation of the cardiac sodium channel Na v 1.5: Recent insights from experimental studies. Cardiovasc Res. 2007, 76, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, M.; Shimizu, W.; Albert, C.M. The spectrum of epidemiology underlying sudden cardiac death. Circ. Res. 2015, 116, 1887–1906. [Google Scholar] [CrossRef]
- Kline, C.F.; Mohler, P.J. Evolving form to fit function: Cardiomyocyte intercalated disc and transverse-tubule membranes. Curr. Top. Membr 2013, 72, 121–158. [Google Scholar] [CrossRef]
- Palatinus, J.A.; Rhett, J.M.; Gourdie, R.G. The connexin43 carboxyl terminus and cardiac gap junction organization. Biochim. Biophys Acta 2012, 1818, 1831–1843. [Google Scholar] [CrossRef] [Green Version]
- Vermij, S.H.; Abriel, H.; van Veen, T.A.B. Refining the molecular organization of the cardiac intercalated disc. Cardiovasc. Res. 2017, 113, 259–275. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lin, J.L.; Wu, K.H.; Wang, D.Z.; Reiter, R.S.; Sinn, H.W.; Lin, C.I.; Lin, C.J. Xin proteins and intercalated disc maturation, signaling and diseases. Front. Biosci. 2012, 17, 2566–2593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhett, J.M.; Veeraraghavan, R.; Poelzing, S.; Gourdie, R.G. The perinexus: Sign-post on the path to a new model of cardiac conduction? Trends Cardiovasc Med. 2013, 23, 222–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balijepalli, R.C.; Lokuta, A.J.; Maertz, N.A.; Buck, J.M.; Haworth, R.A.; Valdivia, H.H.; Kamp, T.J. Depletion of T-tubules and specific subcellular changes in sarcolemmal proteins in tachycardia-induced heart failure. Cardiovasc. Res. 2003, 59, 67–77. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Conklin, M.W.; Foell, J.D.; Wolff, M.R.; Haworth, R.A.; Coronado, R.; Kamp, T.J. Reduction in density of transverse tubules and L-type Ca2+ channels in canine tachycardia-induced heart failure. Cardiovasc. Res. 2001, 49, 298–307. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.B.; de Lange, E.; Garfinkel, A.; Weiss, J.N.; Qu, Z. Delayed afterdepolarizations generate both triggers and a vulnerable substrate promoting reentry in cardiac tissue. Heart Rhythm. 2015, 12, 2115–2124. [Google Scholar] [CrossRef] [Green Version]
- Boukens, B.J.; Christoffels, V.M.; Coronel, R.; Moorman, A.F. Developmental basis for electrophysiological heterogeneity in the ventricular and outflow tract myocardium as a substrate for life-threatening ventricular arrhythmias. Circ. Res. 2009, 104, 19–31. [Google Scholar] [CrossRef]
- Kong, H.; Jones, P.P.; Koop, A.; Zhang, L.; Duff, H.J.; Chen, S.R. Caffeine induces Ca2+ release by reducing the threshold for luminal Ca2+ activation of the ryanodine receptor. Biochem. J. 2008, 414, 441–452. [Google Scholar] [CrossRef] [Green Version]
- Toyofuku, T.; Akamatsu, Y.; Zhang, H.; Kuzuya, T.; Tada, M.; Hori, M. c-Src regulates the interaction between connexin-43 and ZO-1 in cardiac myocytes. J. Biol. Chem. 2001, 276, 1780–1788. [Google Scholar] [CrossRef] [Green Version]
- Rohr, S. Role of gap junctions in the propagation of the cardiac action potential. Cardiovasc. Res. 2004, 62, 309–322. [Google Scholar] [CrossRef]
- Rohr, S.; Kucera, J.P.; Kleber, A.G. Slow conduction in cardiac tissue, I: Effects of a reduction of excitability versus a reduction of electrical coupling on microconduction. Circ. Res. 1998, 83, 781–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wit, A.L.; Hoffman, B.F.; Cranefield, P.F. Slow conduction and reentry in the ventricular conducting system. I. Return extrasystole in canine Purkinje fibers. Circ. Res. 1972, 30, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.L. Murine Electrophysiological Models of Cardiac Arrhythmogenesis. Physiol. Rev. 2017, 97, 283–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, M.R.; Wit, A.L.; Hoffman, B.F. Electrophysiology and pharmacology of cardiac arrhythmias. IV. Cardiac antiarrhythmic and toxic effects of digitalis. Am. Heart J. 1975, 89, 391–399. [Google Scholar] [CrossRef]
- Korb, D.; Tng, P.Y.; Milenkovic, V.M.; Reichhart, N.; Strauss, O.; Ritter, O.; Fischer, T.; Benz, P.M.; Schuh, K. Identification of PDZ Domain Containing Proteins Interacting with Cav1.2 and PMCA4b. Isrn. Cell Biol. 2013, 2013, 265182. [Google Scholar] [CrossRef]
- Maltsev, V.A.; Sabbah, H.N.; Higgins, R.S.; Silverman, N.; Lesch, M.; Undrovinas, A.I. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation 1998, 98, 2545–2552. [Google Scholar] [CrossRef]
- Toischer, K.; Hartmann, N.; Wagner, S.; Fischer, T.H.; Herting, J.; Danner, B.C.; Sag, C.M.; Hund, T.J.; Mohler, P.J.; Belardinelli, L.; et al. Role of late sodium current as a potential arrhythmogenic mechanism in the progression of pressure-induced heart disease. J. Mol. Cell Cardiol. 2013, 61, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Valdivia, C.R.; Chu, W.W.; Pu, J.; Foell, J.D.; Haworth, R.A.; Wolff, M.R.; Kamp, T.J.; Makielski, J.C. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J. Mol. Cell Cardiol. 2005, 38, 475–483. [Google Scholar] [CrossRef]
- Belardinelli, L.; Giles, W.R.; Rajamani, S.; Karagueuzian, H.S.; Shryock, J.C. Cardiac late Na(+) current: Proarrhythmic effects, roles in long QT syndromes, and pathological relationship to CaMKII and oxidative stress. Heart Rhythm 2015, 12, 440–448. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Refaey, M.; Coles, S.; Musa, H.; Stevens, T.L.; Wallace, M.J.; Murphy, N.P.; Antwi-Boasiako, S.; Young, L.J.; Manring, H.R.; Curran, J.; et al. Altered Expression of Zonula occludens-1 Affects Cardiac Na+ Channels and Increases Susceptibility to Ventricular Arrhythmias. Cells 2022, 11, 665. https://doi.org/10.3390/cells11040665
El Refaey M, Coles S, Musa H, Stevens TL, Wallace MJ, Murphy NP, Antwi-Boasiako S, Young LJ, Manring HR, Curran J, et al. Altered Expression of Zonula occludens-1 Affects Cardiac Na+ Channels and Increases Susceptibility to Ventricular Arrhythmias. Cells. 2022; 11(4):665. https://doi.org/10.3390/cells11040665
Chicago/Turabian StyleEl Refaey, Mona, Sara Coles, Hassan Musa, Tyler L. Stevens, Michael J. Wallace, Nathaniel P. Murphy, Steve Antwi-Boasiako, Lindsay J. Young, Heather R. Manring, Jerry Curran, and et al. 2022. "Altered Expression of Zonula occludens-1 Affects Cardiac Na+ Channels and Increases Susceptibility to Ventricular Arrhythmias" Cells 11, no. 4: 665. https://doi.org/10.3390/cells11040665
APA StyleEl Refaey, M., Coles, S., Musa, H., Stevens, T. L., Wallace, M. J., Murphy, N. P., Antwi-Boasiako, S., Young, L. J., Manring, H. R., Curran, J., Makara, M. A., Sas, K., Han, M., Koenig, S. N., Skaf, M., Kline, C. F., Janssen, P. M. L., Accornero, F., Borzok, M. A., & Mohler, P. J. (2022). Altered Expression of Zonula occludens-1 Affects Cardiac Na+ Channels and Increases Susceptibility to Ventricular Arrhythmias. Cells, 11(4), 665. https://doi.org/10.3390/cells11040665