
CRISPR/Cas9 Mediated Knockout of Cyclooxygenase-2 Gene Inhibits Invasiveness in A2058 Melanoma Cells

 , , and
, , and {kind=link}
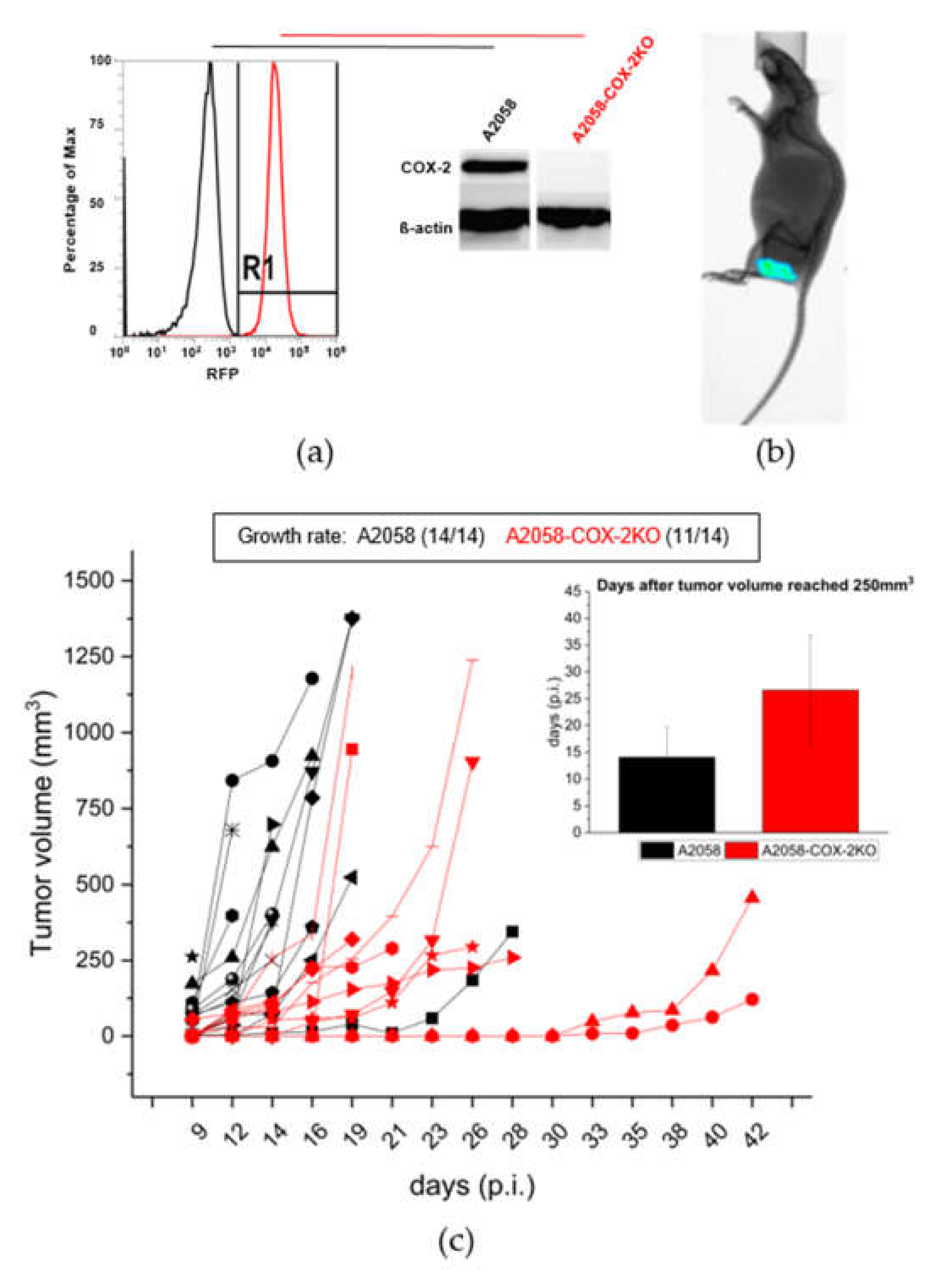{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Spheroid Cell Culture and Culture Conditions
2.2. Knockdown of COX-2 with CRISPR/Cas9
2.3. PGE2 Measurements
2.4. Growth Rate Analysis
2.5. Scratch Wound Cell Migration and Invasion Assay Using the IncuCyte® Live-Cell Monitoring System
2.6. Clonogenic Assay
2.7. Cell Viability Assay
2.8. [18F]FDG and [18F]FMISO Radiotracer Uptake
2.9. Western Blot Analysis
2.10. Animals and Generation of A2058 and A2058-COX-2KO Melanoma Xenografts
2.11. Optical In Vivo Imaging
2.12. Protein Phosphorylation Assay
2.13. Statistical Analysis
3. Results
3.1. COX-2 Knockout Slowes Subcutaneous Tumor Growth
3.2. Cox-2 Knockout Changes the Metabolic Activity under Normoxia and under Conditions of Experimental Hypoxia
3.3. COX-2 Knockout Reduces Cell Invasion and Motility
3.4. Cox-2 Knockout Alters the Tumor Spheroid Formation Capability and Changes the Metabolic Activity and Intrinsic Hypoxia
3.5. COX-2 Knockout Reduces the Plating Efficiency but Does Not Effect Chemosensitivity and Radiosensitivity
3.6. Altered COX-2 Downstream Signaling in COX-2 Knockout Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, N.; Grivennikov, S.I.; Karin, M. The Unholy Trinity: Inflammation, Cytokines, and STAT3 Shape the Cancer Microenvironment. Cancer Cell 2011, 19, 429–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munn, L.L. Cancer and inflammation. WIREs Syst. Biol. Med. 2016, 9, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dannenberg, A.J.; Lippman, S.M.; Mann, J.R.; Subbaramaiah, K.; Dubois, R.N. Cyclooxygenase-2 and Epidermal Growth Factor Receptor: Pharmacologic Targets for Chemoprevention. J. Clin. Oncol. 2005, 23, 254–266. [Google Scholar] [CrossRef] [Green Version]
- Kaidi, A.; Qualtrough, D.; Williams, A.; Paraskeva, C. Direct Transcriptional Up-regulation of Cyclooxygenase-2 by Hypoxia-Inducible Factor (HIF)-1 Promotes Colorectal Tumor Cell Survival and Enhances HIF-1 Transcriptional Activity during Hypoxia. Cancer Res. 2006, 66, 6683–6691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtsuka, J.; Oshima, H.; Ezawa, I.; Abe, R.; Oshima, M.; Ohki, R. Functional loss of p53 cooperates with the in vivo microenvironment to promote malignant progression of gastric cancers. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef]
- Tandler, N.; Mosch, B.; Pietzsch, J. Protein and non-protein biomarkers in melanoma: A critical update. Amino Acids 2012, 43, 2203–2230. [Google Scholar] [CrossRef]
- Goradel, N.H.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in cancer: A review. J. Cell. Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef]
- Tudor, D.V.; Bâldea, I.; Lupu, M.; Kacso, T.; Kutasi, E.; Hopârtean, A.; Stretea, R.; Filip, A.G. COX-2 as a potential biomarker and therapeutic target in melanoma. Cancer Biol. Med. 2020, 17, 20–31. [Google Scholar] [CrossRef]
- Hosseini, F.; Mahdian-Shakib, A.; Jadidi-Niaragh, F.; Enderami, S.E.; Mohammadi, H.; Hemmatzadeh, M.; Mohammed, H.A.; Anissian, A.; Kokhaei, P.; Mirshafiey, A.; et al. Anti-inflammatory and anti-tumor effects of α-l-guluronic acid (G2013) on cancer-related inflammation in a murine breast cancer model. Biomed. Pharmacother. 2018, 98, 793–800. [Google Scholar] [CrossRef]
- Majumder, M.; Dunn, L.; Liu, L.; Hasan, A.; Vincent, K.; Brackstone, M.; Hess, D.; Lala, P.K. COX-2 induces oncogenic micro RNA miR655 in human breast cancer. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Denkert, C.; Köbel, M.; Berger, S.; Siegert, A.; LeClere, A.; Trefzer, U.; Hauptmann, S. Expression of cyclooxygenase 2 in human malignant melanoma. Cancer Res. 2001, 61, 303–308. [Google Scholar] [PubMed]
- Panza, E.; De Cicco, P.; Ercolano, G.; Armogida, C.; Scognamiglio, G.; Anniciello, A.M.; Botti, G.; Cirino, G.; Ianaro, A. Differential expression of cyclooxygenase-2 in metastatic melanoma affects progression free survival. Oncotarget 2016, 7, 57077–57085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuźbicki, Ł.; Lange, D.; Stanek-Widera, A.; Chwirot, B.W. Intratumoral expression of cyclooxygenase-2 (COX-2) is a negative prognostic marker for patients with cutaneous melanoma. Melanoma Res. 2016, 26, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Cahlin, C.; Gelin, J.; Delbro, D.; Lönnroth, C.; Doi, C.; Lundholm, K. Effect of cyclooxygenase and nitric oxide synthase inhibitors on tumor growth in mouse tumor models with and without cancer cachexia related to prostanoids. Cancer Res. 2000, 60, 1742–1749. [Google Scholar]
- Kim, S.-H.; Roszik, J.; Cho, S.-N.; Ogata, D.; Milton, D.R.; Peng, W.; Menter, D.G.; Ekmekcioglu, S.; Grimm, E.A. The COX2 Effector Microsomal PGE2 Synthase 1 is a Regulator of Immunosuppression in Cutaneous Melanoma. Clin. Cancer Res. 2019, 25, 1650–1663. [Google Scholar] [CrossRef] [Green Version]
- Daphu, I.; Horn, S.; Stieber, D.; Varughese, J.K.; Spriet, E.; Dale, H.A.; Skaftnesmo, K.O.; Bjerkvig, R.; Thorsen, F. In Vitro Treatment of Melanoma Brain Metastasis by Simultaneously Targeting the MAPK and PI3K Signaling Pathways. Int. J. Mol. Sci. 2014, 15, 8773–8794. [Google Scholar] [CrossRef] [Green Version]
- Nymark Aasen, S.; Parajuli, H.; Hoang, T.; Feng, Z.; Stokke, K.; Wang, J.; Roy, K.; Bjerkvig, R.; Knappskog, S.; Thorsen, F.; et al. Effective Treatment of Metastatic Melanoma by Combining MAPK and PI3K Signaling Pathway Inhibitors. Int. J. Mol. Sci. 2019, 20, 4235. [Google Scholar] [CrossRef] [Green Version]
- Cheki, M.; Yahyapour, R.; Farhood, B.; Rezaeyan, A.; Shabeeb, D.; Amini, P.; Rezapoor, S.; Najafi, M. COX-2 in Radiotherapy: A Potential Target for Radioprotection and Radiosensitization. Curr. Mol. Pharmacol. 2018, 11, 173–183. [Google Scholar] [CrossRef]
- Lin, F.; Luo, J.; Gao, W.; Wu, J.; Shao, Z.; Wang, Z.; Meng, J.; Ou, Z.; Yang, G. COX-2 promotes breast cancer cell radioresistance via p38/MAPK-mediated cellular anti-apoptosis and invasiveness. Tumor Biol. 2013, 34, 2817–2826. [Google Scholar] [CrossRef]
- Laube, M.; Kniess, T.; Pietzsch, J. Development of Antioxidant COX-2 Inhibitors as Radioprotective Agents for Radiation Therapy—A Hypothesis-Driven Review. Antioxidants 2016, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Bell, E.; Ponthan, F.; Whitworth, C.; Tweddle, D.A.; Lunec, J.; Redfern, C.P.F. COX2 expression in neuroblastoma increases tumorigenicity but does not affect cell death in response to the COX2 inhibitor celecoxib. Clin. Exp. Metastasis 2014, 31, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Grösch, S.; Maier, T.J.; Schiffmann, S.; Geisslinger, G. Cyclooxygenase-2 (COX-2)–Independent Anticarcinogenic Effects of Selective COX-2 Inhibitors. JNCI J. Natl. Cancer Inst. 2006, 98, 736–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, C.; Leng, J.; Demetris, A.J.; Wu, T. Cyclooxygenase-2 Promotes Human Cholangiocarcinoma Growth: Evidence for cyclooxygenase-2-independent mechanism in celecoxib-mediated induction of p21waf1/cip1 and p27kip1 and cell cycle arrest. Cancer Res. 2004, 64, 1369–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, T.J.; Schilling, K.; Schmidt, R.; Geisslinger, G.; Groesch, S. Cyclooxygenase-2 (COX-2)-dependent and -independent anticarcinogenic effects of celecoxib in human colon carcinoma cells. Biochem. Pharmacol. 2004, 67, 1469–1478. [Google Scholar] [CrossRef]
- Schönthal, A.H. Direct non-cyclooxygenase-2 targets of celecoxib and their potential relevance for cancer therapy. Br. J. Cancer 2007, 97, 1465–1468. [Google Scholar] [CrossRef]
- Shin, Y.K.; Park, J.S.; Kim, H.S.; Jun, H.J.; Kim, G.E.; Suh, C.O.; Yun, Y.S.; Pyo, H. Radiosensitivity Enhancement by Celecoxib, a Cyclooxygenase (COX)-2 Selective Inhibitor, via COX-2–Dependent Cell Cycle Regulation on Human Cancer Cells Expressing Differential COX-2 Levels. Cancer Res. 2005, 65, 9501–9509. [Google Scholar] [CrossRef] [Green Version]
- Schönthal, A.H. Induction of Apoptosis by Celecoxib in Cell Culture: An Uncertain Role for Cyclooxygenase-2: Figure 1. Cancer Res. 2007, 67, 5575–5576. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Shi, A.; Han, B.; Li, S.; Wu, D.; Jia, H.; Zheng, C.; Ren, L.; Fan, Z. COX-2 silencing enhances tamoxifen antitumor activity in breast cancer in vivo and in vitro. Int. J. Oncol. 2014, 44, 1385–1393. [Google Scholar] [CrossRef]
- Ercolano, G.; De Cicco, P.; Rubino, V.; Terrazzano, G.; Ruggiero, G.; Carriero, R.; Kunderfranco, P.; Ianaro, A. Knockdown of PTGS2 by CRISPR/CAS9 System Designates a New Potential Gene Target for Melanoma Treatment. Front. Pharmacol. 2019, 10, 1456. [Google Scholar] [CrossRef]
- Ma, C.; Zheng, C.; Bai, E.; Yang, K. miR-101 inhibits glioma cell invasion via the downregulation of COX-2. Oncol. Lett. 2016, 12, 2538–2544. [Google Scholar] [CrossRef] [Green Version]
- Panza, E.; Ercolano, G.; De Cicco, P.; Armogida, C.; Scognamiglio, G.; Botti, G.; Cirino, G.; Ianaro, A. MicroRNA-143-3p inhibits growth and invasiveness of melanoma cells by targeting cyclooxygenase-2 and inversely correlates with malignant melanoma progression. Biochem. Pharmacol. 2018, 156, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Silveira, T.L.; Pang, L.Y.; Di Domenico, A.; Veloso, E.S.; Silva, I.L.D.; Del Puerto, H.L.; Ferreria, E.; Argyle, D.J. COX-2 Silencing in Canine Malignant Melanoma Inhibits Malignant Behaviour. Front. Vet. Sci. 2021, 8, 633170. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sanjana, N.; Zheng, K.; Shalem, O.; Lee, K.; Shi, X.; Scott, D.A.; Song, J.; Pan, J.Q.; Weissleder, R.; et al. Genome-wide CRISPR Screen in a Mouse Model of Tumor Growth and Metastasis. Cell 2015, 160, 1246–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.-S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Reissenweber, B.; Mosch, B.; Pietzsch, J. Experimental hypoxia does not influence gene expression and protein synthesis of Eph receptors and ephrin ligands in human melanoma cells in vitro. Melanoma Res. 2013, 23, 85–95. [Google Scholar] [CrossRef]
- Mosch, B.; Pietzsch, D.; Pietzsch, J. Irradiation affects cellular properties and Eph receptor expression in human melanoma cells. Cell Adh Migr. 2012, 6, 113–125. [Google Scholar] [CrossRef] [Green Version]
- Bechmann, N.; Hauser, S.; Hofheinz, F.; Kniess, T.; Pietzsch, J. Nitric oxide-releasing selective cyclooxygenase-2 inhibitors as promising radiosensitizers in melanoma cells in vitro. Ann. Radiat. Ther. Oncol. 2017, 1, 1010. [Google Scholar]
- Neuber, C.; Belter, B.; Meister, S.; Hofheinz, F.; Bergmann, R.; Pietzsch, H.-J.; Pietzsch, J. Overexpression of Receptor Tyrosine Kinase EphB4 Triggers Tumor Growth and Hypoxia in A375 Melanoma Xenografts: Insights from Multitracer Small Animal Imaging Experiments ‡. Molecules 2018, 23, 444. [Google Scholar] [CrossRef] [Green Version]
- Sobolewski, C.; Cerella, C.; Dicato, M.; Ghibelli, L.; Diederich, M. The Role of Cyclooxygenase-2 in Cell Proliferation and Cell Death in Human Malignancies. Int. J. Cell Biol. 2010, 2010, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Herlyn, M.; Fukunaga-Kalabis, M. What Is a Good Model for Melanoma? J. Investig. Dermatol. 2010, 130, 911–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herwig, N.; Belter, B.; Wolf, S.; Haase-Kohn, C.; Pietzsch, J. Interaction of extracellular S100A4 with RAGE prompts prometastatic activation of A375 melanoma cells. J. Cell. Mol. Med. 2016, 20, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Campillo, N.; Torres, M.; Vilaseca, A.; Nonaka, P.N.; Gozal, D.; Roca-Ferrer, J.; Picado, C.; Montserrat, J.M.; Farre, R.; Navajas, D.; et al. Role of Cyclooxygenase-2 on Intermittent Hypoxia-Induced Lung Tumor Malignancy in a Mouse Model of Sleep Apnea. Sci. Rep. 2017, 7, 44693. [Google Scholar] [CrossRef] [PubMed]
- Kniess, T.; Laube, M.; Bergmann, R.; Sehn, F.; Graf, F.; Steinbach, J.; Wuest, F.; Pietzsch, J. Radiosynthesis of a 18F-labeled 2,3-diarylsubstituted indole via McMurry coupling for functional characterization of cyclooxygenase-2 (COX-2) in vitro and in vivo. Bioorganic Med. Chem. 2012, 20, 3410–3421. [Google Scholar] [CrossRef]
- Doctor, A.; Seifert, V.; Ullrich, M.; Hauser, S.; Pietzsch, J. Three-Dimensional Cell Culture Systems in Radiopharmaceutical Cancer Research. Cancers 2020, 12, 2765. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Han, C.; Guo, D.; Wang, D.; Chen, C.-S.; D’Ambrosio, S.M. OSU03012 activates Erk1/2 and Cdks leading to the accumulation of cells in the S-phase and apoptosis. Int. J. Cancer 2008, 123, 2923–2930. [Google Scholar] [CrossRef]
- Huang, J.W.; Tseng, P.H.; Yang, Y.T.; Fowble, J.; Shiau, C.W.; Shaw, Y.J.; Kulp, S.K.; Chen, C.S. From the Cyclooxygenase-2 Inhibitor Celecoxib to a Novel Class of 3-Phosphoinositide-Dependent Protein Kinase-1 Inhibitors. Cancer Res. 2004, 64, 4309–4318. [Google Scholar] [CrossRef] [Green Version]
- Maier, T.J.; Tausch, L.; Hoernig, M.; Coste, O.; Schmidt, R.; Angioni, C.; Metzner, J.; Groesch, S.; Pergola, C.; Steinhilber, D.; et al. Celecoxib inhibits 5-lipoxygenase. Biochem. Pharmacol. 2008, 76, 862–872. [Google Scholar] [CrossRef]
- Patel, M.I.; Subbaramaiah, K.; Du, B.; Chang, M.; Yang, P.; Newman, R.A.; Cordon-Cardo, C.; Thaler, H.T.; Dannenberg, A.J. Celecoxib Inhibits Prostate Cancer Growth: Evidence of a Cyclooxygenase-2-Independent Mechanism. Clin. Cancer Res. 2005, 11, 1999–2007. [Google Scholar] [CrossRef] [Green Version]
- Niu, K.; Chen, X.-W.; Qin, Y.; Zhang, L.-P.; Liao, R.-X.; Sun, J.-G. Celecoxib Blocks Vasculogenic Mimicry via an Off-Target Effect to Radiosensitize Lung Cancer Cells: An Experimental Study. Front. Oncol. 2021, 11, 697227. [Google Scholar] [CrossRef] [PubMed]
- Ramu, A.; Kathiresan, S.; Ramadoss, H.; Nallu, A.; Kaliyan, R.; Azamuthu, T. Gramine attenuates EGFR-mediated inflammation and cell proliferation in oral carcinogenesis via regulation of NF-κB and STAT3 signaling. Biomed. Pharmacother. 2017, 98, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Todoric, J.; Antonucci, L.; Karin, M. Targeting Inflammation in Cancer Prevention and Therapy. Cancer Prev. Res. 2016, 9, 895–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Lao, X.; Zheng, H. Influencing COX-2 Activity by COX Related Pathways in Inflammation and Cancer. Mini-Rev. Med. Chem. 2016, 16, 1230–1243. [Google Scholar] [CrossRef]
- Zhou, P.; Qin, J.; Li, Y.; Li, G.; Wang, Y.; Zhang, N.; Chen, P.; Li, C. Combination therapy of PKCζ and COX-2 inhibitors synergistically suppress melanoma metastasis. J. Exp. Clin. Cancer Res. 2017, 36, 115. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haase-Kohn, C.; Laube, M.; Donat, C.K.; Belter, B.; Pietzsch, J. CRISPR/Cas9 Mediated Knockout of Cyclooxygenase-2 Gene Inhibits Invasiveness in A2058 Melanoma Cells. Cells 2022, 11, 749. https://doi.org/10.3390/cells11040749
Haase-Kohn C, Laube M, Donat CK, Belter B, Pietzsch J. CRISPR/Cas9 Mediated Knockout of Cyclooxygenase-2 Gene Inhibits Invasiveness in A2058 Melanoma Cells. Cells. 2022; 11(4):749. https://doi.org/10.3390/cells11040749
Chicago/Turabian StyleHaase-Kohn, Cathleen, Markus Laube, Cornelius K. Donat, Birgit Belter, and Jens Pietzsch. 2022. "CRISPR/Cas9 Mediated Knockout of Cyclooxygenase-2 Gene Inhibits Invasiveness in A2058 Melanoma Cells" Cells 11, no. 4: 749. https://doi.org/10.3390/cells11040749
APA StyleHaase-Kohn, C., Laube, M., Donat, C. K., Belter, B., & Pietzsch, J. (2022). CRISPR/Cas9 Mediated Knockout of Cyclooxygenase-2 Gene Inhibits Invasiveness in A2058 Melanoma Cells. Cells, 11(4), 749. https://doi.org/10.3390/cells11040749