
Pseudotime Analysis Reveals Exponential Trends in DNA Methylation Aging with Mortality Associated Timescales


Abstract
:1. Introduction
2. Materials and Methods
2.1. Methylation Array Processing
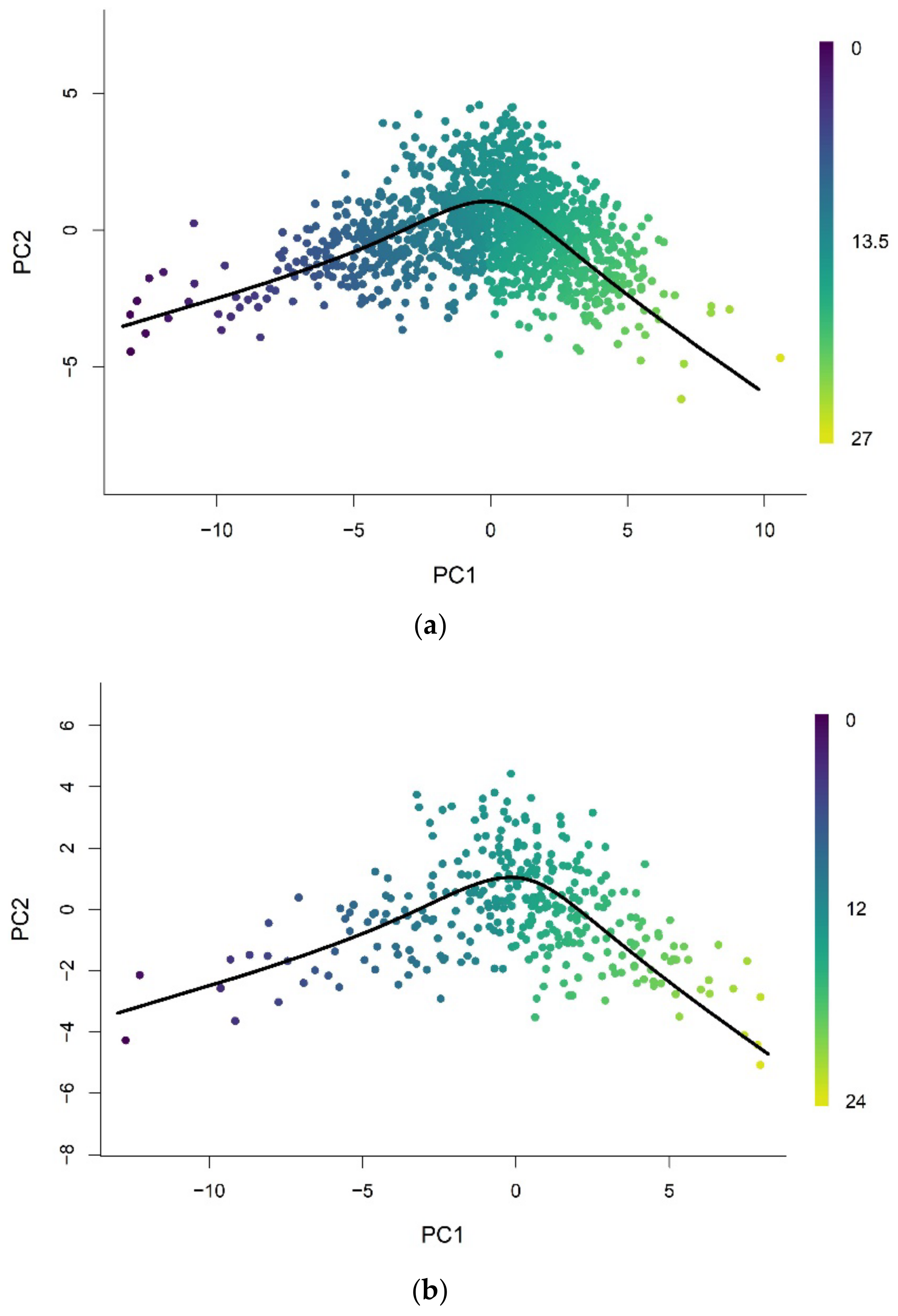
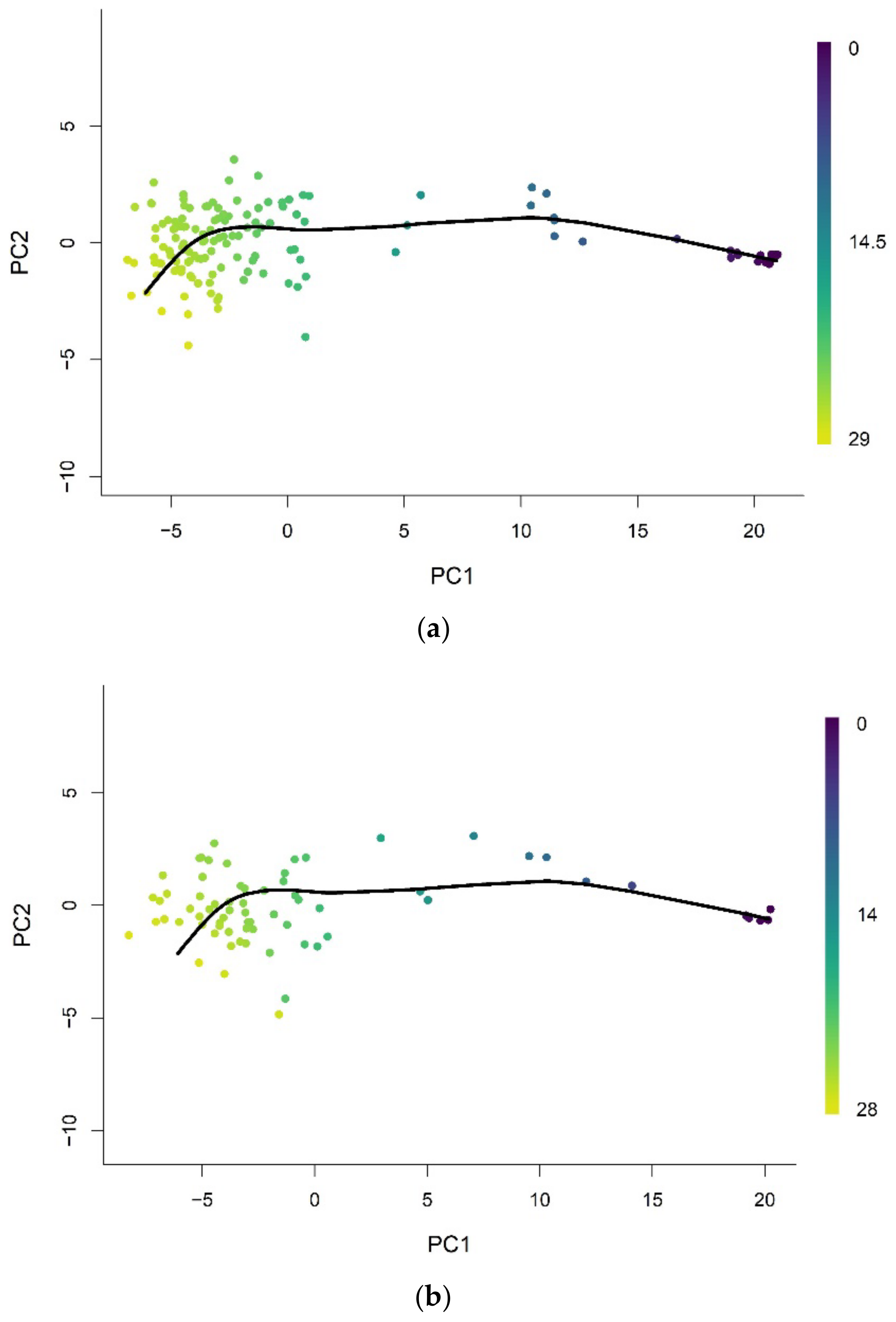
2.2. Pseudotime Analysis
2.3. Epigenetic Pacemaker (EPM)
- is the initial methylation value,
- is the rate of change,
- is the epigenetic state,
- is the normally distributed error term.
2.4. Determining the Functional Form of Methylation Pseudotime
- is methylation pseudotime value,
- is chronological age,
- , , , and are coefficients.
2.5. Analysis Environment
3. Results
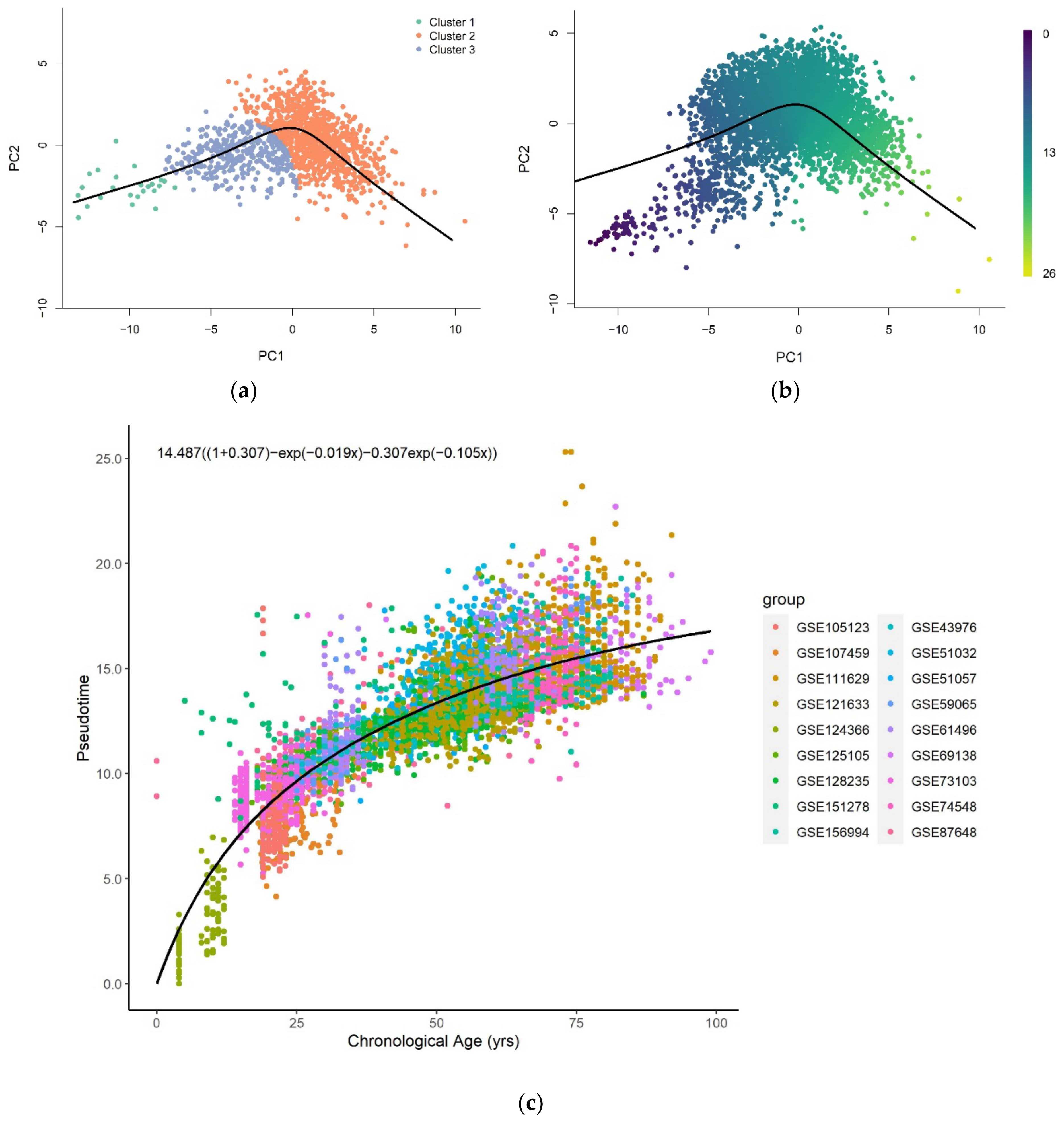
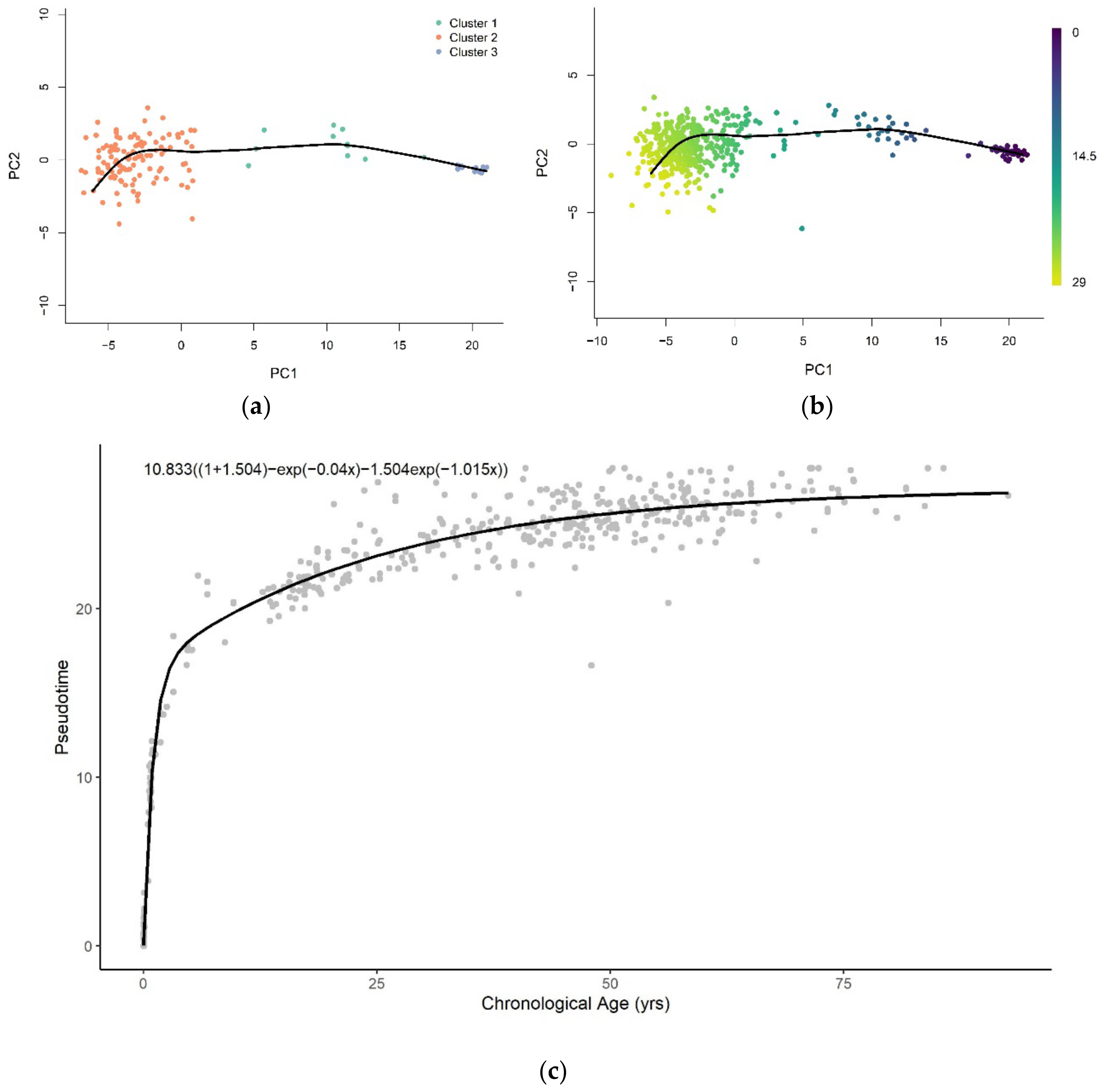
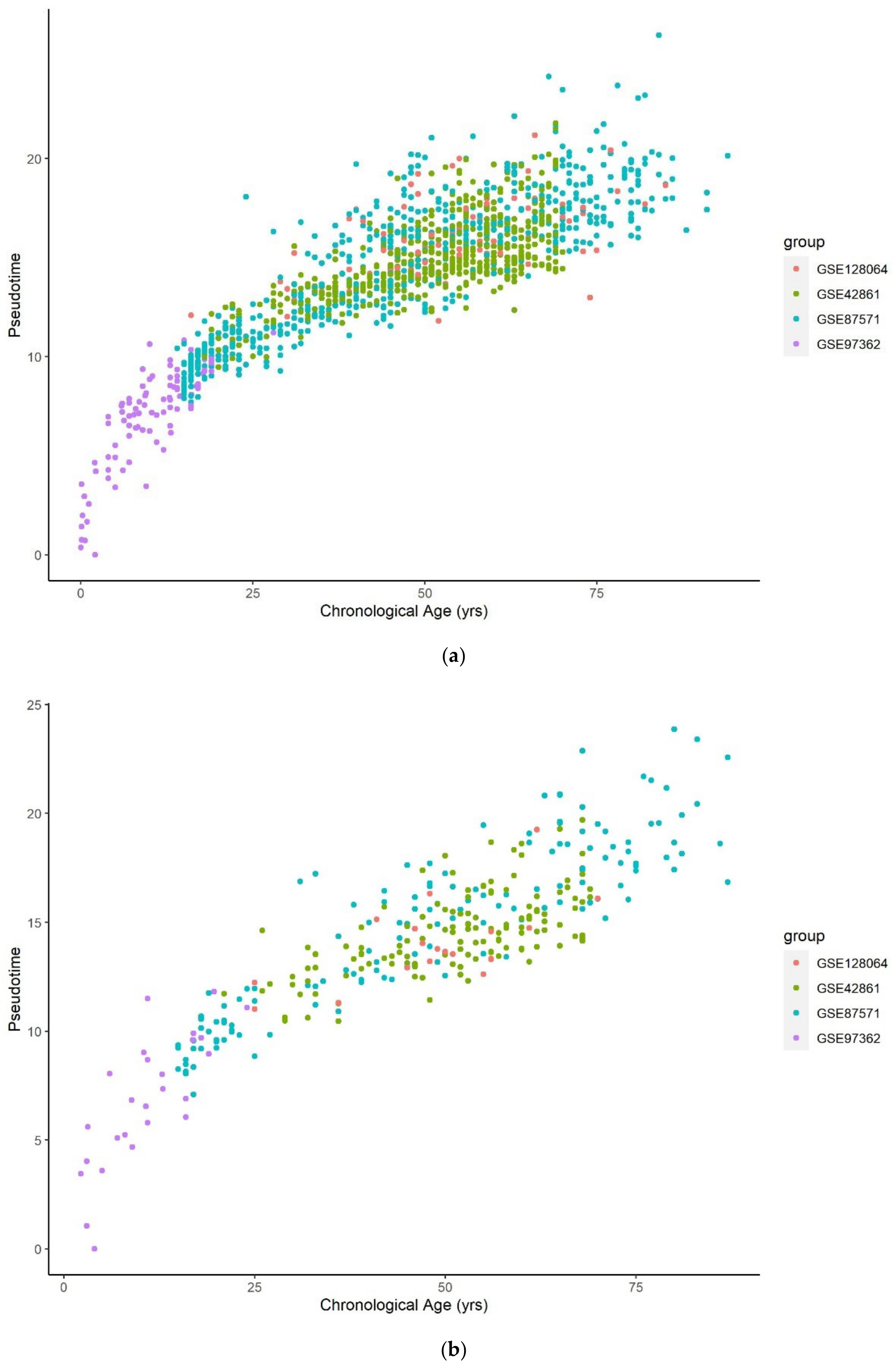
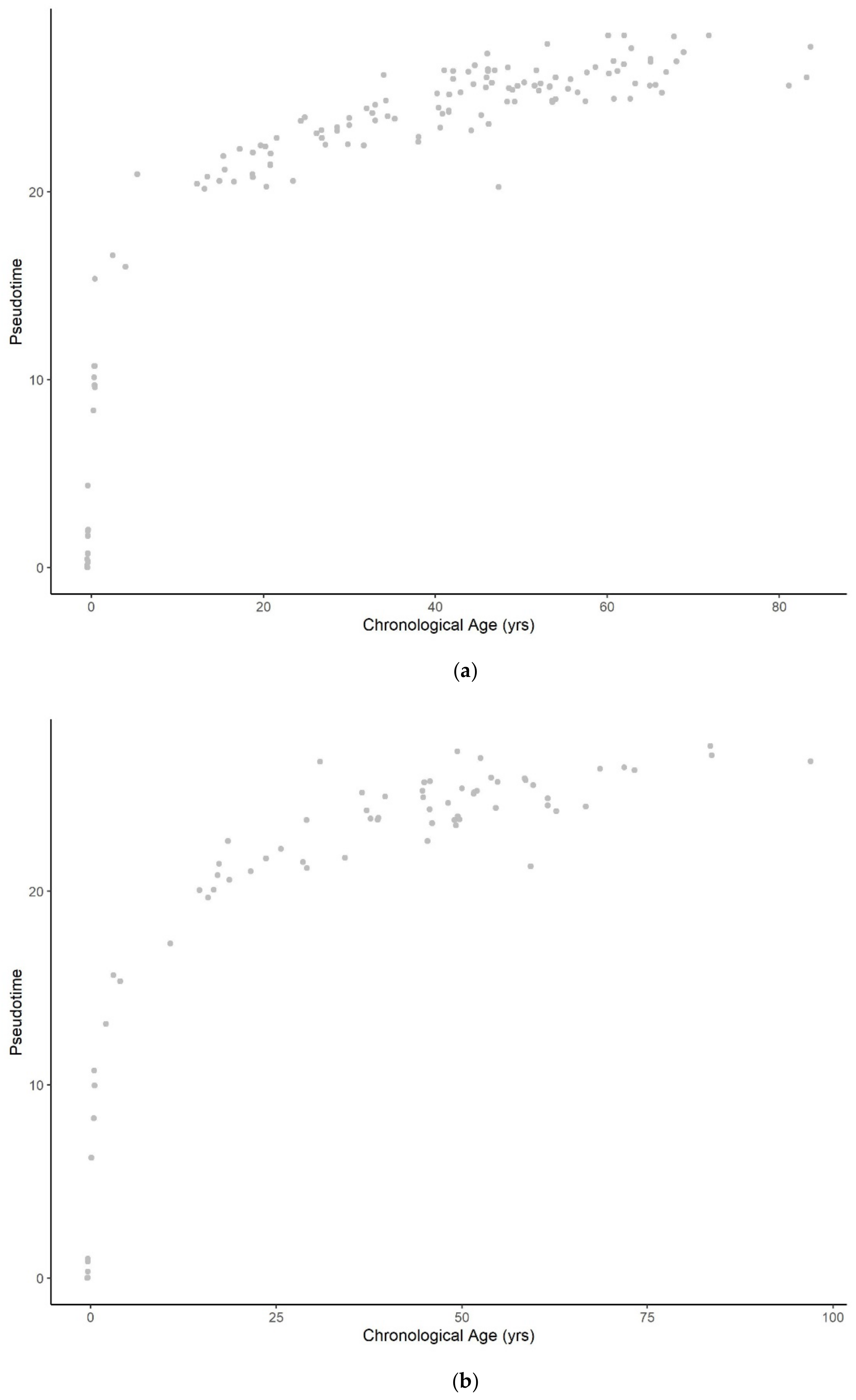
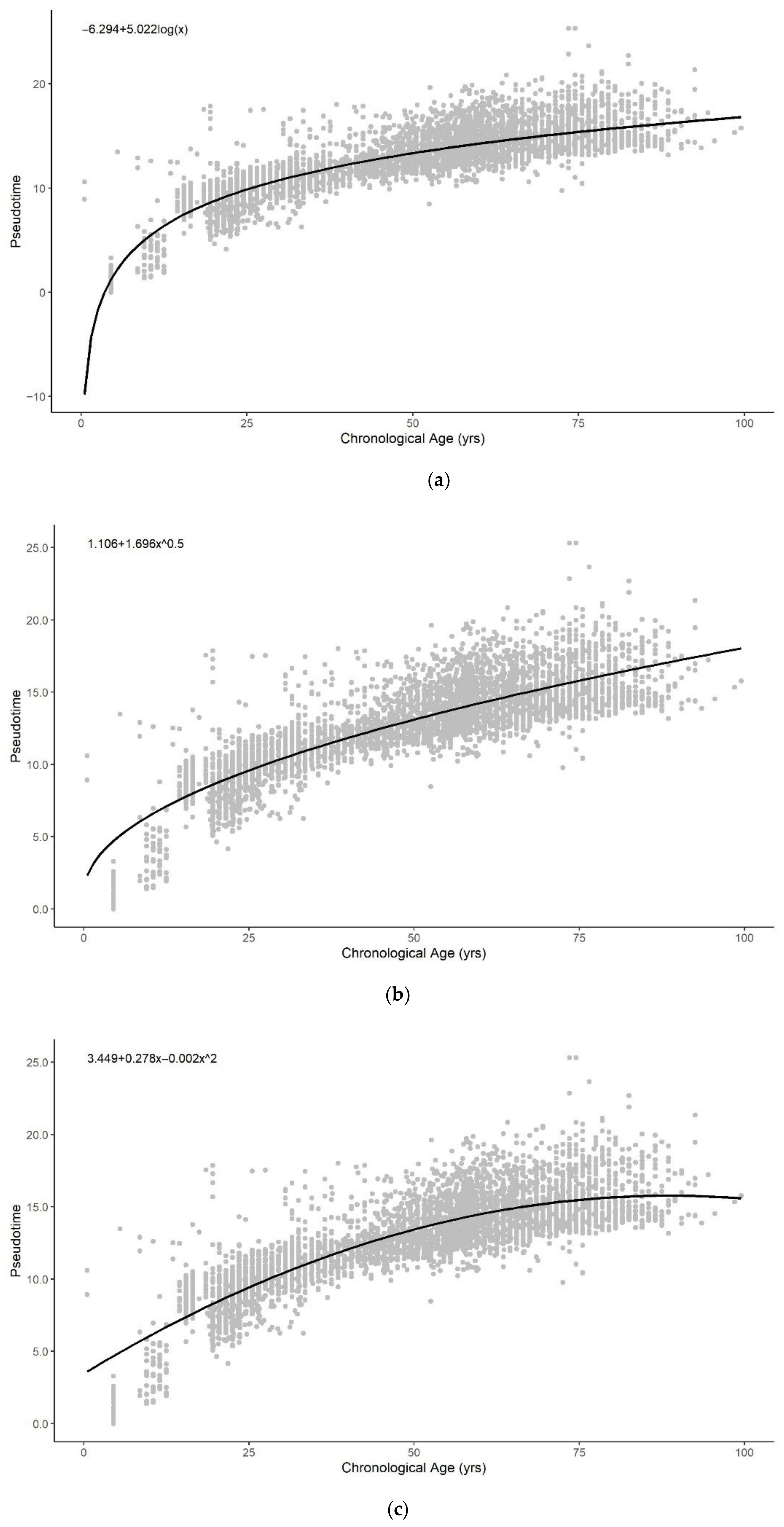
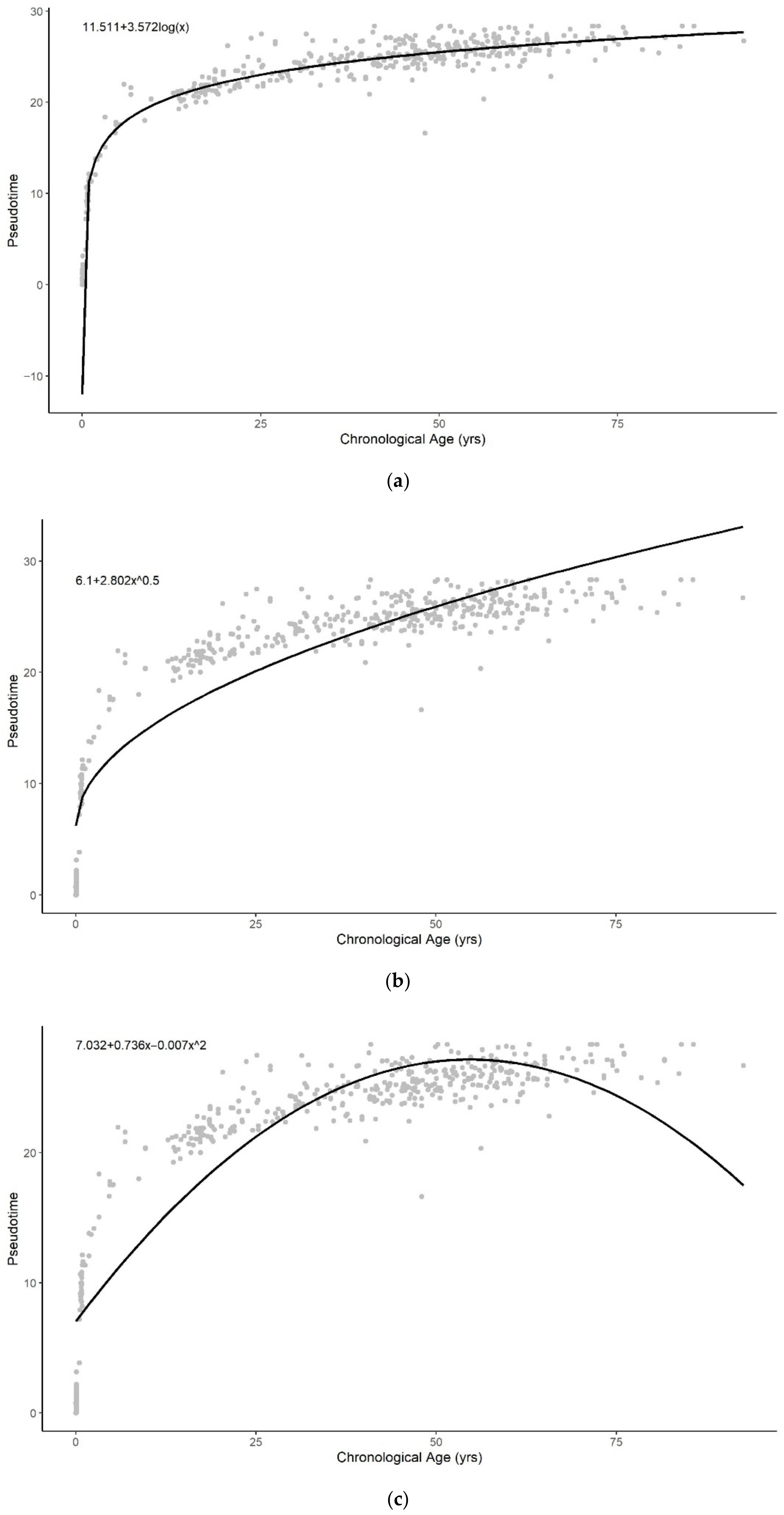
3.1. Methylation Pseudotime Is Nonlinear across the Lifespan
3.1.1. Methylation Pseudotime of Whole Blood Tissue
3.1.2. Methylation Pseudotime of Brain Tissue
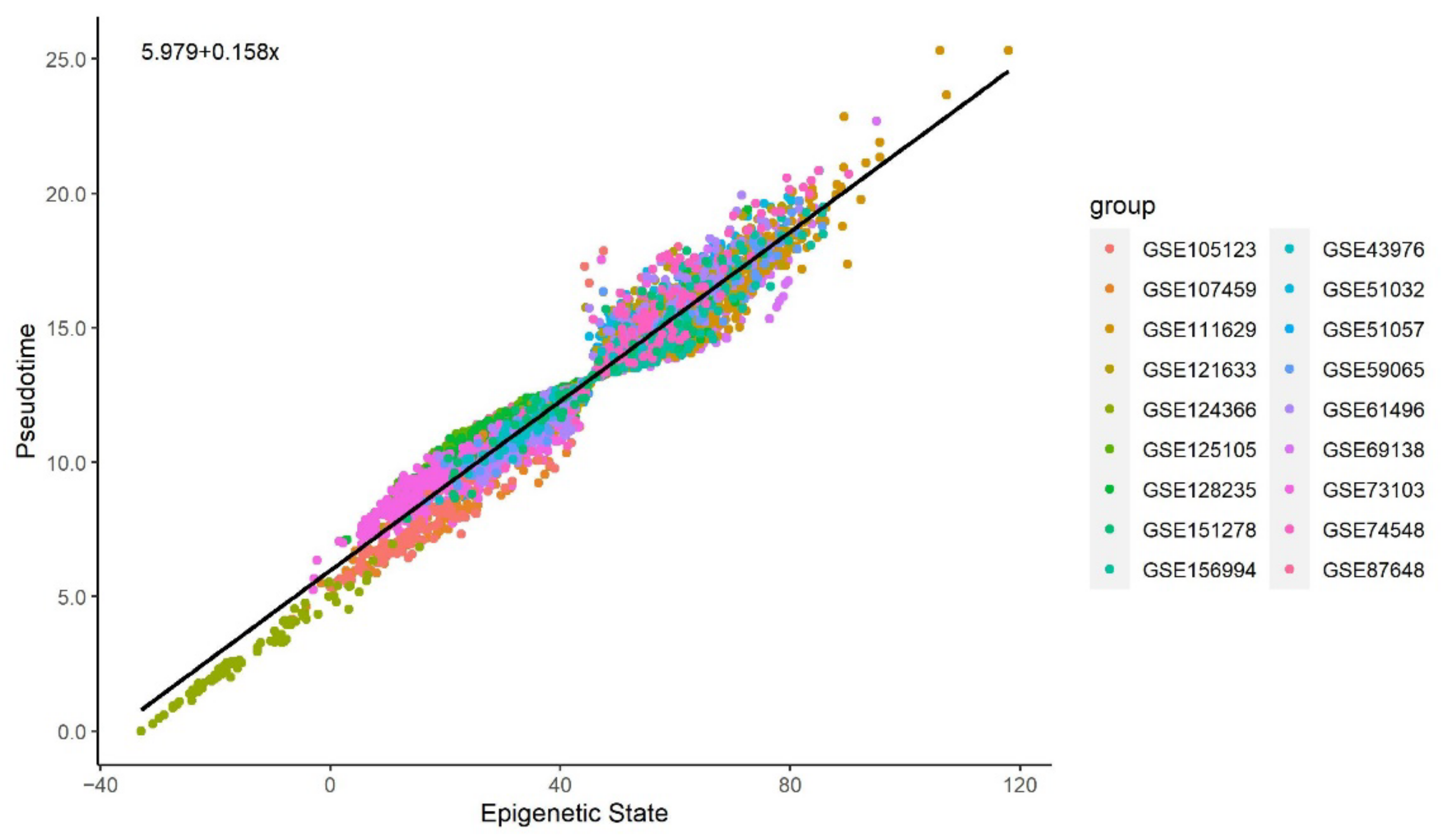
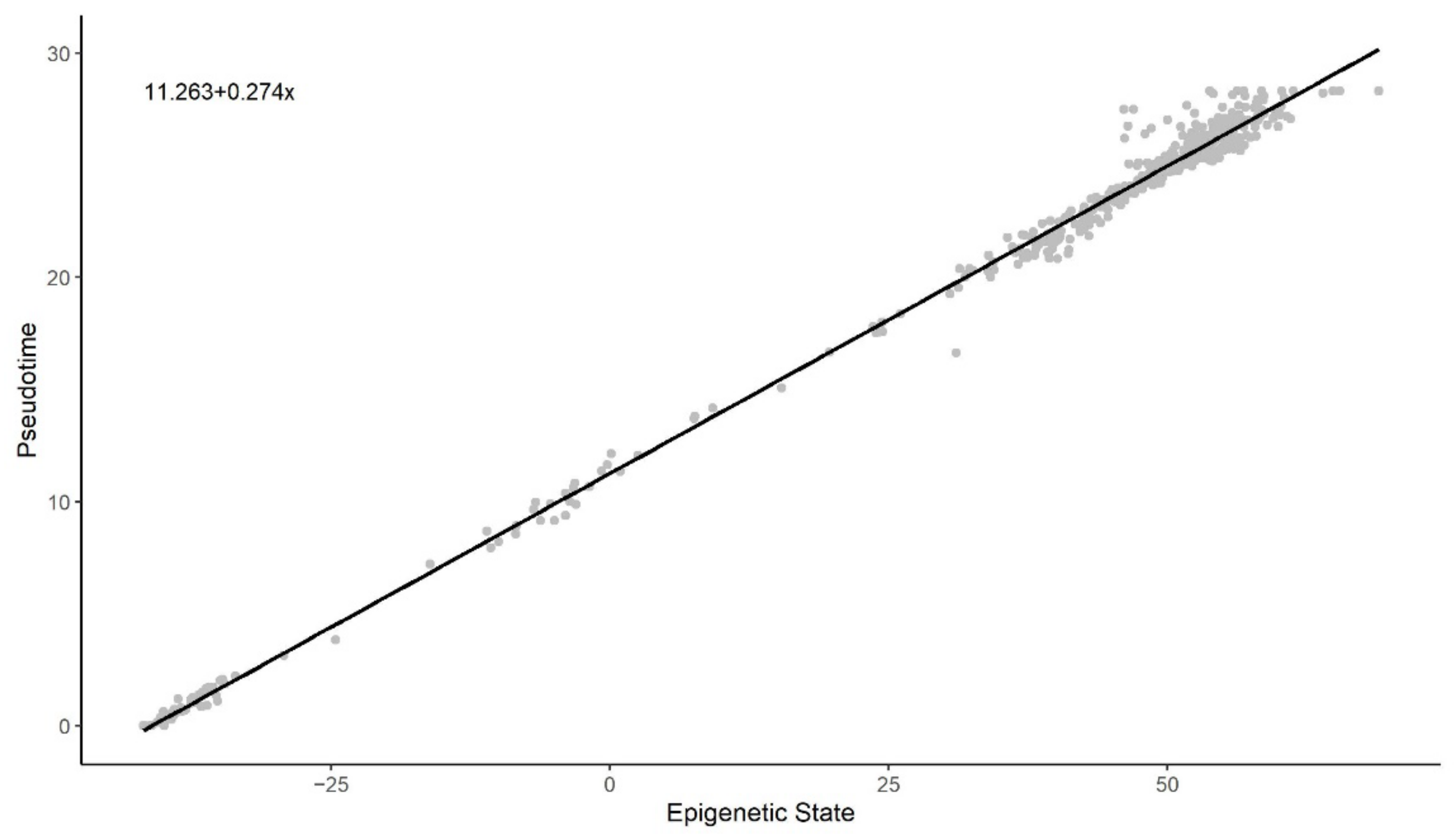
3.2. Methylation Pseudotime Is Linearly Correlated to Epigenetic States Estimated by the EPM
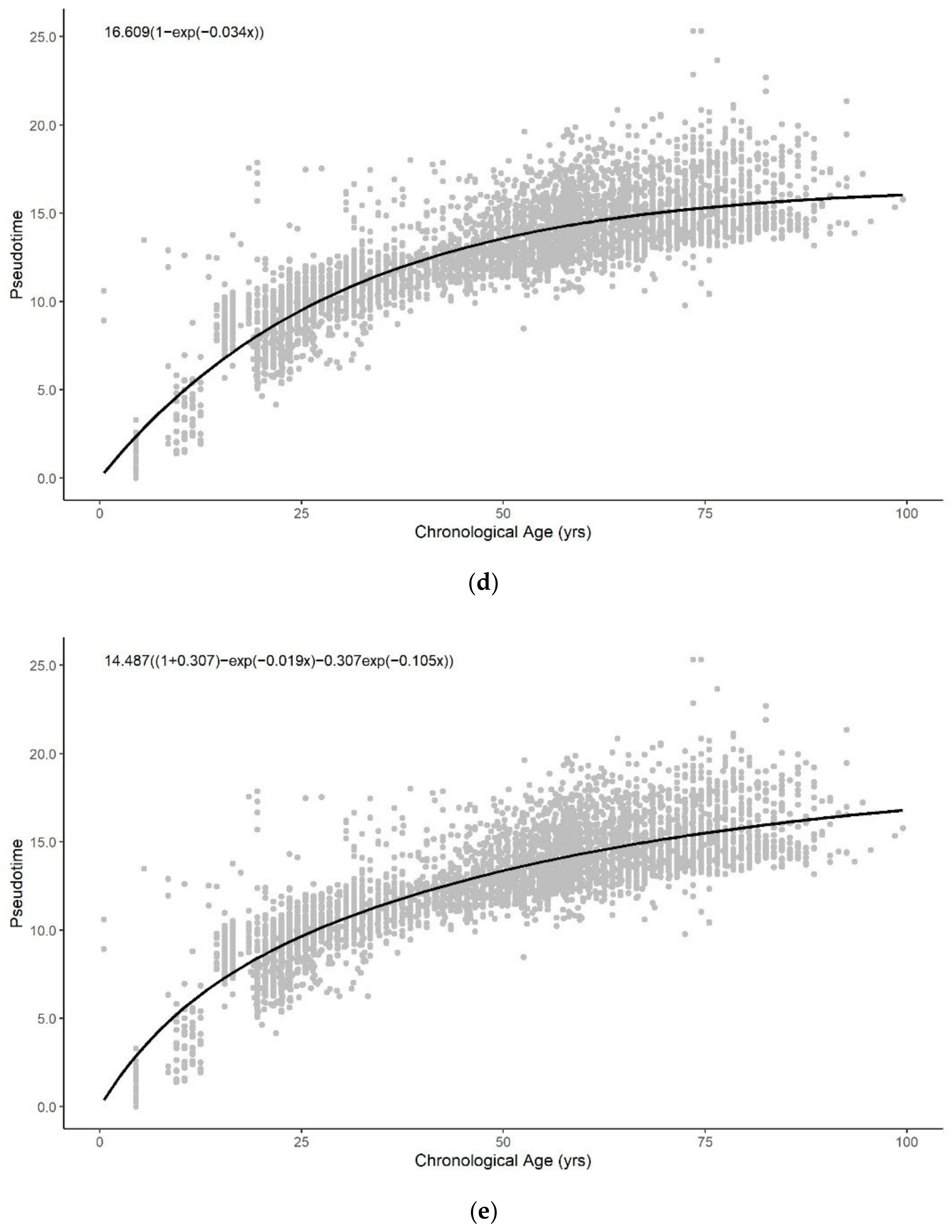
3.3. Sum of Two Exponentials Best Describe Methylation Pseudotime across the Lifespan
3.3.1. Functional Form of Methylation Pseudotime of Whole Blood Tissue
3.3.2. Functional Form of Methylation Pseudotime of Brain Tissue
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Computer Code and Software:
Appendix A








References
- Slagboom, P.E.; van den Berg, N.; Deelen, J. Phenome and Genome Based Studies into Human Ageing and Longevity: An Overview. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2742–2751. [Google Scholar] [CrossRef] [PubMed]
- McCormick, M.A.; Kennedy, B.K. Genome-Scale Studies of Aging: Challenges and Opportunities. Curr. Genom. 2012, 13, 500–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Avraham, D.; Muzumdar, R.H.; Atzmon, G. Epigenetic Genome-Wide Association Methylation in Aging and Longevity. Epigenomics 2012, 4, 503–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-Wide Evolutionary Analysis of Eukaryotic DNA Methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziller, M.J.; Gu, H.; Müller, F.; Donaghey, J.; Tsai, L.T.-Y.; Kohlbacher, O.; De Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E.; et al. Charting a Dynamic DNA Methylation Landscape of the Human Genome. Nature 2013, 500, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.E.; Gao, Y.; Deep-Soboslay, A.; Tao, R.; Hyde, T.M.; Weinberger, D.R.; Kleinman, J.E. Mapping DNA Methylation across Development, Genotype and Schizophrenia in the Human Frontal Cortex. Nat. Neurosci. 2016, 19, 40–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snir, S.; Farrell, C.; Pellegrini, M. Human Epigenetic Ageing Is Logarithmic with Time across the Entire Lifespan. Epigenetics 2019, 14, 912–926. [Google Scholar] [CrossRef]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y.; et al. Genome-Wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Bocklandt, S.; Lin, W.; Sehl, M.E.; Sánchez, F.J.; Sinsheimer, J.S.; Horvath, S.; Vilain, E. Epigenetic Predictor of Age. PLoS ONE 2011, 6, e14821. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S. DNA Methylation Age of Human Tissues and Cell Types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [Green Version]
- Stubbs, T.M.; Bonder, M.J.; Stark, A.-K.; Krueger, F.; BI Ageing Clock Team; von Meyenn, F.; Stegle, O.; Reik, W. Multi-Tissue DNA Methylation Age Predictor in Mouse. Genome Biol. 2017, 18, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Q.; Weidner, C.I.; Costa, I.G.; Marioni, R.E.; Ferreira, M.R.P.; Deary, I.J.; Wagner, W. DNA Methylation Levels at Individual Age-Associated CpG Sites Can Be Indicative for Life Expectancy. Aging 2016, 8, 394–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galkin, F.; Mamoshina, P.; Kochetov, K.; Sidorenko, D.; Zhavoronkov, A. DeepMAge: A Methylation Aging Clock Developed with Deep Learning. Aging Dis. 2021, 12, 1252–1262. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Raj, K. DNA Methylation-Based Biomarkers and the Epigenetic Clock Theory of Ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Marioni, R.E.; Shah, S.; McRae, A.F.; Chen, B.H.; Colicino, E.; Harris, S.E.; Gibson, J.; Henders, A.K.; Redmond, P.; Cox, S.R.; et al. DNA Methylation Age of Blood Predicts All-Cause Mortality in Later Life. Genome Biol. 2015, 16, 25. [Google Scholar] [CrossRef] [Green Version]
- Perna, L.; Zhang, Y.; Mons, U.; Holleczek, B.; Saum, K.-U.; Brenner, H. Epigenetic Age Acceleration Predicts Cancer, Cardiovascular, and All-Cause Mortality in a German Case Cohort. Clin. Epigenet. 2016, 8, 64. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.; Erhart, W.; Brosch, M.; Ammerpohl, O.; von Schönfels, W.; Ahrens, M.; Heits, N.; Bell, J.T.; Tsai, P.-C.; Spector, T.D.; et al. Obesity Accelerates Epigenetic Aging of Human Liver. Proc. Natl. Acad. Sci. USA 2014, 111, 15538–15543. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.; Ritz, B.R. Increased Epigenetic Age and Granulocyte Counts in the Blood of Parkinson’s Disease Patients. Aging 2015, 7, 1130–1142. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.-C.; Lillycrop, K.A.; Beilin, L.J.; Godfrey, K.M.; Anderson, D.; Mori, T.A.; Rauschert, S.; Craig, J.M.; Oddy, W.H.; Ayonrinde, O.T.; et al. Epigenetic Age Acceleration in Adolescence Associates With BMI, Inflammation, and Risk Score for Middle Age Cardiovascular Disease. J. Clin. Endocrinol. Metab. 2019, 104, 3012–3024. [Google Scholar] [CrossRef]
- Armstrong, N.J.; Mather, K.A.; Thalamuthu, A.; Wright, M.J.; Trollor, J.N.; Ames, D.; Brodaty, H.; Schofield, P.R.; Sachdev, P.S.; Kwok, J.B. Aging, Exceptional Longevity and Comparisons of the Hannum and Horvath Epigenetic Clocks. Epigenomics 2017, 9, 689–700. [Google Scholar] [CrossRef]
- Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Gori, D.; Giuliani, C.; Mari, D.; Di Blasio, A.M.; Gentilini, D.; Vitale, G.; Collino, S.; et al. Methylation of ELOVL2 Gene as a New Epigenetic Marker of Age. Aging Cell 2012, 11, 1132–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jöckel, K.-H.; Erbel, R.; Mühleisen, T.W.; et al. Aging of Blood Can Be Tracked by DNA Methylation Changes at Just Three CpG Sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alisch, R.S.; Barwick, B.G.; Chopra, P.; Myrick, L.K.; Satten, G.A.; Conneely, K.N.; Warren, S.T. Age-Associated DNA Methylation in Pediatric Populations. Genome Res. 2012, 22, 623–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergsma, T.; Rogaeva, E. DNA Methylation Clocks and Their Predictive Capacity for Aging Phenotypes and Healthspan. Neurosci. Insights 2020, 15, 2633105520942221. [Google Scholar] [CrossRef]
- Farrell, C.; Lapborisuth, K.; Hu, C.; Pu, K.; Snir, S.; Pellegrini, M. The Epigenetic Pacemaker Is a More Sensitive Tool than Penalized Regression for Identifying Moderators of Epigenetic Aging. bioRxiv 2021. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef] [Green Version]
- Farrell, C.; Snir, S.; Pellegrini, M. The Epigenetic Pacemaker: Modeling Epigenetic States under an Evolutionary Framework. Bioinformatics 2020, 36, 4662–4663. [Google Scholar] [CrossRef]
- Saelens, W.; Cannoodt, R.; Todorov, H.; Saeys, Y. A Comparison of Single-Cell Trajectory Inference Methods. Nat. Biotechnol. 2019, 37, 547–554. [Google Scholar] [CrossRef]
- Reid, J.E.; Wernisch, L. Pseudotime Estimation: Deconfounding Single Cell Time Series. Bioinformatics 2016, 32, 2973–2980. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C. Defining Cell Types and States with Single-Cell Genomics. Genome Res. 2015, 25, 1491–1498. [Google Scholar] [CrossRef] [Green Version]
- Cannoodt, R.; Saelens, W.; Saeys, Y. Computational Methods for Trajectory Inference from Single-Cell Transcriptomics. Eur. J. Immunol. 2016, 46, 2496–2506. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.N.; Bader, G.D. Tempora: Cell Trajectory Inference Using Time-Series Single-Cell RNA Sequencing Data. PLoS Comput. Biol. 2020, 16, e1008205. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for Functional Genomics Data Sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A Flexible and Comprehensive Bioconductor Package for the Analysis of Infinium DNA Methylation Microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Butcher, D.T.; Cytrynbaum, C.; Turinsky, A.L.; Siu, M.T.; Inbar-Feigenberg, M.; Mendoza-Londono, R.; Chitayat, D.; Walker, S.; Machado, J.; Caluseriu, O.; et al. CHARGE and Kabuki Syndromes: Gene-Specific DNA Methylation Signatures Identify Epigenetic Mechanisms Linking These Clinically Overlapping Conditions. Am. J. Hum. Genet. 2017, 100, 773–788. [Google Scholar] [CrossRef]
- Johansson, A.; Enroth, S.; Gyllensten, U. Continuous Aging of the Human DNA Methylome Throughout the Human Lifespan. PLoS ONE 2013, 8, e67378. [Google Scholar] [CrossRef] [Green Version]
- Dámaso, E.; González-Acosta, M.; Vargas-Parra, G.; Navarro, M.; Balmaña, J.; Ramon, Y.; Cajal, T.; Tuset, N.; Thompson, B.A.; Marín, F.; et al. Comprehensive Constitutional Genetic and Epigenetic Characterization of Lynch-Like Individuals. Cancers 2020, 12, 1799. [Google Scholar] [CrossRef]
- Liu, Y.; Aryee, M.J.; Padyukov, L.; Fallin, M.D.; Hesselberg, E.; Runarsson, A.; Reinius, L.; Acevedo, N.; Taub, M.; Ronninger, M.; et al. Epigenome-Wide Association Data Implicate DNA Methylation as an Intermediary of Genetic Risk in Rheumatoid Arthritis. Nat. Biotechnol. 2013, 31, 142–147. [Google Scholar] [CrossRef]
- Ventham, N.T.; Kennedy, N.A.; Adams, A.T.; Kalla, R.; Heath, S.; O’Leary, K.R.; Drummond, H.; IBD BIOM consortium; IBD CHARACTER consortium; Wilson, D.C.; et al. Integrative Epigenome-Wide Analysis Demonstrates That DNA Methylation May Mediate Genetic Risk in Inflammatory Bowel Disease. Nat. Commun. 2016, 7, 13507. [Google Scholar] [CrossRef] [Green Version]
- Demetriou, C.A.; Chen, J.; Polidoro, S.; van Veldhoven, K.; Cuenin, C.; Campanella, G.; Brennan, K.; Clavel-Chapelon, F.; Dossus, L.; Kvaskoff, M.; et al. Methylome Analysis and Epigenetic Changes Associated with Menarcheal Age. PLoS ONE 2013, 8, e79391. [Google Scholar] [CrossRef] [Green Version]
- Cordero, F.; Ferrero, G.; Polidoro, S.; Fiorito, G.; Campanella, G.; Sacerdote, C.; Mattiello, A.; Masala, G.; Agnoli, C.; Frasca, G.; et al. Differentially Methylated microRNAs in Prediagnostic Samples of Subjects Who Developed Breast Cancer in the European Prospective Investigation into Nutrition and Cancer (EPIC-Italy) Cohort. Carcinogenesis 2015, 36, 1144–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arloth, J.; Eraslan, G.; Andlauer, T.F.M.; Martins, J.; Iurato, S.; Kühnel, B.; Waldenberger, M.; Frank, J.; Gold, R.; Hemmer, B.; et al. DeepWAS: Multivariate Genotype-Phenotype Associations by Directly Integrating Regulatory Information Using Deep Learning. PLoS Comput. Biol. 2020, 16, e1007616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriano-Tárraga, C.; Giralt-Steinhauer, E.; Mola-Caminal, M.; Ois, A.; Rodríguez-Campello, A.; Cuadrado-Godia, E.; Fernández-Cadenas, I.; Cullell, N.; Roquer, J.; Jiménez-Conde, J. Biological Age Is a Predictor of Mortality in Ischemic Stroke. Sci. Rep. 2018, 8, 4148. [Google Scholar] [CrossRef]
- Zannas, A.S.; Jia, M.; Hafner, K.; Baumert, J.; Wiechmann, T.; Pape, J.C.; Arloth, J.; Ködel, M.; Martinelli, S.; Roitman, M.; et al. Epigenetic Upregulation of FKBP5 by Aging and Stress Contributes to NF-κB-Driven Inflammation and Cardiovascular Risk. Proc. Natl. Acad. Sci. USA 2019, 116, 11370–11379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilaru, V.; Knight, A.K.; Katrinli, S.; Cobb, D.; Lori, A.; Gillespie, C.F.; Maihofer, A.X.; Nievergelt, C.M.; Dunlop, A.L.; Conneely, K.N.; et al. Critical Evaluation of Copy Number Variant Calling Methods Using DNA Methylation. Genet. Epidemiol. 2020, 44, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Kurushima, Y.; Tsai, P.-C.; Castillo-Fernandez, J.; Couto Alves, A.; El-Sayed Moustafa, J.S.; Le Roy, C.; Spector, T.D.; Ide, M.; Hughes, F.J.; Small, K.S.; et al. Epigenetic Findings in Periodontitis in UK Twins: A Cross-Sectional Study. Clin. Epigenet. 2019, 11, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voisin, S.; Almén, M.S.; Zheleznyakova, G.Y.; Lundberg, L.; Zarei, S.; Castillo, S.; Eriksson, F.E.; Nilsson, E.K.; Blüher, M.; Böttcher, Y.; et al. Many Obesity-Associated SNPs Strongly Associate with DNA Methylation Changes at Proximal Promoters and Enhancers. Genome Med. 2015, 7, 103. [Google Scholar] [CrossRef] [Green Version]
- Subudhi, A.W.; Bourdillon, N.; Bucher, J.; Davis, C.; Elliott, J.E.; Eutermoster, M.; Evero, O.; Fan, J.-L.; Jameson-Van Houten, S.; Julian, C.G.; et al. AltitudeOmics: The Integrative Physiology of Human Acclimatization to Hypobaric Hypoxia and Its Retention upon Reascent. PLoS ONE 2014, 9, e92191. [Google Scholar] [CrossRef]
- Tan, Q.; Frost, M.; Heijmans, B.T.; von Bornemann Hjelmborg, J.; Tobi, E.W.; Christensen, K.; Christiansen, L. Epigenetic Signature of Birth Weight Discordance in Adult Twins. BMC Genom. 2014, 15, 1062. [Google Scholar] [CrossRef] [Green Version]
- Tserel, L.; Kolde, R.; Limbach, M.; Tretyakov, K.; Kasela, S.; Kisand, K.; Saare, M.; Vilo, J.; Metspalu, A.; Milani, L.; et al. Age-Related Profiling of DNA Methylation in CD8+ T Cells Reveals Changes in Immune Response and Transcriptional Regulator Genes. Sci. Rep. 2015, 5, 13107. [Google Scholar] [CrossRef] [Green Version]
- Dabin, L.C.; Guntoro, F.; Campbell, T.; Bélicard, T.; Smith, A.R.; Smith, R.G.; Raybould, R.; Schott, J.M.; Lunnon, K.; Sarkies, P.; et al. Altered DNA Methylation Profiles in Blood from Patients with Sporadic Creutzfeldt-Jakob Disease. Acta Neuropathol. 2020, 140, 863–879. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.A.; Goodman, S.J.; MacIsaac, J.L.; Obradović, J.; Barr, R.G.; Boyce, W.T.; Kobor, M.S. Integration of DNA Methylation Patterns and Genetic Variation in Human Pediatric Tissues Help Inform EWAS Design and Interpretation. Epigenet. Chromatin 2019, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Kok, D.E.G.; Dhonukshe-Rutten, R.A.M.; Lute, C.; Heil, S.G.; Uitterlinden, A.G.; van der Velde, N.; van Meurs, J.B.J.; van Schoor, N.M.; Hooiveld, G.J.E.J.; de Groot, L.C.P.G.M.; et al. The Effects of Long-Term Daily Folic Acid and Vitamin B12 Supplementation on Genome-Wide DNA Methylation in Elderly Subjects. Clin. Epigenet. 2015, 7, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marabita, F.; Almgren, M.; Lindholm, M.E.; Ruhrmann, S.; Fagerström-Billai, F.; Jagodic, M.; Sundberg, C.J.; Ekström, T.J.; Teschendorff, A.E.; Tegnér, J.; et al. An Evaluation of Analysis Pipelines for DNA Methylation Profiling Using the Illumina HumanMethylation450 BeadChip Platform. Epigenetics 2013, 8, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Ovejero-Benito, M.C.; Cabaleiro, T.; Sanz-García, A.; Llamas-Velasco, M.; Saiz-Rodríguez, M.; Prieto-Pérez, R.; Talegón, M.; Román, M.; Ochoa, D.; Reolid, A.; et al. Epigenetic Biomarkers Associated with Antitumour Necrosis Factor Drug Response in Moderate-to-Severe Psoriasis. Br. J. Dermatol. 2018, 178, 798–800. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Scrucca, L.; Fop, M.; Murphy, T.B.; Raftery, A.E. Mclust 5: Clustering, Classification and Density Estimation Using Gaussian Finite Mixture Models. R J. 2016, 8, 289–317. [Google Scholar] [CrossRef] [Green Version]
- Street, K.; Risso, D.; Fletcher, R.B.; Das, D.; Ngai, J.; Yosef, N.; Purdom, E.; Dudoit, S. Slingshot: Cell Lineage and Pseudotime Inference for Single-Cell Transcriptomics. BMC Genom. 2018, 19, 477. [Google Scholar] [CrossRef] [Green Version]
- Kluyver, T.; Ragan-Kelley, B.; Pérez, F.; Granger, B.; Bussonnier, M.; Frederic, J.; Kelley, K.; Hamrick, J.; Grout, J.; Corlay, S.; et al. Jupyter Notebooks—A Publishing Format for Reproducible Computational Workflows. In Proceedings of the Positioning and Power in Academic Publishing: Players, Agents and Agendas, Göttingen, Germany, 7–9 June 2016; Loizides, F., Scmidt, B., Eds.; IOS Press: Amsterdam, The Netherlands, 2016; pp. 87–90. [Google Scholar]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array Programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- McKinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference SciPy, Austin, TX, USA, 28 June–3 July 2010. [Google Scholar]
- Varoquaux, G.; Grisel, O. Joblib: Running Python Function as Pipeline Jobs; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- R Studio Team. R Studio: Integrated Development for R; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Neuwirth, E. R Color Brewer: Color Brewer Palettes; R Foundation for Statistical Computing: Vienna, Austria, 2014. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009; ISBN 9780387981406. [Google Scholar]
- Garnier, S.; Ross, N.; Rudis, B.; Sciaini, M.; Camargo, P.A.; Scherer, C. Viridis—Colorblind-Friendly Color Maps for R; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Mayer, M. splitTools: Tools for Data Splitting; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Murdoch, D.; Nash, J.C. nlsr: Functions for Nonlinear Least Squares Solutions; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Gompertz, B., XXIV. On the Nature of the Function Expressive of the Law of Human Mortality, and on a New Mode of Determining the Value of Life Contingencies. In A Letter to Francis Baily, Esq. F.R S. &c. Philos. Trans. R. Soc. Lond. 1825, 115, 513–583. [Google Scholar]
- Sacher, G.A. Handbook of the Biology of Aging; Finch, C.E., Hayflick, L., Eds.; Van Nostrand Reinhold Company: New York, NY, USA, 1977; p. 612. [Google Scholar]
- Kirkwood, T.B.L. Deciphering Death: A Commentary on Gompertz (1825) “On the Nature of the Function Expressive of the Law of Human Mortality, and on a New Mode of Determining the Value of Life Contingencies”. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rushton, J.P.; Ankney, C.D. Whole brain size and general mental ability: A review. Int. J. Neurosci. 2009, 119, 691–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functions with Estimated Coefficients | AIC | RMSE | |
|---|---|---|---|
| 18,258.27 | 1.773 | 0.671 | |
| 17,991.60 | 1.722 | 0.690 | |
| 17,896.36 | 1.704 | 0.697 | |
| 17,913.38 | 1.708 | 0.695 | |
| 17,826.79 | 1.690 | 0.700 |
| Functions with Estimated Coefficients | AIC | RMSE | |
|---|---|---|---|
| 1824.60 | 1.696 | 0.953 | |
| 2422.50 | 3.217 | 0.831 | |
| 2446.07 | 3.292 | 0.823 | |
| 2007.40 | 2.062 | 0.931 | |
| 1555.68 | 1.266 | 0.974 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lapborisuth, K.; Farrell, C.; Pellegrini, M. Pseudotime Analysis Reveals Exponential Trends in DNA Methylation Aging with Mortality Associated Timescales. Cells 2022, 11, 767. https://doi.org/10.3390/cells11050767
Lapborisuth K, Farrell C, Pellegrini M. Pseudotime Analysis Reveals Exponential Trends in DNA Methylation Aging with Mortality Associated Timescales. Cells. 2022; 11(5):767. https://doi.org/10.3390/cells11050767
Chicago/Turabian StyleLapborisuth, Kalsuda, Colin Farrell, and Matteo Pellegrini. 2022. "Pseudotime Analysis Reveals Exponential Trends in DNA Methylation Aging with Mortality Associated Timescales" Cells 11, no. 5: 767. https://doi.org/10.3390/cells11050767
APA StyleLapborisuth, K., Farrell, C., & Pellegrini, M. (2022). Pseudotime Analysis Reveals Exponential Trends in DNA Methylation Aging with Mortality Associated Timescales. Cells, 11(5), 767. https://doi.org/10.3390/cells11050767