
Effects of the Mutant TP53 Reactivator APR-246 on Therapeutic Sensitivity of Pancreatic Cancer Cells in the Presence and Absence of WT-TP53

 ,
,  , ,
, ,  ,
,  , ,
, ,  and
and
Abstract
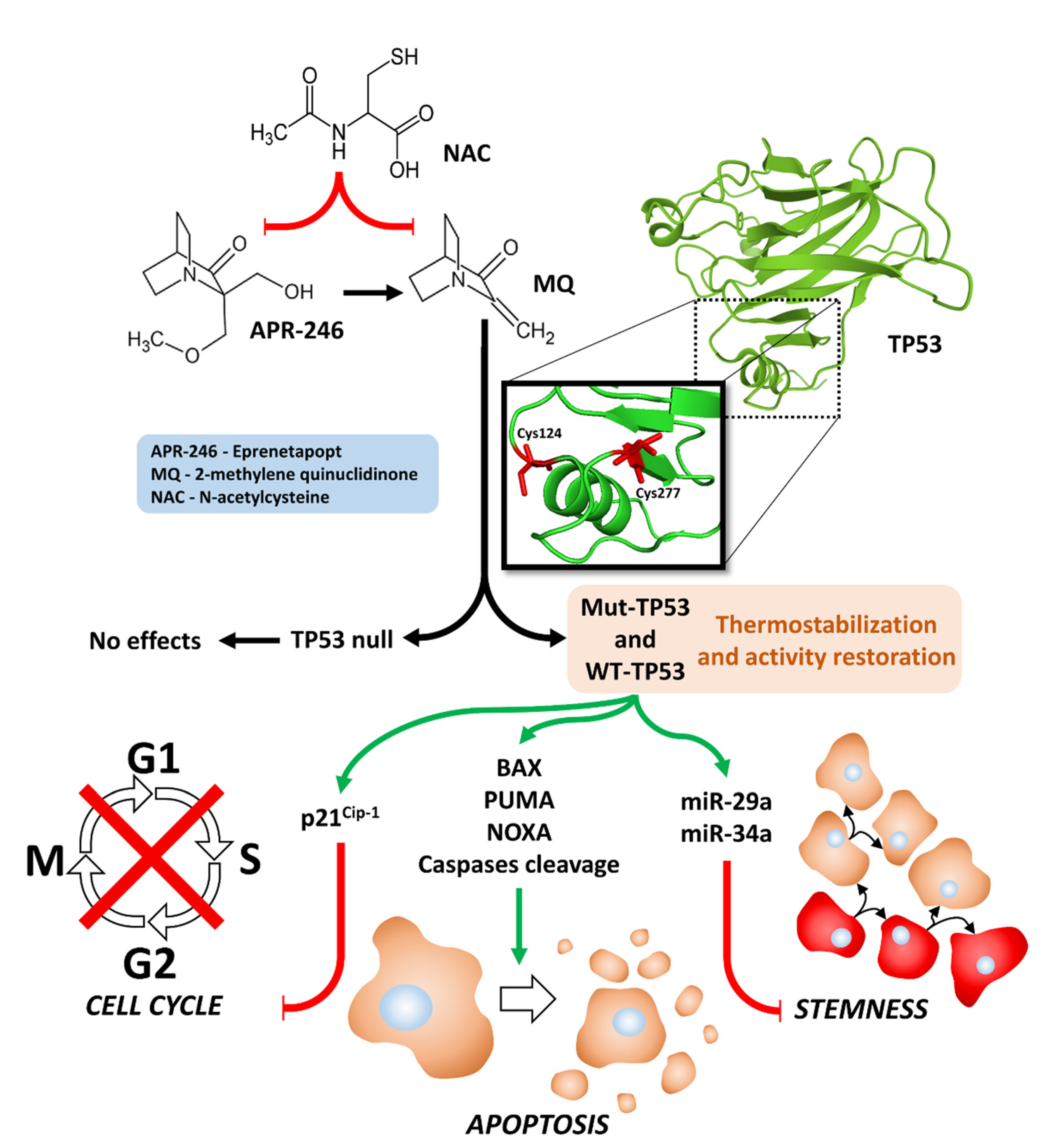
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture
2.2. Chemotherapeutic Drugs and Small Molecule Signal Transduction Inhibitors
2.3. Introduction of Either WT-TP53 or a Control Plasmid into MIA-PaCa-2 and PANC-28 Cells
2.4. Cell Proliferation Assays in the Presence of Chemotherapeutic Drugs and Signal Transduction Inhibitors
2.5. Clonogenicity Assays
3. Results
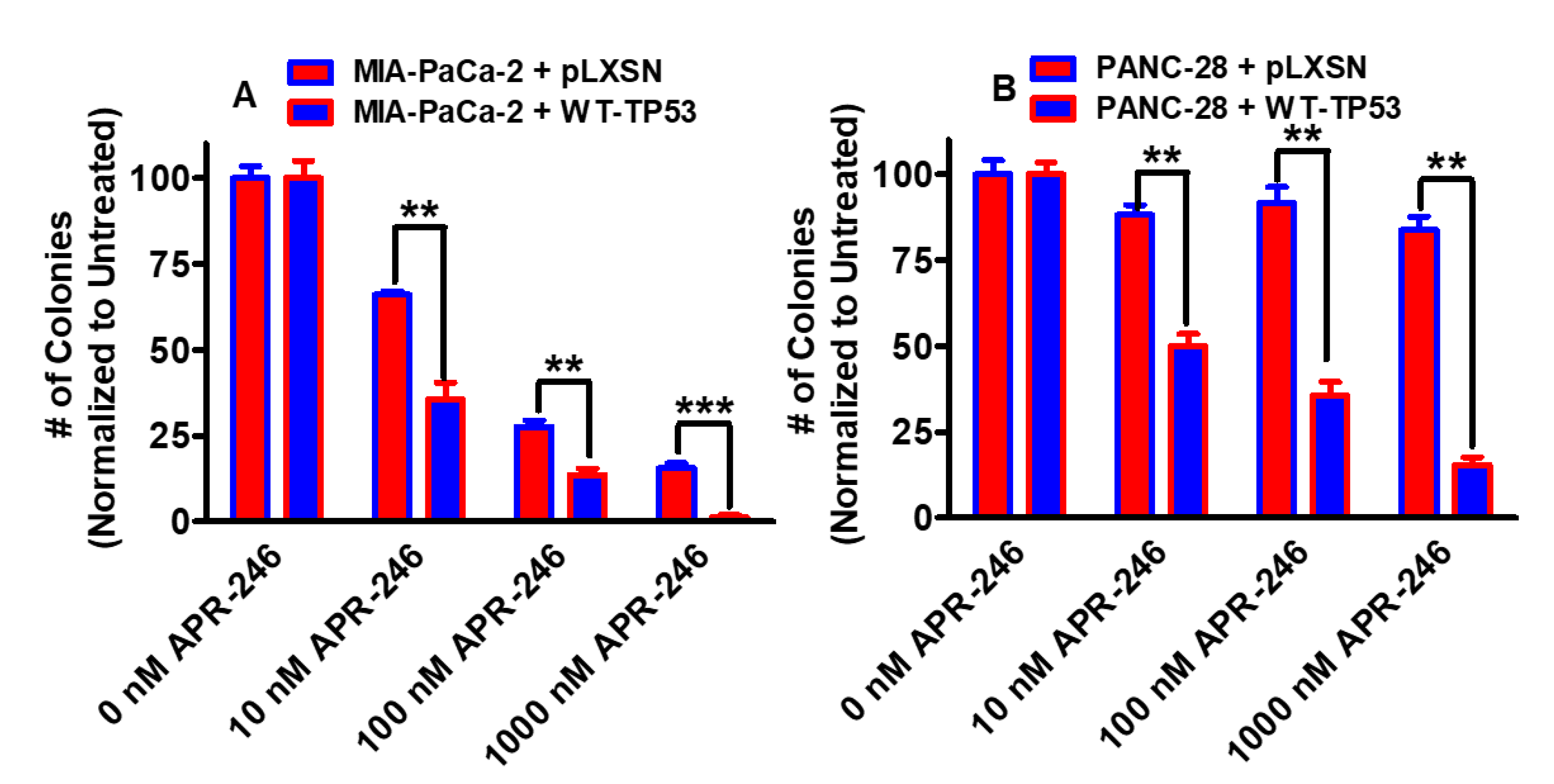
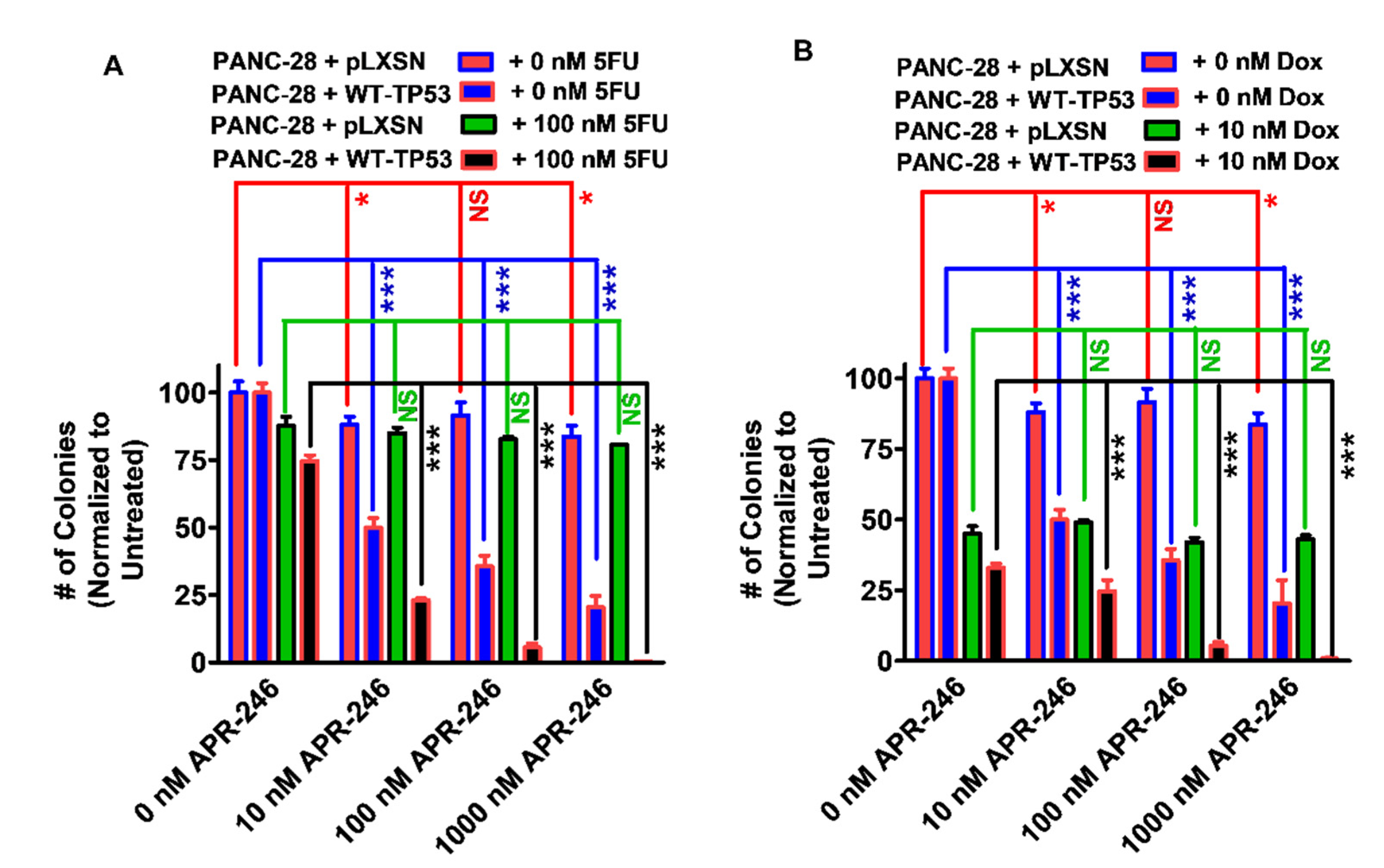
3.1. Effects of APR-246 on Clonogenicity of MIA-PaCa-2 and PANC-28 Cells Containing and Lacking WT-TP53
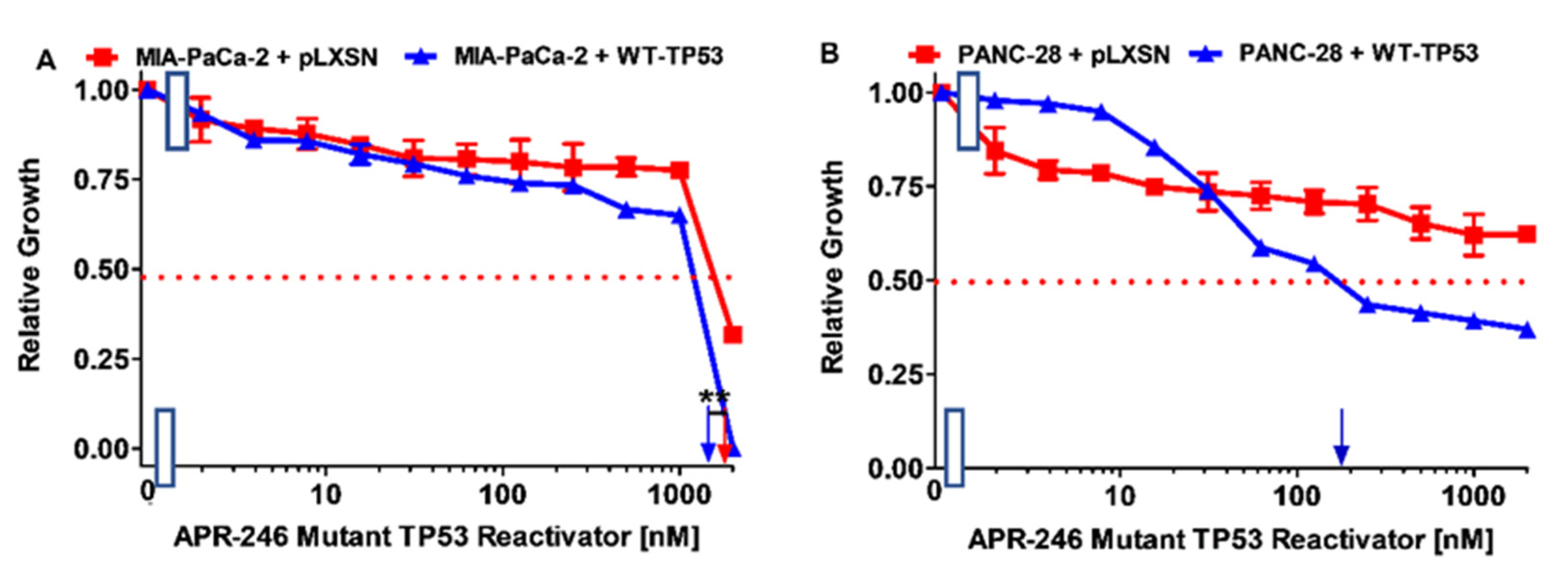
3.2. Effects of APR-246 on Cell Growth in MIA-PaCa-2 and PANC-28 Cells Containing and Lacking WT-TP53
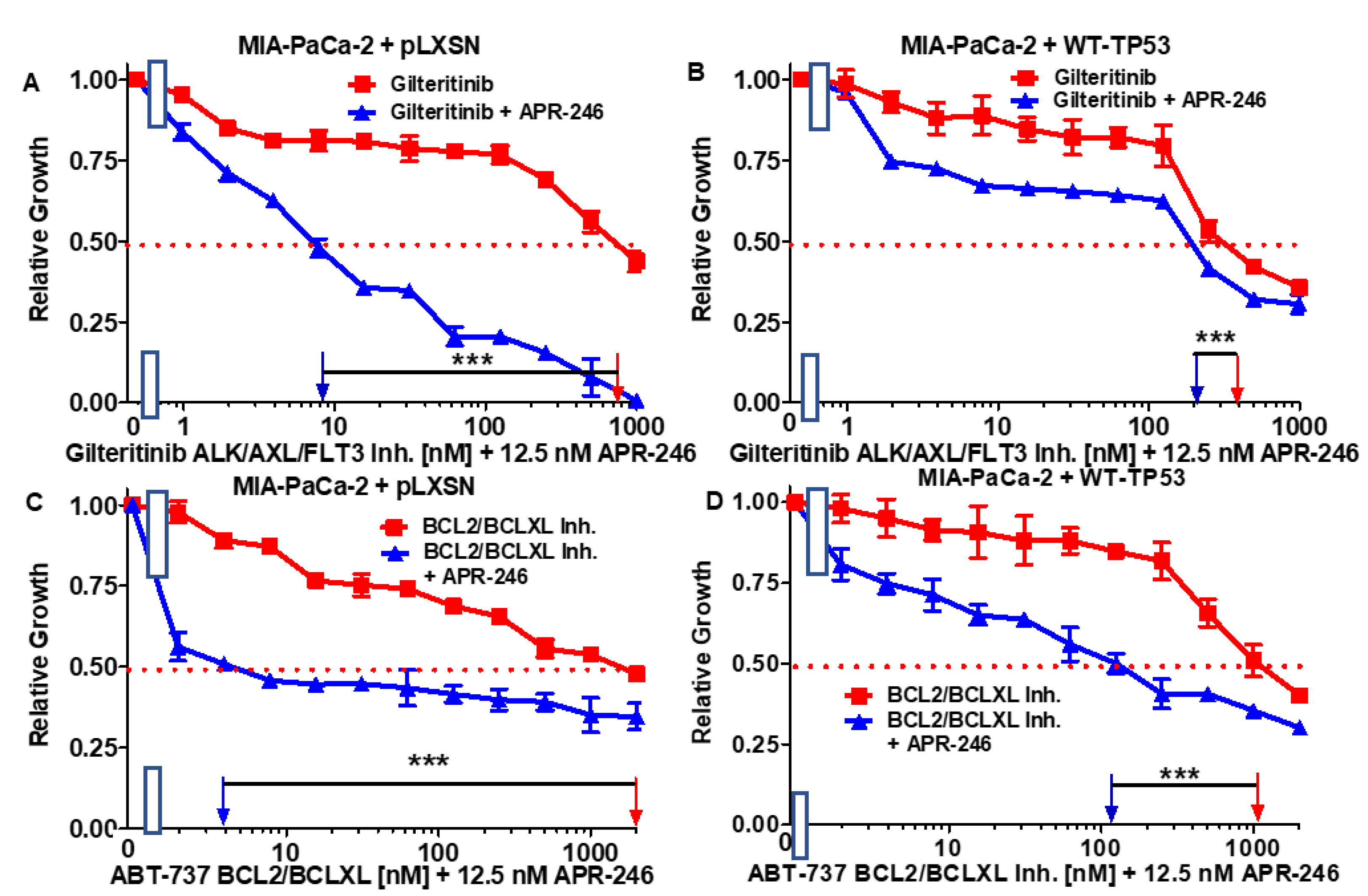
3.3. Abilities of a Low Dose of APR-246 to Decrease the IC50 Values of Chemotherapeutic Drugs and Signal Transduction Inhibitors of MIA-PaCa-2 Cells Containing and Lacking WT-TP53
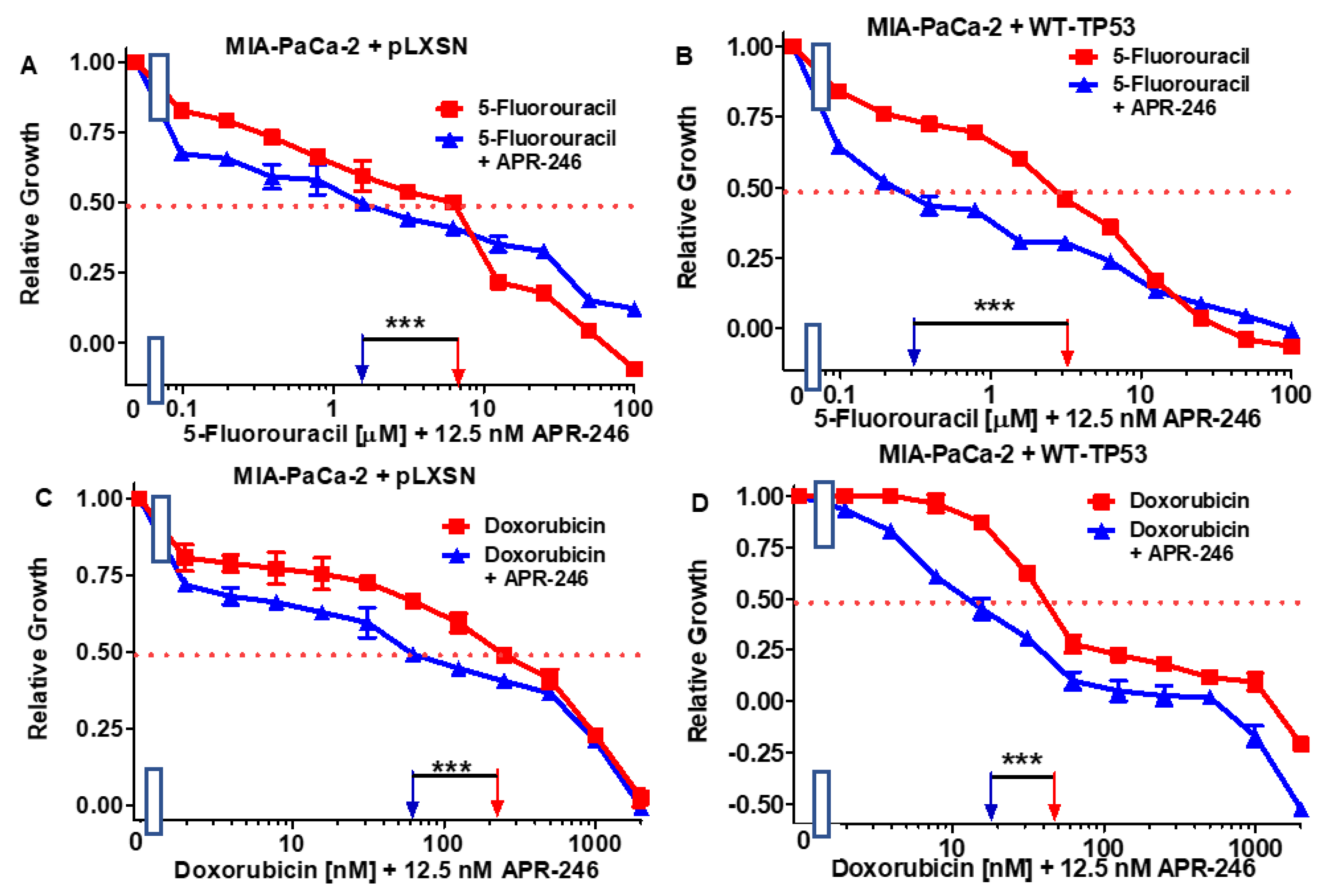
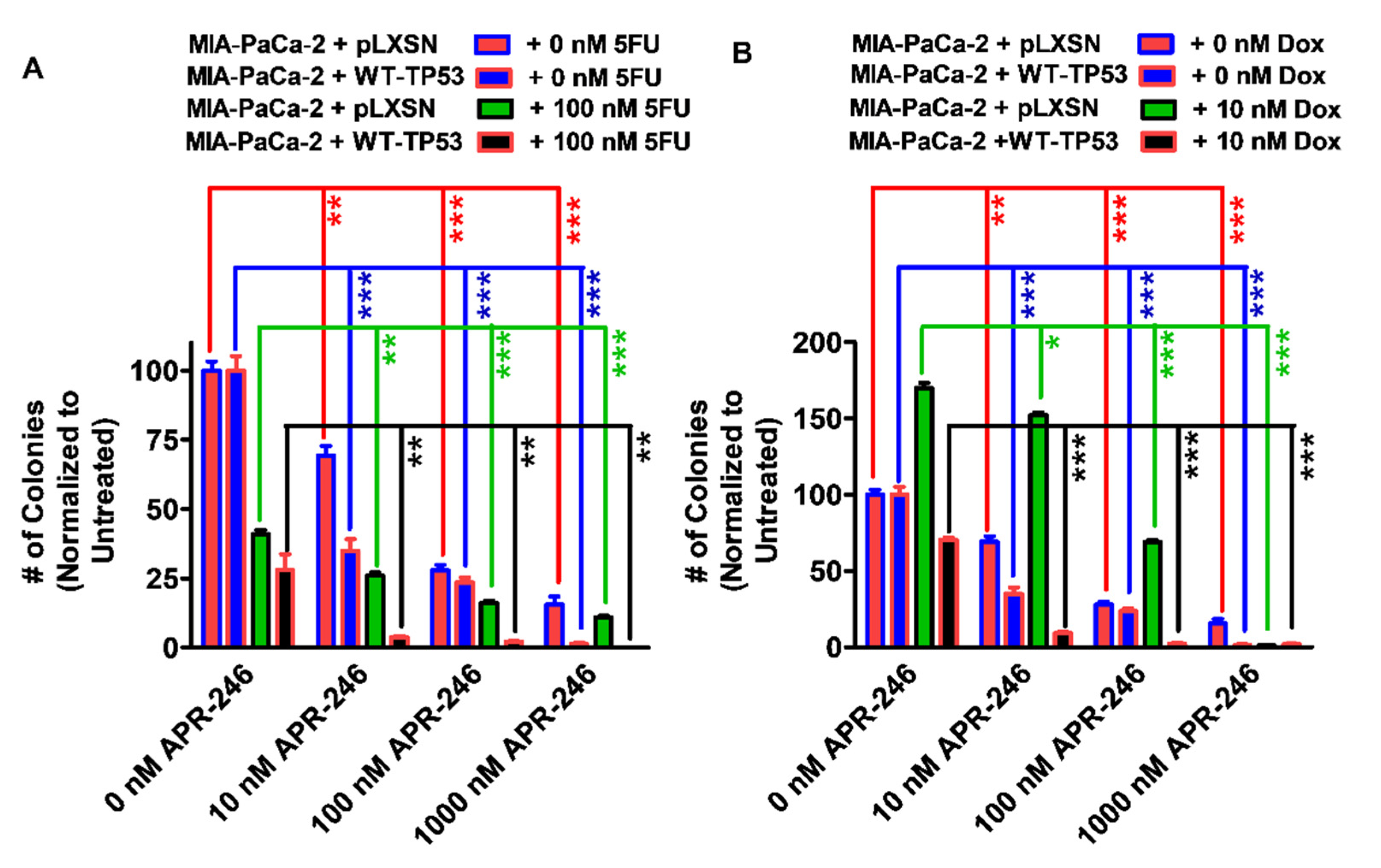
3.4. Abilities of Low Doses of 5FU or Doxorubicin to Increase the Cytotoxicity of APR-246 and Decrease Clonogenicity of MIA-PaCa-2 Cells Containing and Lacking WT-TP53
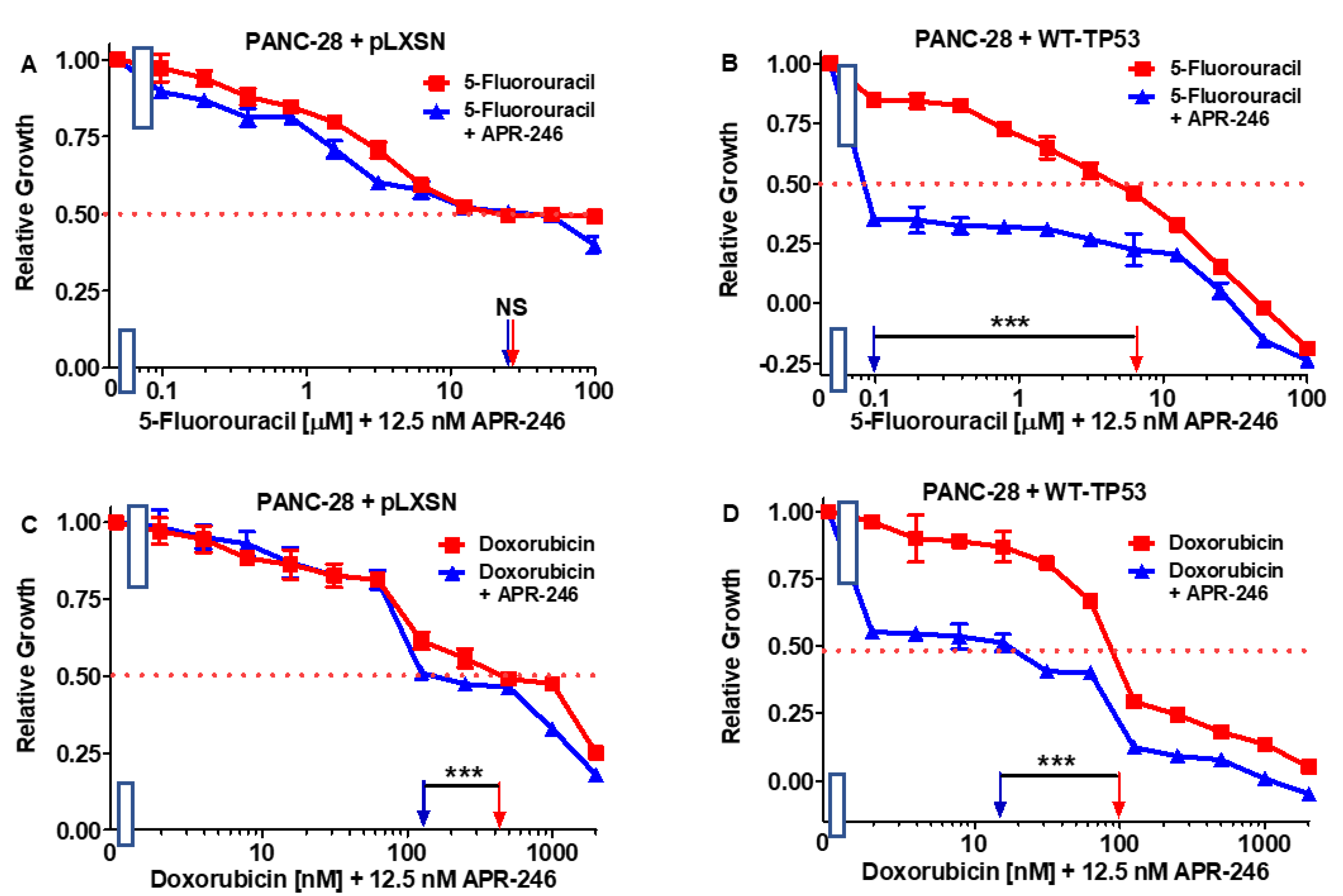
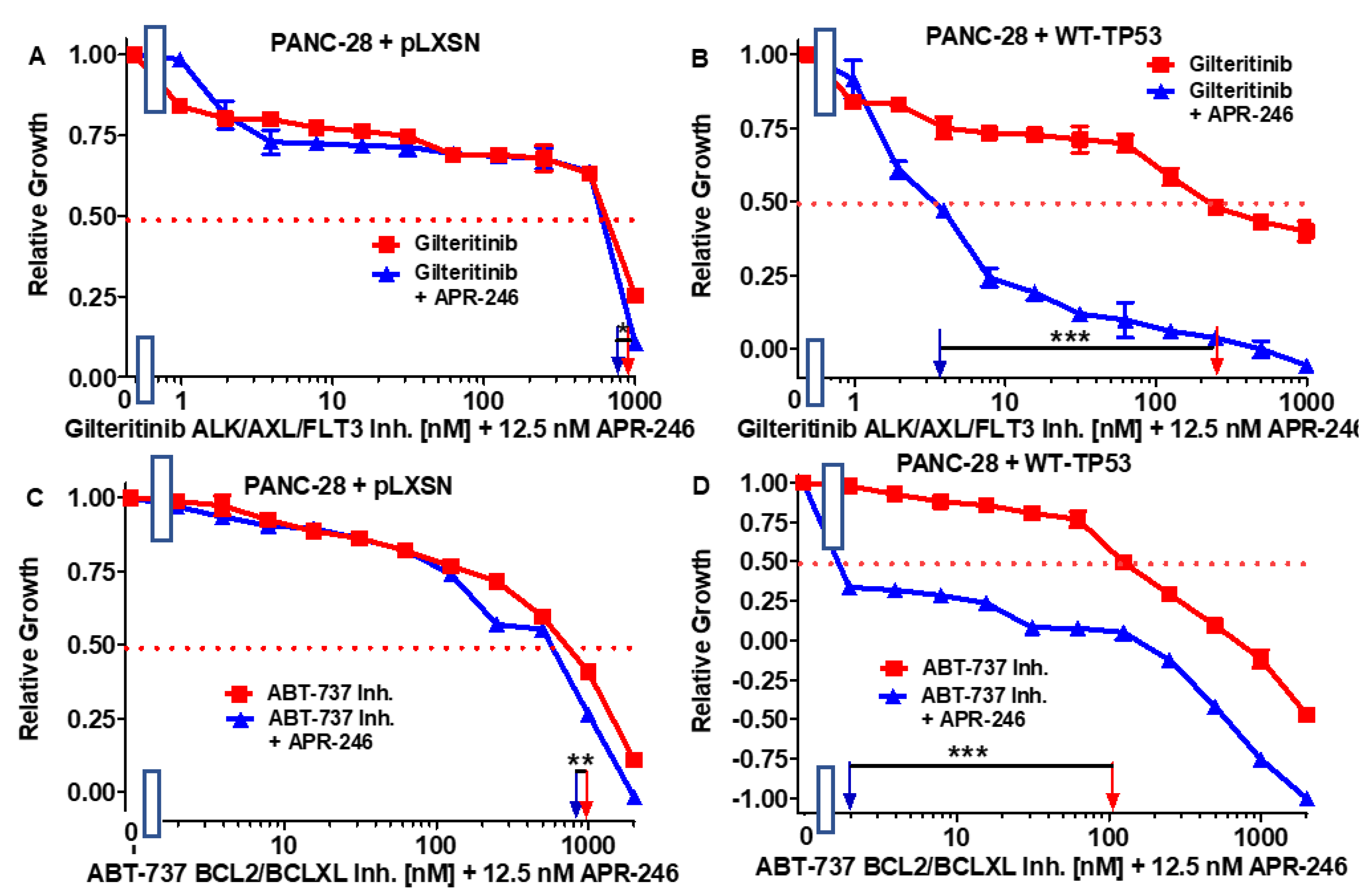
3.5. Abilities of a Low Dose of APR-246 to Decrease the IC50 Values of Chemotherapeutic Drugs and Signal Transduction Inhibitors of PANC-28 Cells Containing and Lacking WT-TP53
3.6. Abilities of a Low Dose of Either 5FU or Doxorubicin to Increase the Cytotoxicity of APR-246 in PANC-28 Cells Containing and Lacking WT-TP53
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Muniraj, T.; Jamidar, P.A.; Aslanian, H.R. Pancreatic cancer: A comprehensive review and update. Dis.-A-Mon. DM. 2013, 59, 368–402. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kommalapati, A.; Tella, S.H.; Goyal, G.; Ma, W.W.; Mahipal, A. Contemporary management of localized resectable pancreatic cancer. Cancers 2018, 10, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruarus, A.; Vroomen, L.; Puijk, R.; Scheffer, H.; Meijerink, M. Locally advanced pancreatic cancer: A review of local ablative therapies. Cancers 2018, 10, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, P.C.; Frey, M.C.; Ruzza, C.M.; Nickel, F.; Jost, C.; Gwerder, C.; Hackert, T.; Z’graggen, K.; Kessler, U. Neoadjuvant chemotherapy in pancreatic cancer: An appraisal of the current high-level evidence. Pharmacology 2021, 106, 143–153. [Google Scholar] [CrossRef]
- Pu, N.; Chen, Q.; Gao, S.; Liu, G.; Zhu, Y.; Yin, L.; Hu, H.; Wei, L.; Wu, Y.; Maeda, S.; et al. Genetic landscape of prognostic value in pancreatic ductal adenocarcinoma microenvironment. Ann. Trans. Med. 2019, 7, 645. [Google Scholar] [CrossRef]
- Qian, Y.; Gong, Y.; Fan, Z.; Luo, G.; Huang, Q.; Deng, S.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. J. Hemat. Oncol. 2020, 13, 130. [Google Scholar] [CrossRef]
- Waters, A.M.; Der, C.J. KRAS: The critical driver and therapeutic target for pancreatic cancer. Cold Spring Harb. Pre. Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- Grant, T.J.; Hua, K.; Singh, A. Molecular pathogenesis of pancreatic cancer. Prog. Mole. Biol. Trans. Sci. 2016, 144, 241–275. [Google Scholar]
- Escobar-Hoyos, L.F.; Penson, A.; Kannan, R.; Cho, H.; Pan, C.H.; Singh, R.K.; Apken, L.H.; Hobbs, G.A.; Luo, R.; Lecomte, N.; et al. Altered RNA splicing by mutant p53 activates oncogenic RAS signaling in pancreatic cancer. Cancer Cell 2020, 38, 198–211. [Google Scholar] [CrossRef]
- Bykov, V.J.; Issaeva, N.; Selivanova, G.; Wiman, K.G. Mutant p53-dependent growth suppression distinguishes PRIMA-1 from known anticancer drugs: A statistical analysis of information in the National Cancer Institute database. Carcinogenesis 2002, 23, 2011–2018. [Google Scholar] [CrossRef] [Green Version]
- Zache, N.; Lambert, J.M.; Wiman, K.G.; Bykov, V.J. PRIMA-1MET inhibits growth of mouse tumors carrying mutant p53. Cell. Oncol. 2008, 30, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Zandi, R.; Selivanova, G.; Christensen, C.L.; Gerds, T.A.; Willumsen, B.M.; Poulsen, H.S. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin. Cancer Res. 2011, 17, 2830–2841. [Google Scholar] [CrossRef] [Green Version]
- Fujihara, K.M.; Corrales Benitez, M.; Cabalag, C.S.; Zhang, B.Z.; Ko, H.S.; Liu, D.S.; Simpson, K.J.; Haupt, Y.; Lipton, L.; Haupt, S.; et al. SLC7A11 Is a Superior Determinant of APR-246 (Eprenetapopt) Response than TP53 Mutation Status. Mol. Cancer Ther. 2021, 20, 1858–1867. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef] [PubMed]
- Perdrix, A.; Najem, A.; Saussez, S.; Awada, A.; Journe, F.; Ghanem, G.; Krayem, M. PRIMA-1 and PRIMA-1Met (APR-246): From mutant/wild type p53 reactivation to unexpected mechanisms underlying their potent anti-tumor effect in combinatorial therapies. Cancers 2017, 9, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, Z. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018, 9, 439. [Google Scholar] [CrossRef] [Green Version]
- Rökaeus, N.; Shen, J.; Eckhardt, I.; Bykov, V.J.; Wiman, K.G.; Wilhelm, M.T. PRIMA-1(MET)/APR-246 targets mutant forms of p53 family members p63 and p73. Oncogene 2010, 29, 6442–6451. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.J.; Zhang, Q.; Zhang, M.; Ceder, S.; Abrahmsen, L.; Wiman, K.G. Targeting of mutant p53 and the cellular redox balance by APR-246 as a strategy for efficient cancer therapy. Front. Oncol. 2016, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Synnott, N.C.; Madden, S.F.; Bykov, V.; Crown, J.; Wiman, K.G.; Duffy, M.J. The mutant p53-targeting compound APR-246 induces ROS-modulating genes in breast cancer cells. Trans. Oncol. 2018, 11, 1343–1349. [Google Scholar] [CrossRef]
- Yin, Z.X.; Hang, W.; Liu, G.; Wang, Y.S.; Shen, X.F.; Sun, Q.H.; Li, D.D.; Jian, Y.P.; Zhang, Y.H.; Quan, C.S.; et al. PARP-1 inhibitors sensitize HNSCC cells to APR-246 by inactivation of thioredoxin reductase 1 (TrxR1) and promotion of ROS accumulation. Oncotarget 2018, 9, 1885–1897. [Google Scholar] [CrossRef] [Green Version]
- Haffo, L.; Lu, J.; Bykov, V.; Martin, S.S.; Ren, X.; Coppo, L.; Wiman, K.G.; Holmgren, A. Inhibition of the glutaredoxin and thioredoxin systems and ribonucleotide reductase by mutant p53-targeting compound APR-246. Sci. Rep. 2018, 8, 12671. [Google Scholar] [CrossRef] [PubMed]
- Deer, E.L.; González-Hernández, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klemke, L.; Fehlau, C.F.; Winkler, N.; Toboll, F.; Singh, S.K.; Moll, U.M.; Schulz-Heddergott, R. The gain-of-function p53 R248W mutant promotes migration by STAT3 deregulation in human pancreatic cancer cells. Front. Oncol. 2021, 11, 642603. [Google Scholar] [CrossRef] [PubMed]
- Frazier, M.L.; Fernández, E.; de Llorens, R.; Brown, N.M.; Pathak, S.; Cleary, K.R.; Abbruzzese, J.L.; Berry, K.; Olive, M.; Le Maistre, A.; et al. Pancreatic adenocarcinoma cell line, MDAPanc-28, with features of both acinar and ductal cells. Int. J. Pancreatol. 1996, 19, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Abbruzzese, J.L.; Izzo, J.; Hittelman, W.N.; Li, D. AURKA amplification, chromosome instability, and centrosome abnormality in human pancreatic carcinoma cells. Cancer Genet. Cytogenet. 2005, 159, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Eliyahu, D.; Michalovitz, D.; Eliyahu, S.; Pinhasi-Kimhi, O.; Oren, M. Wild-type p53 can inhibit oncogene-mediated focus formation. Proc. Natl. Acad. Sci. USA 1989, 86, 8763–9767. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; McCubrey, J.A.; Jefferson, H.S.; Paine, M.S.; Chappell, W.H.; Terrian, D.M. A dominant role for p53-dependent cellular senescence in radiosensitization of human prostate cancer cells. Cell Cycle (Georget. Tex.) 2007, 6, 595–605. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.D.; Rosman, G.J. Improved retroviral vectors for gene transfer and expression. Biotechniques 1989, 7, 980–982. [Google Scholar]
- Abrams, S.L.; Lertpiriyapong, K.; Yang, L.V.; Martelli, A.M.; Cocco, L.; Ratti, S.; Falasca, M.; Murata, R.M.; Rosalen, P.L.; Lombardi, P.; et al. Introduction of WT-TP53 into pancreatic cancer cells alters sensitivity to chemotherapeutic drugs, targeted therapeutics and nutraceuticals. Adv. Biol. Regul. 2018, 69, 16–34. [Google Scholar] [CrossRef]
- Abrams, S.L.; Akula, S.M.; Martelli, A.M.; Cocco, L.; Ratti, S.; Libra, M.; Candido, S.; Montalto, G.; Cervello, M.; Gizak, A.; et al. Sensitivity of pancreatic cancer cells to chemotherapeutic drugs, signal transduction inhibitors and nutraceuticals can be regulated by WT-TP53. Adv. Biol. Regul. 2021, 79, 100780. [Google Scholar] [CrossRef] [PubMed]
- Abrams, S.L.; Akula, S.M.; Meher, A.K.; Steelman, L.S.; Gizak, A.; Duda, P.; Rakus, D.; Martelli, A.M.; Ratti, S.; Cocco, L.; et al. GSK-3β can regulate the sensitivity of MIA-PaCa-2 pancreatic and MCF-7 breast cancer cells to chemotherapeutic drugs, targeted therapeutics and nutraceuticals. Cells 2021, 10, 816. [Google Scholar] [CrossRef] [PubMed]
- Sokolosky, M.; Chappell, W.H.; Stadelman, K.; Abrams, S.L.; Davis, N.M.; Steelman, L.S.; McCubrey, J.A. Inhibition of GSK-3β activity can result in drug and hormonal resistance and alter sensitivity to targeted therapy in MCF-7 breast cancer cells. Cell Cycle 2014, 13, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Kitago, M.; Aiura, K.; Shinoda, M.; Yagi, H.; Abe, Y.; Oshima, G.; Hori, S.; Nakano, Y.; Itano, O.; et al. Efficacy and safety of preoperative 5-fluorouracil, cisplatin, and mitomycin C in combination with radiotherapy in patients with resectable and borderline resectable pancreatic cancer: A long-term follow-up study. World J. Sur. Oncol. 2019, 17, 145. [Google Scholar] [CrossRef] [PubMed]
- Syrigos, K.N.; Michalaki, B.; Alevyzaki, F.; Machairas, A.; Mandrekas, D.; Kindilidis, K.; Karatzas, G. A phase-II study of liposomal doxorubicin and docetaxel in patients with advanced pancreatic cancer. Anticancer Res. 2002, 22, 3583–3588. [Google Scholar]
- Lee, L.Y.; Hernandez, D.; Rajkhowa, T.; Smith, S.C.; Raman, J.R.; Nguyen, B.; Small, D.; Levis, M. Preclinical studies of gilteritinib, a next-generation FLT3 inhibitor. Blood 2017, 129, 257–260. [Google Scholar] [CrossRef] [Green Version]
- Pedro, J.M.; Wei, Y.; Sica, V.; Maiuri, M.C.; Zou, Z.; Kroemer, G.; Levine, B. BAX and BAK1 are dispensable for ABT-737-induced dissociation of the BCL2-BECN1 complex and autophagy. Autophagy 2015, 11, 452–459. [Google Scholar] [CrossRef] [Green Version]
- McCubrey, J.A.; Abrams, S.L.; Ligresti, G.; Misaghian, N.; Wong, E.T.; Basecke, J.; Troppmair, J.; Libra, N.; Nicoletti, F.; Molton, S.; et al. Involvement of p53 and Raf/MEK/ERK pathways in hematopoietic drug resistance. Leukemia 2008, 22, 2080–2090. [Google Scholar] [CrossRef] [Green Version]
- Steelman, L.S.; Navolanic, P.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.T.; Martelli, A.M.; Cocco, L.; Stivala, F.; Libra, M.; Nicoletti, F.; et al. Involvement of Akt and mTOR in chemotherapeutic- and hormonal-based drug resistance and response to radiation in breast cancer cells. Cell Cycle 2011, 10, 3003–3015. [Google Scholar] [CrossRef]
- Chappell, W.H.; Candid, S.; Abrams, S.L.; Akula, S.M.; Steelman, L.S.; Martelli, A.M.; Ratti, S.; Cocco, L.; Cervello, M.; Montalto, G.; et al. Influences of TP53 and the anti-aging DDR1 receptor in controlling Raf/MEK/ERK and PI3K/Akt expression and chemotherapeutic drug sensitivity in prostate cancer cell lines. Aging (Albany NY) 2020, 12, 10194–10210. [Google Scholar] [CrossRef]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P., 4th; Wagenblast, E.; Davis, C.A.; Moon, S.H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor β signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddalena, M.; Mallel, G.; Nataraj, N.B.; Shreberk-Shaked, M.; Hassin, O.; Mukherjee, S.; Arandkar, S.; Rotkopf, R.; Kapsack, A.; Lambiase, G.; et al. TP53 missense mutations in PDAC are associated with enhanced fibrosis and an immunosuppressive microenvironment. Proc. Nat. Acad. Sci. USA 2021, 118, e2025631118. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Furukawa, S.; Hashimoto, A.; Tsutaho, A.; Fukao, A.; Sakamura, Y.; Parajuli, G.; Onodera, Y.; Otsuka, Y.; Handa, H.; et al. ARF6 and AMAP1 are major targets of KRAS and TP53 mutations to promote invasion, PD-L1 dynamics, and immune evasion of pancreatic cancer. Proc. Nat. Acad. Sci. USA 2019, 116, 17450–17459. [Google Scholar] [CrossRef] [Green Version]
- Sabe, H.; Hashimoto, S.; Morishige, M.; Ogawa, E.; Hashimoto, A.; Nam, J.M.; Miura, K.; Yano, H.; Onodera, Y. The EGFR-GEP100-Arf6-AMAP1 signaling pathway specific to breast cancer invasion and metastasis. Traffic (Cph. Den.) 2009, 10, 982–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, J.; Dong, K.; Lin, F.; Long, M.; Ouyang, Y.; Wei, J.; Chen, X.; Weng, Y.; He, T.; et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell. Signal 2015, 27, 443–452. [Google Scholar] [CrossRef]
- Akula, S.M.; Ruvolo, P.P.; McCubrey, J.A. TP53/miR-34a-associated signaling targets SERPINE1 expression in human pancreatic cancer. Aging 2020, 12, 2777–2797. [Google Scholar] [CrossRef]
- Du, Y.; Liu, Z.; You, L.; Hou, P.; Ren, X.; Jiao, T.; Zhao, W.; Li, Z.; Shu, H.; Liu, C.; et al. Pancreatic cancer progression relies upon mutant p53-induced oncogenic signaling mediated by NOP14. Cancer Res. 2017, 77, 2661–2673. [Google Scholar] [CrossRef] [Green Version]
- Meirelles, K.; Benedict, L.A.; Dombkowski, D.; Pepin, D.; Preffer, F.I.; Teixeira, J.; Tanwar, P.S.; Young, R.H.; MacLaughlin, D.T.; Donahoe, P.K.; et al. Human ovarian cancer stem/progenitor cells are stimulated by doxorubicin but inhibited by Mullerian inhibiting substance. Proc. Natl. Acad. Sci. USA 2012, 109, 2358–2363. [Google Scholar] [CrossRef] [Green Version]
- Cole, A.; Dwight, T.; Gill, A.; Dickson, K.A.; Zhu, Y.; Clarkson, A.; Gard, G.B.; Maidens, J.; Valmadre, S.; Clifton-Bligh, R.; et al. Assessing mutant p53 in primary high-grade serous ovarian cancer using immunohistochemistry and massively parallel sequencing. Sci. Rep. 2016, 6, 26191. [Google Scholar] [CrossRef]
- Macleod, K.F.; Sherry, N.; Hannon, G.; Beach, D.; Tokino, T.; Kinzler, K.; Vogelstein, B.; Jacks, T. p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes Devel. 1995, 9, 935–944. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.H.; Lee, W.H.; Park, K.-Y.; Zhang, L. p53-independent Induction of p21 (WAF1/CIP1), Reduction of cyclin B1 and G2/ M arrest by the isoflavone genistein in human prostate carcinoma cells. Jpn. J. Cancer Res. 2000, 91, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Emerling, B.M.; Hurov, J.B.; Poulogiannis, G.; Tsukazawa, K.S.; Choo-Wing, R.; Wulf, G.M.; Bell, E.L.; Shim, H.S.; Lamia, K.A.; Rameh, L.E.; et al. Depletion of a putatively druggable class of phosphatidylinositol kinases inhibits growth of p53-null tumors. Cell 2013, 155, 844–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.S.; Read, M.; Cullinane, C.; Azar, W.J.; Fennell, C.M.; Montgomery, K.G.; Haupt, S.; Haupt, Y.; Wiman, K.G.; Duong, C.P.; et al. APR-246 potently inhibits tumour growth and overcomes chemoresistance in preclinical models of oesophageal adenocarcinoma. Gut 2015, 64, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Mohell, N.; Alfredsson, J.; Fransson, Å.; Uustalu, M.; Byström, S.; Gullbo, J.; Hallberg, A.; Bykov, V.J.; Björklund, U.; Wiman, K.G. APR-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis. 2015, 6, e1794. [Google Scholar] [CrossRef] [Green Version]
- Fransson, Å.; Glaessgen, D.; Alfredsson, J.; Wiman, K.G.; Bajalica-Lagercrantz, S.; Mohell, N. Strong synergy with APR-246 and DNA-damaging drugs in primary cancer cells from patients with TP53 mutant High-Grade Serous ovarian cancer. J. Ovar. Res. 2016, 9, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanchina, M.; Soong, D.; Zheng-Lin, B.; Watts, J.M.; Taylor, J. Advances in acute myeloid leukemia: Recently approved therapies and drugs in development. Cancers 2020, 12, 3225. [Google Scholar] [CrossRef]
- Bold, R.J.; Virudachalam, S.; McConkey, D.J. BCL2 expression correlates with metastatic potential in pancreatic cancer cell lines. Cancer 2001, 92, 1122–1129. [Google Scholar] [CrossRef]
- Menichini, P.; Monti, P.; Speciale, A.; Cutrona, G.; Matis, S.; Fais, F.; Taiana, E.; Neri, A.; Bomben, R.; Gentile, M.; et al. Antitumor effects of PRIMA-1 and PRIMA-1Met (APR246) in hematological halignancies: Still a mutant P53-dependent affair? Cells 2021, 10, 98. [Google Scholar] [CrossRef]
- Bykov, V.J.; Wiman, K.G. Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS Let. 2014, 588, 2622–2627. [Google Scholar] [CrossRef] [Green Version]
- Deneberg, S.; Cherif, H.; Lazarevic, V.; Andersson, P.O.; von Euler, M.; Juliusson, G.; Lehmann, S. An open-label phase I dose-finding study of APR-246 in hematological Malignancies. Blood Cancer J. 2016, 6, e447. [Google Scholar] [CrossRef] [Green Version]
- Krayem, M.; Journe, F.; Wiedig, M.; Morandini, R.; Najem, A.; Salès, F.; van Kempen, L.C.; Sibille, C.; Awada, A.; Marine, J.C.; et al. p53 Reactivation by PRIMA-1(Met) (APR-246) sensitizes (V600E/K) BRAF melanoma to vemurafenib. Eur. J. Cancer 2016, 55, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Maslah, N.; Salomao, N.; Drevon, L.; Verger, E.; Partouche, N.; Ly, P.; Aubin, P.; Naoui, N.; Schlageter, M.H.; Bally, C.; et al. Synergistic effects of PRIMA-1Met (APR-246) and 5-azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2020, 105, 1539–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Bève, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt plus azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia: A phase II study by the groupe Francophone des myélodysplasies (GFM). J. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug/Agent | MIA-PaCa-2 + pLXSN (−APR-246) | MIA-PaCa-2 + pLXSN (+12.5 nM APR-246) | Fold Change +/− APR-246 | MIA-PaCa-2 + WT-TP53 (−APR-246) | MIA-PaCa-2 + WT-TP53 (+12.5 nM APR-246) | Fold Change +/− APR-246 |
|---|---|---|---|---|---|---|
| 5FU (nucleoside analogue) | 7 µM | 1.8 µM | 3.9 × ↓ | 3.2 µM | 0.3 µM | 10.7 × ↓ |
| Doxorubicin (topoisomerase inh.) | 220 nM | 60 nM | 3.6 × ↓ | 50 nM | 18 nM | 2.8 × ↓ |
| Gilteritinib (ALK/AXL/FLT3 inh.) | 750 nM | 8.5 nM | 88.2 × ↓ | 400 nM | 200 nM | 2 × ↓ |
| ABT-737 (BCL2/BCLXL inh.) | 2000 nM | 4 nM | 500 × ↓ | 1000 nM | 110 nM | 9.1 × ↓ |
| Drug/Agent | PANC-28 + pLXSN (−APR-246) | PANC-28 + pLXSN (+12.5 nM APR-246) | Fold Change +/− APR-246 | PANC-28 + WT-TP53 (−APR-246) | PANC-28 + WT-TP53 (+12.5 nM AP-246) | Fold Change +/− APR-246 |
|---|---|---|---|---|---|---|
| 5FU (nucleoside analogue) | 30 µM | 30 µM | 1 × | 6.5 µM | 0.1 µM | 65 × ↓ |
| Doxorubicin (topoisomerase inh.) | 210 nM | 170 nM | 1.2 × ↓ | 100 nM | 15 nM | 6.7 × ↓ |
| Gilteritinib (ALK/AXL/FLT3 inh.) | 900 nM | 800 nM | 1.1 × ↓ | 280 nM | 3.8 nM | 73.7 × ↓ |
| ABT-737 (BCL2/BCLXL inh.) | 1000 nM | 800 nM | 1.3 × ↓ | 100 nM | 2 nM | 50 × ↓ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abrams, S.L.; Duda, P.; Akula, S.M.; Steelman, L.S.; Follo, M.L.; Cocco, L.; Ratti, S.; Martelli, A.M.; Montalto, G.; Emma, M.R.; et al. Effects of the Mutant TP53 Reactivator APR-246 on Therapeutic Sensitivity of Pancreatic Cancer Cells in the Presence and Absence of WT-TP53. Cells 2022, 11, 794. https://doi.org/10.3390/cells11050794
Abrams SL, Duda P, Akula SM, Steelman LS, Follo ML, Cocco L, Ratti S, Martelli AM, Montalto G, Emma MR, et al. Effects of the Mutant TP53 Reactivator APR-246 on Therapeutic Sensitivity of Pancreatic Cancer Cells in the Presence and Absence of WT-TP53. Cells. 2022; 11(5):794. https://doi.org/10.3390/cells11050794
Chicago/Turabian StyleAbrams, Stephen L., Przemysław Duda, Shaw M. Akula, Linda S. Steelman, Matilde L. Follo, Lucio Cocco, Stefano Ratti, Alberto M. Martelli, Giuseppe Montalto, Maria Rita Emma, and et al. 2022. "Effects of the Mutant TP53 Reactivator APR-246 on Therapeutic Sensitivity of Pancreatic Cancer Cells in the Presence and Absence of WT-TP53" Cells 11, no. 5: 794. https://doi.org/10.3390/cells11050794
APA StyleAbrams, S. L., Duda, P., Akula, S. M., Steelman, L. S., Follo, M. L., Cocco, L., Ratti, S., Martelli, A. M., Montalto, G., Emma, M. R., Cervello, M., Rakus, D., Gizak, A., & McCubrey, J. A. (2022). Effects of the Mutant TP53 Reactivator APR-246 on Therapeutic Sensitivity of Pancreatic Cancer Cells in the Presence and Absence of WT-TP53. Cells, 11(5), 794. https://doi.org/10.3390/cells11050794