
The Role of MeCP2 in Regulating Synaptic Plasticity in the Context of Stress and Depression



Abstract
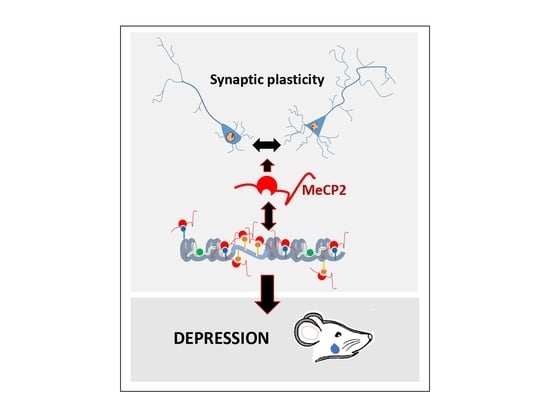
:
1. Chromatin and Epigenetics: A Brief Overview
2. The Unique Epigenetics of the Brain
3. Stress and Synaptic Plasticity
4. MeCP2 and Synaptic Plasticity
5. MeCP2 and Stress-Related Pathology: Focus on Depression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject | Experimental Paradigm | MeCP2 Involvement | References |
|---|---|---|---|
| Mice | MeCP2 knock-in and chronic social defeat stress (CSDS) Imipramine treatment | pMeCP2 is required for the effects of chronic imipramine on depressive-like behaviors induced by chronic social defeat stress. | [123] |
| Early life stress (ELS) by maternal separation (MS) | They suggest that MeCP2 acts in association with the chromatin modifiers HDAC2 and DNMT1, to repress Pomc gene expression in the ELS paradigm. | [112] | |
| Post-stroke depression (PSD) fluoxetine treatment(FLX) | Fluoxetine improved depression-like behaviors of PSD mice and upregulated the expression of BDNF in the hippocampus but depletion of BDNF suppressed the effect of fluoxetine. FLX treatment also disassociated the MeCP2-CREB-Bdnf promoter IV complex via phosphorylation of MeCP2 at Ser421 by PKA. | [96] | |
| Social isolation stress (SI) | Decreased PPAR-α expression in the hippocampus of SI mice was associated with increased MeCP2, which favored hypermethylation and was also associated with increased TLR-4 and pro-inflammatory markers, mediated by NF-κB signaling in the hippocampus of aggressive mice. | [119] | |
| Chronic social defeat stress | Nrf2 induces BDNF transcription via upregulation of Nrf2 and downregulation of MeCP2 in microglia, which is associated with changes in the morphology of damaged dendritic spines in stressed mice. | [117] | |
| Chronic social defeat stress | Activation of Nrf2 by sulforaphane showed fast-acting antidepressant-like effects in mice by activating BDNF and inhibiting MeCP2 but not in Nrf2 knockout mice. In contrast, levels of MeCP2 in the CSDS-susceptible mice were higher than those of control and CSDS-resilient mice. | [126] | |
| Chronic social defeat stress (R)-ketamine treatment | (R)-ketamine fast-acting antidepressant effects are suggested to be mediated by an increase in BDNF transcription induced by the activation of CREB and MeCP2 suppression in microglia in a CSDS model of Depression. | [97] | |
| Rats | Chronic unpredictable stress (CUS) rat model of depression | Knockdown of MeCP2 expression in primary hippocampal neurons increased miR-132 expression and decreased BDNF expression. CUS-induced depressive-like behaviors correlated with an increase in miR-132 and decreased levels of MeCP2 and BDNF in the hippocampus. | [115] |
| Social defeat stress (SD) Exercise treatment | Moderate exercise rescued social defeat induced anxiety-like behavior and memory impairment, and normalized SD-induced increase in oxidative stress, leading to decreased MeCP2 protein levels in the hippocampus. | [116] | |
| Early life stress by maternal separation and adult restraint stress (RS). Escitalopram(ESC) treatment. | Both the MS and RS groups had increased MeCP2 levels at hippocampal BDNF promoter IV with a greater effect by combining MS and RS. This was associated with an increased despair-like behavior measured with the forced swim test. Chronic escitalopram treatment recovered these alterations. | [113] | |
| Early life stress by maternal separation (MS). Escitalopram treatment. | Escitalopram treatment decreased MeCP2 binding to the BDNF promoter exon I in the hippocampus of MS animals; however, MS had no effects on MeCP2 binding levels, compared with controls. MS animals treated with ESC revealed significant increases in BDNF protein and significant decreases in MeCP2 mRNA levels. | [114] | |
| Chronic unpredictable mild stress (CUMS) rat model of depression. Escitalopram treatment. | CUMS increased depressive-like behavior but did not change MeCP2 expression, compared with controls. CUMS reduced BDNF levels in the hippocampus and increased them in the frontal lobe. ESC increased BDNF levels in the hippocampus and increased MeCP2 levels in the hippocampus and the frontal lobe. | [124] | |
| Humans | Major Depression | MeCP2 and BDNF negatively correlated with miR-132 expression levels in the blood of depression patients. | [115] |
| Nondiagnosed psychiatric suicide | Suicide samples have decreased BDNF, increased H3K27me2, Sin3a levels, and decreased p-S421-MeCP2/ MeCP2 protein ratio. They suggest a role in MeCP2 in lowering BDNF protein levels in suicide patients. | [125] |
6. MeCP2 and Reelin
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Kornberg, R.D.; Thomas, J.O. Chromatin structure; oligomers of the histones. Science 1974, 184, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Olins, A.L.; Olins, D.E. Spheroid chromatin units (v bodies). Science 1974, 183, 330–332. [Google Scholar] [CrossRef] [PubMed]
- Oudet, P.; Gross-Bellard, M.; Chambon, P. Electron microscopic and biochemical evidence that chromatin structure is a repeating unit. Cell 1975, 4, 281–300. [Google Scholar] [CrossRef]
- Van Holde, K.E. Chromatin; Springer: New York, NY, USA, 1988. [Google Scholar]
- Kasinsky, H.E.; Lewis, J.D.; Dacks, J.B.; Ausió, J. Origin of H1 linker histones. FASEB J. 2001, 15, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Allan, J.; Hartman, P.G.; Crane-Robinson, C.; Aviles, F.X. The structure of histone H1 and its location in chromatin. Nature 1980, 288, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Vogirala, V.K.; Soman, A.; Berezhnoy, N.V.; Liu, Z.B.; Wong, A.S.W.; Korolev, N.; Su, C.J.; Sandin, S.; Nordenskiold, L. Linker histone defines structure and self-association behaviour of the 177 bp human chromatosome. Sci. Rep. 2021, 11, 380. [Google Scholar] [CrossRef]
- Hao, F.; Kale, S.; Dimitrov, S.; Hayes, J.J. Unraveling linker histone interactions in nucleosomes. Curr. Opin. Struct. Biol. 2021, 71, 87–93. [Google Scholar] [CrossRef]
- Howe, L.; Iskandar, M.; Ausio, J. Folding of chromatin in the presence of heterogeneous histone H1 binding to nucleosomes. J. Biol. Chem. 1998, 273, 11625–11629. [Google Scholar] [CrossRef] [Green Version]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, V.; Finch, J.T.; Graziano, V.; Lee, P.L.; Sweet, R.M. Crystal structure of globular domain of his-tone H5 and its implications for nucleosome binding. Nature 1993, 362, 219–223. [Google Scholar] [CrossRef]
- Arents, G.; Moudrianakis, E.N. The histone fold: A ubiquitous architectural motif utilized in DNA compaction and protein dimerization. Proc. Natl. Acad. Sci. USA 1995, 92, 11170–11174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and Methylation of Histones and Their Possible Role in the Regulation of Rna Synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Wang, R.Y. 5-Methylcytosine in eukaryotic DNA. Science 1981, 212, 1350–1357. [Google Scholar] [CrossRef]
- Adams, R.L.; Burdon, R.H. DNA methylation in eukaryotes. CRC Crit. Rev. Biochem. 1982, 13, 349–384. [Google Scholar] [CrossRef]
- Busslinger, M.; Hurst, J.; Flavell, R.A. DNA methylation and the regulation of globin gene expression. Cell 1983, 34, 197–206. [Google Scholar] [CrossRef]
- Meehan, R.R.; Lewis, J.D.; McKay, S.; Kleiner, E.L.; Bird, A.P. Identification of a mammalian protein that binds specifically to DNA containing methylated CpGs. Cell 1989, 58, 499–507. [Google Scholar] [CrossRef]
- Hendrich, B.; Bird, A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol. 1998, 18, 6538–6547. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.D.; Meehan, R.R.; Henzel, W.J.; Maurer-Fogy, I.; Jeppesen, P.; Klein, F.; Bird, A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 1992, 69, 905–914. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [Green Version]
- Head, J.A. Patterns of DNA methylation in animals: An ecotoxicological perspective. Integr. Comp. Biol. 2014, 54, 77–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tammen, S.A.; Friso, S.; Choi, S.W. Epigenetics: The link between nature and nurture. Mol. Asp. Med. 2013, 34, 753–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marmorstein, R.; Zhou, M.M. Writers and readers of histone acetylation: Structure, mechanism, and inhibition. Cold Spring Harb. Perspect. Biol. 2014, 6, a018762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Ecker, J.R. Epigenetics. Exceptional epigenetics in the brain. Science 2015, 348, 1094–1095. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef] [Green Version]
- Thambirajah, A.A.; Ng, M.K.; Frehlick, L.J.; Li, A.; Serpa, J.J.; Petrotchenko, E.V.; Silva-Moreno, B.; Missiaen, K.K.; Borchers, C.H.; Adam Hall, J.; et al. MeCP2 binds to nucleosome free (linker DNA) regions and to H3K9/H3K27 methylated nucleosomes in the brain. Nucleic Acids Res. 2012, 40, 2884–2897. [Google Scholar] [CrossRef] [Green Version]
- Skene, P.J.; Illingworth, R.S.; Webb, S.; Kerr, A.R.; James, K.D.; Turner, D.J.; Andrews, R.; Bird, A.P. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell 2010, 37, 457–468. [Google Scholar] [CrossRef] [Green Version]
- Gabel, H.W.; Kinde, B.; Stroud, H.; Gilbert, C.S.; Harmin, D.A.; Kastan, N.R.; Hemberg, M.; Ebert, D.H.; Green-berg, M.E. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 2015, 522, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Chen, K.; Lavery, L.A.; Baker, S.A.; Shaw, C.A.; Li, W.; Zoghbi, H.Y. MeCP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome. Proc. Natl. Acad. Sci. USA 2015, 112, 5509–5514. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, A.; Papin, C.; Mohideen-Abdul, K.; le Gras, S.; Stoll, I.; Bronner, C.; Dimitrov, S.; Klaholz, B.P.; Hamiche, A. MeCP2 is a microsatellite binding protein that protects CA repeats from nucleosome invasion. Science 2021, 372. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.P.; Nikitina, T.; Horowitz-Scherer, R.A.; Gierasch, L.M.; Uversky, V.N.; Hite, K.; Hansen, J.C.; Wood-cock, C.L. Unique physical properties and interactions of the domains of methylated DNA binding protein 2. Biochemistry 2010, 49, 4395–4410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ausió, J.; de Paz, A.; Esteller, M. MeCP2: The long trip from a chromatin protein to neurological disorders. Trends Mol. Med. 2014, 20, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Martínez de Paz, A.; Khajavi, L.; Martin, H.; Claveria-Gimeno, R.; Tom Dieck, S.; Cheema, M.S.; Sanchez-Mut, J.V.; Moksa, M.M.; Carles, A.; Brodie, N.I.; et al. MeCP2-E1 isoform is a dynamically expressed, weakly DNA-bound protein with different protein and DNA interactions compared to MeCP2-E2. Epigenet. Chromatin 2019, 12, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ausio, J. Role of MeCP2 in neurological disorders: Current status and future perspectives. Epigenomics 2018, 10, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Farrelly, L.A.; Thompson, R.E.; Zhao, S.; Lepack, A.E.; Lyu, Y.; Bhanu, N.V.; Zhang, B.; Loh, Y.E.; Ramakrishnan, A.; Vadodaria, K.C.; et al. Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Nature 2019, 567, 535–539. [Google Scholar] [CrossRef]
- Maze, I.; Wenderski, W.; Noh, K.-M.; Bagot, R.C.; Tzavaras, N.; Purushothaman, I.; Elsässer, S.J.; Guo, Y.; Io-nete, C.; Hurd, Y.L.; et al. Critical Role of Histone Turnover in Neuronal Transcription and Plasticity. Neuron 2015, 87, 77–94. [Google Scholar] [CrossRef] [Green Version]
- Johnston, M.V.; Jeon, O.H.; Pevsner, J.; Blue, M.E.; Naidu, S. Neurobiology of Rett syndrome: A genetic disorder of synapse development. Brain Dev. 2001, 23 (Suppl. S1), S206–S213. [Google Scholar] [CrossRef]
- Kishi, N.; Macklis, J.D. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol. Cell. Neurosci. 2004, 27, 306–321. [Google Scholar] [CrossRef]
- Armstrong, D.D. Neuropathology of Rett syndrome. J. Child Neurol. 2005, 20, 747–753. [Google Scholar] [CrossRef]
- Duman, R.S.; Aghajanian, G.K. Synaptic dysfunction in depression: Potential therapeutic targets. Science 2012, 338, 68–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duman, R.S.; Aghajanian, G.K.; Sanacora, G.; Krystal, J.H. Synaptic plasticity and depression: New insights from stress and rapid-acting antidepressants. Nat. Med. 2016, 22, 238–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Liu, J.; Wang, M.; Zhang, Y.; Li, L. From Serotonin to Neuroplasticity: Evolvement of Theories for Major Depressive Disorder. Front. Cell. Neurosci. 2017, 11, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, C.; Castren, E.; Kasper, S.; Lanzenberger, R. Serotonin and neuroplasticity—Links between molecular, functional and structural pathophysiology in depression. Neurosci. Biobehav. Rev. 2017, 77, 317–326. [Google Scholar] [CrossRef] [Green Version]
- Petazzi, P.; Sandoval, J.; Szczesna, K.; Jorge, O.C.; Roa, L.; Sayols, S.; Gomez, A.; Huertas, D.; Esteller, M. Dysregulation of the long non-coding RNA transcriptome in a Rett syndrome mouse model. RNA Biol. 2013, 10, 1197–1203. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, S.S.; Pelka, G.J.; Tam, P.P.; El-Osta, A. Chromatin context and ncRNA highlight targets of MeCP2 in brain. RNA Biol. 2013, 10, 1741–1757. [Google Scholar] [CrossRef] [Green Version]
- Good, K.V.; Vincent, J.B.; Ausio, J. MeCP2: The Genetic Driver of Rett Syndrome Epigenetics. Front. Genet. 2021, 12, 620859. [Google Scholar] [CrossRef]
- Lin, R.; Turecki, G. Noncoding RNAs in Depression. Adv. Exp. Med. Biol. 2017, 978, 197–210. [Google Scholar] [CrossRef]
- Huang, X.; Luo, Y.L.; Mao, Y.S.; Ji, J.L. The link between long noncoding RNAs and depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 73, 73–78. [Google Scholar] [CrossRef]
- Obiols-Guardia, A.; Guil, S.; Obiols-Guardia, A.; Guil, S. The Role of Noncoding RNAs in Neurodevelopmental Disorders: The Case of Rett Syndrome. Adv. Exp. Med. Biol. 2017, 978, 23–37. [Google Scholar]
- Mendes-Silva, A.P.; Fujimura, P.T.; da Silva, J.R.C.; Teixeira, A.L.; Vieira, E.M.; Guedes, P.H.G.; Barroso, L.S.S.; de Souza Nicolau, M.; Ferreira, J.D.R.; Bertola, L.; et al. Brain-enriched MicroRNA-184 is downregulated in older adults with major depressive disorder: A translational study. J. Psychiatr. Res. 2019, 111, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Lepack, A.E.; Bagot, R.C.; Pena, C.J.; Loh, Y.E.; Farrelly, L.A.; Lu, Y.; Powell, S.K.; Lorsch, Z.S.; Issler, O.; Cates, H.M.; et al. Aberrant H3.3 dynamics in NAc promote vulnerability to depressive-like behavior. Proc. Natl. Acad. Sci. USA 2016, 113, 12562–12567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Sun, T. Molecular regulation of hypothalamic development and physiological functions. Mol. Neurobiol. 2016, 53, 4275–4285. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.P.; McKlveen, J.M.; Ghosal, S.; Kopp, B.; Wulsin, A.; Makinson, R.; Scheimann, J.; Myers, B. Regulation of the Hypothalamic-Pituitary-Adrenocortical Stress Response. Compr. Physiol. 2016, 6, 603–621. [Google Scholar] [CrossRef] [Green Version]
- Makino, S.; Shibasaki, T.; Yamauchi, N.; Nishioka, T.; Mimoto, T.; Wakabayashi, I.; Gold, P.W.; Hashimoto, K. Psychological stress increased corticotropin-releasing hormone mRNA and content in the central nucleus of the amygdala but not in the hypothalamic paraventricular nucleus in the rat. Brain Res. 1999, 850, 136–143. [Google Scholar] [CrossRef]
- Rivier, C.; Vale, W. Modulation of stress-induced ACTH release by corticotropin-releasing factor, catecholamines and vasopressin. Nature 1983, 305, 325–327. [Google Scholar] [CrossRef]
- Cain, D.W.; Cidlowski, J.A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247. [Google Scholar] [CrossRef]
- Nicolaides, N.C.; Kyratzi, E.; Lamprokostopoulou, A.; Chrousos, G.P.; Charmandari, E. Stress, the stress system and the role of glucocorticoids. Neuroimmunomodulation 2015, 22, 6–19. [Google Scholar] [CrossRef]
- Goel, N.; Workman, J.L.; Lee, T.T.; Innala, L.; Viau, V. Sex differences in the HPA axis. Compr. Physiol. 2014, 4, 1121–1155. [Google Scholar] [CrossRef]
- Goel, N.; Innala, L.; Viau, V. Sex differences in serotonin (5-HT) 1A receptor regulation of HPA axis and dorsal raphe responses to acute restraint. Psychoneuroendocrinology 2014, 40, 232–241. [Google Scholar] [CrossRef]
- Heck, A.L.; Handa, R.J. Sex differences in the hypothalamic-pituitary-adrenal axis’ response to stress: An important role for gonadal hormones. Neuropsychopharmacology 2019, 44, 45–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, S.M.; Young, E.A.; Kerber, K.B.; Kornstein, S.; Farabaugh, A.H.; Mitchell, J.; Wisniewski, S.R.; Balasubramani, G.K.; Trivedi, M.H.; Rush, A.J. Gender differences in depression: Findings from the STAR*D study. J. Affect. Disord. 2005, 87, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Myers, B.; Mark Dolgas, C.; Kasckow, J.; Cullinan, W.E.; Herman, J.P. Central stress-integrative circuits: Fore-brain glutamatergic and GABAergic projections to the dorsomedial hypothalamus, medial preoptic area, and bed nucleus of the stria terminalis. Brain Struct. Funct. 2014, 219, 1287–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, J.P.; Flak, J.; Jankord, R. Chronic stress plasticity in the hypothalamic paraventricular nucleus. Prog. Brain Res. 2008, 170, 353–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, E.W.M.; Goh, E.L.K. MeCP2 Dysfunction in Rett Syndrome and Neuropsychiatric Disorders. Methods Mol. Biol. 2019, 2011, 573–591. [Google Scholar] [CrossRef]
- Bauman, M.L.; Kemper, T.L.; Arin, D.M. Pervasive neuroanatomic abnormalities of the brain in three cases of Rett’s syndrome. Neurology 1995, 45, 1581–1586. [Google Scholar] [CrossRef]
- Armstrong, D.; Dunn, J.K.; Antalffy, B.; Trivedi, R. Selective dendritic alterations in the cortex of Rett syndrome. J. Neuropathol. Exp. Neurol. 1995, 54, 195–201. [Google Scholar] [CrossRef]
- Belichenko, P.V.; Dahlstrom, A. Confocal laser scanning microscopy and 3-D reconstructions of neuronal structures in human brain cortex. Neuroimage 1995, 2, 201–207. [Google Scholar] [CrossRef]
- Jung, B.P.; Jugloff, D.G.; Zhang, G.; Logan, R.; Brown, S.; Eubanks, J.H. The expression of methyl CpG binding factor MeCP2 correlates with cellular differentiation in the developing rat brain and in cultured cells. J. Neurobiol. 2003, 55, 86–96. [Google Scholar] [CrossRef]
- Kaufmann, W.E.; Johnston, M.V.; Blue, M.E. MeCP2 expression and function during brain development: Implications for Rett syndrome’s pathogenesis and clinical evolution. Brain Dev. 2005, 27 (Suppl. S1), S77–S87. [Google Scholar] [CrossRef]
- Chen, R.Z.; Akbarian, S.; Tudor, M.; Jaenisch, R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 2001, 27, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Asaka, Y.; Jugloff, D.G.; Zhang, L.; Eubanks, J.H.; Fitzsimonds, R.M. Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol. Dis. 2006, 21, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Calfa, G.; Li, W.; Rutherford, J.M.; Pozzo-Miller, L. Excitation/inhibition imbalance and impaired synaptic inhibition in hippocampal area CA3 of Mecp2 knockout mice. Hippocampus 2015, 25, 159–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Peterson, M.; Beyer, B.; Frankel, W.N.; Zhang, Z.W. Loss of MeCP2 from forebrain excitatory neurons leads to cortical hyperexcitation and seizures. J. Neurosci. 2014, 34, 2754–2763. [Google Scholar] [CrossRef] [Green Version]
- Morello, N.; Schina, R.; Pilotto, F.; Phillips, M.; Melani, R.; Plicato, O.; Pizzorusso, T.; Pozzo-Miller, L.; Giustetto, M. Loss of Mecp2 Causes Atypical Synaptic and Molecular Plasticity of Parvalbumin-Expressing Interneurons Reflecting Rett Syndrome-Like Sensorimotor Defects. eNeuro 2018, 5. [Google Scholar] [CrossRef] [Green Version]
- Kishi, N.; Macklis, J.D. MeCP2 functions largely cell-autonomously, but also non-cell-autonomously, in neuronal maturation and dendritic arborization of cortical pyramidal neurons. Exp. Neurol. 2010, 222, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Nelson, E.D.; Kavalali, E.T.; Monteggia, L.M. MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr. Biol. 2006, 16, 710–716. [Google Scholar] [CrossRef] [Green Version]
- Moretti, P.; Levenson, J.M.; Battaglia, F.; Atkinson, R.; Teague, R.; Antalffy, B.; Armstrong, D.; Arancio, O.; Sweatt, J.D.; Zoghbi, H.Y. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J. Neurosci. 2006, 26, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Ip, J.P.K.; Mellios, N.; Sur, M. Rett syndrome: Insights into genetic, molecular and circuit mechanisms. Nat. Rev. Neurosci. 2018, 19, 368–382. [Google Scholar] [CrossRef]
- Van Esch, H. MECP2 Duplication Syndrome. Mol. Syndromol. 2012, 2, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Ramocki, M.B.; Tavyev, Y.J.; Peters, S.U. The MECP2 duplication syndrome. Am. J. Med. Genet. A 2010, 152A, 1079–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, A.A.; White, R.; Siniard, A.; Corneveaux, J.; Huentelman, M.; Duch, C. MECP2 impairs neuronal structure by regulating KIBRA. Neurobiol. Dis. 2016, 91, 284–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, A.L.; Levenson, J.M.; Vilaythong, A.P.; Richman, R.; Armstrong, D.L.; Noebels, J.L.; David Sweatt, J.; Zoghbi, H.Y. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum. Mol. Genet. 2004, 13, 2679–2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Na, E.S.; Nelson, E.D.; Adachi, M.; Autry, A.E.; Mahgoub, M.A.; Kavalali, E.T.; Monteggia, L.M. A mouse model for MeCP2 duplication syndrome: MeCP2 overexpression impairs learning and memory and synaptic transmission. J. Neurosci. 2012, 32, 3109–3117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ash, R.T.; Buffington, S.A.; Park, J.; Suter, B.; Costa-Mattioli, M.; Zoghbi, H.Y.; Smirnakis, S.M. Inhibition of Elevated Ras-MAPK Signaling Normalizes Enhanced Motor Learning and Excessive Clustered Dendritic Spine Stabilization in the MECP2-Duplication Syndrome Mouse Model of Autism. eNeuro 2021, 8, 56–77. [Google Scholar] [CrossRef]
- Ash, R.T.; Park, J.; Suter, B.; Smirnakis, S.M.; Zoghbi, H.Y. Excessive Formation and Stabilization of Dendritic Spine Clusters in the MECP2-Duplication Syndrome Mouse Model of Autism. eNeuro 2021, 8, 1–13. [Google Scholar] [CrossRef]
- Frank, A.C.; Huang, S.; Zhou, M.; Gdalyahu, A.; Kastellakis, G.; Silva, T.K.; Lu, E.; Wen, X.; Poirazi, P.; Trachtenberg, J.T.; et al. Hotspots of dendritic spine turnover facilitate clustered spine addition and learning and memory. Nat. Commun. 2018, 9, 422. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.R.; Fernandes, T.G.; Cabral, J.M.S.; Diogo, M.M. Modeling Rett Syndrome with Human Pluripotent Stem Cells: Mechanistic Outcomes and Future Clinical Perspectives. Int. J. Mol. Sci. 2021, 22, 3751. [Google Scholar] [CrossRef]
- Zhou, Z.; Hong, E.J.; Cohen, S.; Zhao, W.N.; Ho, H.Y.; Schmidt, L.; Chen, W.G.; Lin, Y.; Savner, E.; Griffith, E.C.; et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 2006, 52, 255–269. [Google Scholar] [CrossRef] [Green Version]
- Chapleau, C.A.; Calfa, G.D.; Lane, M.C.; Albertson, A.J.; Larimore, J.L.; Kudo, S.; Armstrong, D.L.; Percy, A.K.; Pozzo-Miller, L. Dendritic spine pathologies in hippocampal pyramidal neurons from Rett syndrome brain and after expression of Rett-associated MECP2 mutations. Neurobiol. Dis. 2009, 35, 219–233. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Kim, J.; Zhou, L.; Wengert, E.; Zhang, L.; Wu, Z.; Carromeu, C.; Muotri, A.R.; Marchetto, M.C.; Gage, F.H.; et al. KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, 751–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchthal, B.; Lau, D.; Weiss, U.; Weislogel, J.M.; Bading, H. Nuclear calcium signaling controls methyl-CpG-binding protein 2 (MeCP2) phosphorylation on serine 421 following synaptic activity. J. Biol. Chem. 2012, 287, 30967–30974. [Google Scholar] [CrossRef] [Green Version]
- Murgatroyd, C.; Patchev, A.V.; Wu, Y.; Micale, V.; Bockmuhl, Y.; Fischer, D.; Holsboer, F.; Wotjak, C.T.; Al-meida, O.F.; Spengler, D. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat. Neurosci. 2009, 12, 1559–1566. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.V.; Rodriguiz, R.M.; Hutchinson, A.N.; Kim, I.H.; Wetsel, W.C.; West, A.E. MeCP2 in the nucleus accumbens contributes to neural and behavioral responses to psychostimulants. Nat. Neurosci. 2010, 13, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.J.; Pei, L.; Li, Y.N.; Zheng, H.; Yang, S.; Wan, Y.; Mao, L.; Xia, Y.P.; He, Q.W.; Li, M.; et al. Alleviative effects of fluoxetine on depressive-like behaviors by epigenetic regulation of BDNF gene transcription in mouse model of post-stroke depression. Sci. Rep. 2017, 7, 14926. [Google Scholar] [CrossRef]
- Yao, W.; Cao, Q.; Luo, S.; He, L.; Yang, C.; Chen, J.; Qi, Q.; Hashimoto, K.; Zhang, J.C. Microglial ERK-NRBP1-CREB-BDNF signaling in sustained antidepressant actions of (R)-ketamine. Mol. Psychiatry 2021, 1476–5578. [Google Scholar] [CrossRef]
- Marchetto, M.C.N.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef] [Green Version]
- Chao, H.T.; Zoghbi, H.Y.; Rosenmund, C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron 2007, 56, 58–65. [Google Scholar] [CrossRef] [Green Version]
- DiFilippo, A.; Ferreira, C.; Engle, J.; Barnhart, T.; Dong, Q.; Chang, Q.; Christian, B. PET imaging of pre-synaptic protein SV2A in Rett syndrome mouse model. J. Nucl. Med. 2019, 60 (Suppl. S1), 117. [Google Scholar]
- Jin, X.; Cui, N.; Zhong, W.; Jin, X.T.; Jiang, C. GABAergic synaptic inputs of locus coeruleus neurons in wild-type and Mecp2-null mice. Am. J. Physiol. Cell Physiol. 2013, 304, C844–C857. [Google Scholar] [CrossRef] [Green Version]
- Bertoldi, M.L.; Zalosnik, M.I.; Fabio, M.C.; Aja, S.; Roth, G.A.; Ronnett, G.V.; Degano, A.L. MeCP2 Deficiency Disrupts Kainate-Induced Presynaptic Plasticity in the Mossy Fiber Projections in the Hippocampus. Front. Cell. Neurosci. 2019, 13, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackman, M.P.; Djukic, B.; Nelson, S.B.; Turrigiano, G.G. A critical and cell-autonomous role for MeCP2 in synaptic scaling up. J. Neurosci. 2012, 32, 13529–13536. [Google Scholar] [CrossRef] [PubMed]
- Lyst, M.J.; Ekiert, R.; Ebert, D.H.; Merusi, C.; Nowak, J.; Selfridge, J.; Guy, J.; Kastan, N.R.; Robinson, N.D.; de Lima Alves, F.; et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat. Neurosci. 2013, 16, 898–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [Green Version]
- Degano, A.L.; Park, M.J.; Penati, J.; Li, Q.; Ronnett, G.V. MeCP2 is required for activity-dependent refinement of olfactory circuits. Mol. Cell. Neurosci. 2014, 59, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.G.; Chang, Q.; Lin, Y.; Meissner, A.; West, A.E.; Griffith, E.C.; Jaenisch, R.; Greenberg, M.E. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 2003, 302, 885–889. [Google Scholar] [CrossRef]
- Ballas, N.; Grunseich, C.; Lu, D.D.; Speh, J.C.; Mandel, G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 2005, 121, 645–657. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Nie, Z.; Shu, H.; Kuang, Y.; Chen, X.; Cheng, J.; Yu, S.; Liu, H. The Role of BDNF on Neural Plasticity in Depression. Front. Cell Neurosci. 2020, 14, 82. [Google Scholar] [CrossRef]
- Menard, C.; Hodes, G.E.; Russo, S.J. Pathogenesis of depression: Insights from human and rodent studies. Neuroscience 2016, 321, 138–162. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, C.A.; Hoffmann, A.; Raabe, F.; Spengler, D. Role of mecp2 in experience-dependent epigenetic programming. Genes 2015, 6, 60–86. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Patchev, A.V.; Daniel, G.; Almeida, O.F.; Spengler, D. Early-life stress reduces DNA methylation of the Pomc gene in male mice. Endocrinology 2014, 155, 1751–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, M.K.; Ly, N.N.; Lee, C.H.; Cho, H.Y.; Choi, C.M.; Nhu, L.H.; Lee, J.G.; Lee, B.J.; Kim, G.M.; Yoon, B.J.; et al. Early life stress increases stress vulnerability through BDNF gene epigenetic changes in the rat hippocampus. Neuropharmacology 2016, 105, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Seo, M.K.; Lee, J.G.; Hien, L.T.; Kim, Y.H. Effects of maternal separation and antidepressant drug on epigenetic regulation of the brain-derived neurotrophic factor exon I promoter in the adult rat hippocampus. Psychiatry Clin. Neurosci. 2018, 72, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Hong, J.; Zhao, Y.; Liu, S.; Xue, X. MeCP2 controls hippocampal brain-derived neurotrophic factor expression via homeostatic interactions with microRNA132 in rats with depression. Mol. Med. Rep. 2015, 12, 5399–5406. [Google Scholar] [CrossRef] [Green Version]
- Patki, G.; Solanki, N.; Atrooz, F.; Ansari, A.; Allam, F.; Jannise, B.; Maturi, J.; Salim, S. Novel mechanistic insights into treadmill exercise based rescue of social defeat-induced anxiety-like behavior and memory impairment in rats. Physiol. Behav. 2014, 130, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Yao, W.; Lin, S.; Su, J.; Cao, Q.; Chen, Y.; Chen, J.; Zhang, Z.; Hashimoto, K.; Qi, Q.; Zhang, J.C. Activation of BDNF by transcription factor Nrf2 contributes to antidepressant-like actions in rodents. Transl. Psychiatry 2021, 11, 140. [Google Scholar] [CrossRef]
- Kim, J.W.; Autry, A.E.; Na, E.S.; Adachi, M.; Bjorkholm, C.; Kavalali, E.T.; Monteggia, L.M. Sustained effects of rapidly acting antidepressants require BDNF-dependent MeCP2 phosphorylation. Nat. Neurosci. 2021, 24, 1100–1109. [Google Scholar] [CrossRef]
- Matrisciano, F.; Pinna, G. PPAR-α Hypermethylation in the Hippocampus of Mice Exposed to Social Isolation Stress Is Associated with Enhanced Neuroinflammation and Aggressive Behavior. Int. J. Mol. Sci. 2021, 22, 10678. [Google Scholar] [CrossRef]
- Filosa, S.; Pecorelli, A.; D’Espositoc, M.; Valacchi, G.; Hajek, J. Exploring the possible link between MeCP2 and oxidative stress in Rett syndrome. Free Radic. Biol. Med. 2015, 88, 81–90. [Google Scholar] [CrossRef]
- Signorini, C.; de Felice, C.; Leoncini, S.; Møller, R.S.; Zollo, G.; Buoni, S.; Cortelazzo, A.; Guerranti, R.; Durand, T.; Ciccoli, L.; et al. MECP2 Duplication Syndrome: Evidence of Enhanced Oxidative Stress. A Comparison with Rett Syndrome. PLoS ONE 2016, 11, e0150101. [Google Scholar] [CrossRef] [Green Version]
- Allen, J.; Romay-Tallon, R.; Brymer, K.J.; Caruncho, H.J.; Kalynchuk, L.E. Mitochondria and Mood: Mitochondrial Dysfunction as a Key Player in the Manifestation of Depression. Front. Neurosci. 2018, 12, 386. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, A.N.; Deng, J.V.; Cohen, S.; West, A.E. Phosphorylation of MeCP2 at Ser421 contributes to chronic antidepressant action. J. Neurosci. 2012, 32, 14355–14363. [Google Scholar] [CrossRef] [PubMed]
- Dionisie, V.; Ciobanu, A.M.; Toma, V.A.; Manea, M.C.; Baldea, I.; Olteanu, D.; Sevastre-Berghian, A.; Clichici, S.; Manea, M.; Riga, S.; et al. Escitalopram Targets Oxidative Stress, Caspase-3, BDNF and MeCP2 in the Hippo-campus and Frontal Cortex of a Rat Model of Depression Induced by Chronic Unpredictable Mild Stress. Int. J. Mol. Sci. 2021, 22, 7483. [Google Scholar] [CrossRef] [PubMed]
- Misztak, P.; Panczyszyn-Trzewik, P.; Nowak, G.; Sowa-Kucma, M. Epigenetic marks and their relationship with BDNF in the brain of suicide victims. PLoS ONE 2020, 15, e0239335. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Cao, Q.Q.; Hu, S.W.; He, L.J.; Du, P.F.; Chen, G.; Fu, R.; Xiao, F.; Sun, Y.R.; Zhang, J.C.; et al. Sulforaphane activates anti-inflammatory microglia, modulating stress resilience associated with BDNF transcription. Acta Pharmacol. Sin. 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Weeber, E.J.; Beffert, U.; Jones, C.; Christian, J.M.; Forster, E.; Sweatt, J.D.; Herz, J. Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J. Biol. Chem. 2002, 277, 39944–39952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beffert, U.; Weeber, E.J.; Durudas, A.; Qiu, S.; Masiulis, I.; Sweatt, J.D.; Li, W.P.; Adelmann, G.; Frotscher, M.; Hammer, R.E.; et al. Modulation of synaptic plasticity and memory by Reelin involves differential splicing of the lipoprotein receptor Apoer2. Neuron 2005, 47, 567–579. [Google Scholar] [CrossRef] [Green Version]
- Qiu, S.; Zhao, L.F.; Korwek, K.M.; Weeber, E.J. Differential reelin-induced enhancement of NMDA and AMPA receptor activity in the adult hippocampus. J. Neurosci. 2006, 26, 12943–12955. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Earle, J.A.; McMenomy, T. Reduction in Reelin immunoreactivity in hippocampus of subjects with schizophrenia, bipolar disorder and major depression. Mol. Psychiatry 2000, 5, 654–663. [Google Scholar] [CrossRef] [Green Version]
- Brymer, K.J.; Fenton, E.Y.; Kalynchuk, L.E.; Caruncho, H.J. Peripheral Etanercept Administration Normalizes Behavior, Hippocampal Neurogenesis, and Hippocampal Reelin and GABAA Receptor Expression in a Preclinical Model of Depression. Front. Pharmacol. 2018, 9, 121. [Google Scholar] [CrossRef]
- Fenton, E.Y.; Fournier, N.M.; Lussier, A.L.; Romay-Tallon, R.; Caruncho, H.J.; Kalynchuk, L.E. Imipramine protects against the deleterious effects of chronic corticosterone on depression-like behavior, hippocampal reelin expression, and neuronal maturation. Prog. Neuropsychopharmacol. Biol. Psychiatry 2015, 60, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Brymer, K.J.; Johnston, J.; Botterill, J.J.; Romay-Tallon, R.; Mitchell, M.A.; Allen, J.; Pinna, G.; Caruncho, H.J.; Kalynchuk, L.E. Fast-acting antidepressant-like effects of Reelin evaluated in the repeated-corticosterone chronic stress paradigm. Neuropsychopharmacology 2020, 45, 1707–1716. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sharma, R.P.; Costa, R.H.; Costa, E.; Grayson, D.R. On the epigenetic regulation of the human reelin promoter. Nucleic Acids Res. 2002, 30, 2930–2939. [Google Scholar] [CrossRef] [PubMed]
- Grayson, D.R.; Chen, Y.; Costa, E.; Dong, E.; Guidotti, A.; Kundakovic, M.; Sharma, R.P. The human reelin gene: Transcription factors (+), repressors (−) and the methylation switch (+/−) in schizophrenia. Pharmacol. Ther. 2006, 111, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Grayson, D.R.; Jia, X.; Chen, Y.; Sharma, R.P.; Mitchell, C.P.; Guidotti, A.; Costa, E. Reelin promoter hypermethylation in schizophrenia. Proc. Natl. Acad. Sci. USA 2005, 102, 9341–9346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kundakovic, M.; Chen, Y.; Costa, E.; Grayson, D.R. DNA methyltransferase inhibitors coordinately induce expression of the human reelin and glutamic acid decarboxylase 67 genes. Mol. Pharmacol. 2007, 71, 644–653. [Google Scholar] [CrossRef] [Green Version]
- Kundakovic, M.; Chen, Y.; Guidotti, A.; Grayson, D.R. The reelin and GAD67 promoters are activated by epige-netic drugs that facilitate the disruption of local repressor complexes. Mol. Pharmacol. 2009, 75, 342–354. [Google Scholar] [CrossRef] [Green Version]
- Covington, H.E., 3rd; Maze, I.; LaPlant, Q.C.; Vialou, V.F.; Ohnishi, Y.N.; Berton, O.; Fass, D.M.; Renthal, W.; Rush, A.J., 3rd; Wu, E.Y.; et al. Antidepressant actions of histone deacetylase inhibitors. J. Neurosci. 2009, 29, 11451–11460. [Google Scholar] [CrossRef]
- Abdolmaleky, H.M.; Cheng, K.H.; Russo, A.; Smith, C.L.; Faraone, S.V.; Wilcox, M.; Shafa, R.; Glatt, S.J.; Nguyen, G.; Ponte, J.F.; et al. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: A preliminary report. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005, 134B, 60–66. [Google Scholar] [CrossRef]
- Palacios-Garcia, I.; Lara-Vasquez, A.; Montiel, J.F.; Diaz-Veliz, G.F.; Sepulveda, H.; Utreras, E.; Montecino, M.; Gonzalez-Billault, C.; Aboitiz, F. Prenatal stress downregulates Reelin expression by methylation of its promoter and induces adult behavioral impairments in rats. PLoS ONE 2015, 10, e0117680. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Tu, W.; Sun, X.; Zhang, J.; Chen, Y.; Zhao, H. Retardation of neurobehavioral development and reelin down-regulation regulated by further DNA methylation in the hippocampus of the rat pups are associated with maternal deprivation. Behav. Brain Res. 2011, 217, 142–147. [Google Scholar] [CrossRef]
- Veldic, M.; Caruncho, H.J.; Liu, W.S.; Davis, J.; Satta, R.; Grayson, D.R.; Guidotti, A.; Costa, E. DNA-methyltransferase 1 mRNA is selectively overexpressed in telencephalic GABAergic interneurons of schizophrenia brains. Proc. Natl. Acad. Sci. USA 2004, 101, 348–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veldic, M.; Kadriu, B.; Maloku, E.; Agis-Balboa, R.C.; Guidotti, A.; Davis, J.M.; Costa, E. Epigenetic mechanisms expressed in basal ganglia GABAergic neurons differentiate schizophrenia from bipolar disorder. Schizophr. Res. 2007, 91, 51–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruzicka, W.B.; Zhubi, A.; Veldic, M.; Grayson, D.R.; Costa, E.; Guidotti, A. Selective epigenetic alteration of lay-er I GABAergic neurons isolated from prefrontal cortex of schizophrenia patients using laser-assisted microdissection. Mol. Psychiatry 2007, 12, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Matrisciano, F.; Tueting, P.; Dalal, I.; Kadriu, B.; Grayson, D.R.; Davis, J.M.; Nicoletti, F.; Guidotti, A. Epigenetic modifications of GABAergic interneurons are associated with the schizophrenia-like phenotype induced by prenatal stress in mice. Neuropharmacology 2013, 68, 184–194. [Google Scholar] [CrossRef] [Green Version]
- Zhubi, A.; Chen, Y.; Dong, E.; Cook, E.H.; Guidotti, A.; Grayson, D.R. Increased binding of MeCP2 to the GAD1 and RELN promoters may be mediated by an enrichment of 5-hmC in autism spectrum disorder (ASD) cerebellum. Transl. Psychiatry 2014, 4, e349. [Google Scholar] [CrossRef] [Green Version]
- Do, H.T.; Bruelle, C.; Tselykh, T.; Jalonen, P.; Korhonen, L.; Lindholm, D. Reciprocal regulation of very low density lipoprotein receptors (VLDLRs) in neurons by brain-derived neurotrophic factor (BDNF) and Reelin: Involvement of the E3 ligase Mylip/Idol. J. Biol. Chem. 2013, 288, 29613–29620. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.H.; D’Arcangelo, G. New Insights into Reelin-Mediated Signaling Pathways. Front. Cell. Neurosci. 2016, 10, 122. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Lafuente, C.L.; Kalynchuk, L.E.; Caruncho, H.J.; Ausió, J. The Role of MeCP2 in Regulating Synaptic Plasticity in the Context of Stress and Depression. Cells 2022, 11, 748. https://doi.org/10.3390/cells11040748
Sánchez-Lafuente CL, Kalynchuk LE, Caruncho HJ, Ausió J. The Role of MeCP2 in Regulating Synaptic Plasticity in the Context of Stress and Depression. Cells. 2022; 11(4):748. https://doi.org/10.3390/cells11040748
Chicago/Turabian StyleSánchez-Lafuente, Carla L., Lisa E. Kalynchuk, Hector J. Caruncho, and Juan Ausió. 2022. "The Role of MeCP2 in Regulating Synaptic Plasticity in the Context of Stress and Depression" Cells 11, no. 4: 748. https://doi.org/10.3390/cells11040748
APA StyleSánchez-Lafuente, C. L., Kalynchuk, L. E., Caruncho, H. J., & Ausió, J. (2022). The Role of MeCP2 in Regulating Synaptic Plasticity in the Context of Stress and Depression. Cells, 11(4), 748. https://doi.org/10.3390/cells11040748