
Baicalein Activates Parkin-Dependent Mitophagy through NDP52 and OPTN


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture, Reagents, and Antibodies
2.2. Construction of Expression Plasmids
2.3. Establishment of Stable Reporter Cells by Lentiviral Gene Delivery
2.4. SDS-PAGE, Western Blotting, and Purification of Mitochondria
2.5. Immunofluorescence (IF), Confocal Microscopy, and Fluorescence-Activated Cell Sorting (FACS) Analysis
2.6. Time-Lapse Live-Cell Imaging
2.7. Transmission Electron Microscopy (TEM)
2.8. Correlative Light and Electron Microscopy (CLEM)
2.9. Generation of Knockout Cells Using Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR)/CRISPR-Associated Protein 9 (Cas9) Gene Editing
3. Results
3.1. Baicalein Induces the Formation of Autophagic Vacuoles
3.2. Baicalein Activates Complete Autophagy throughout Autolysosome Maturation
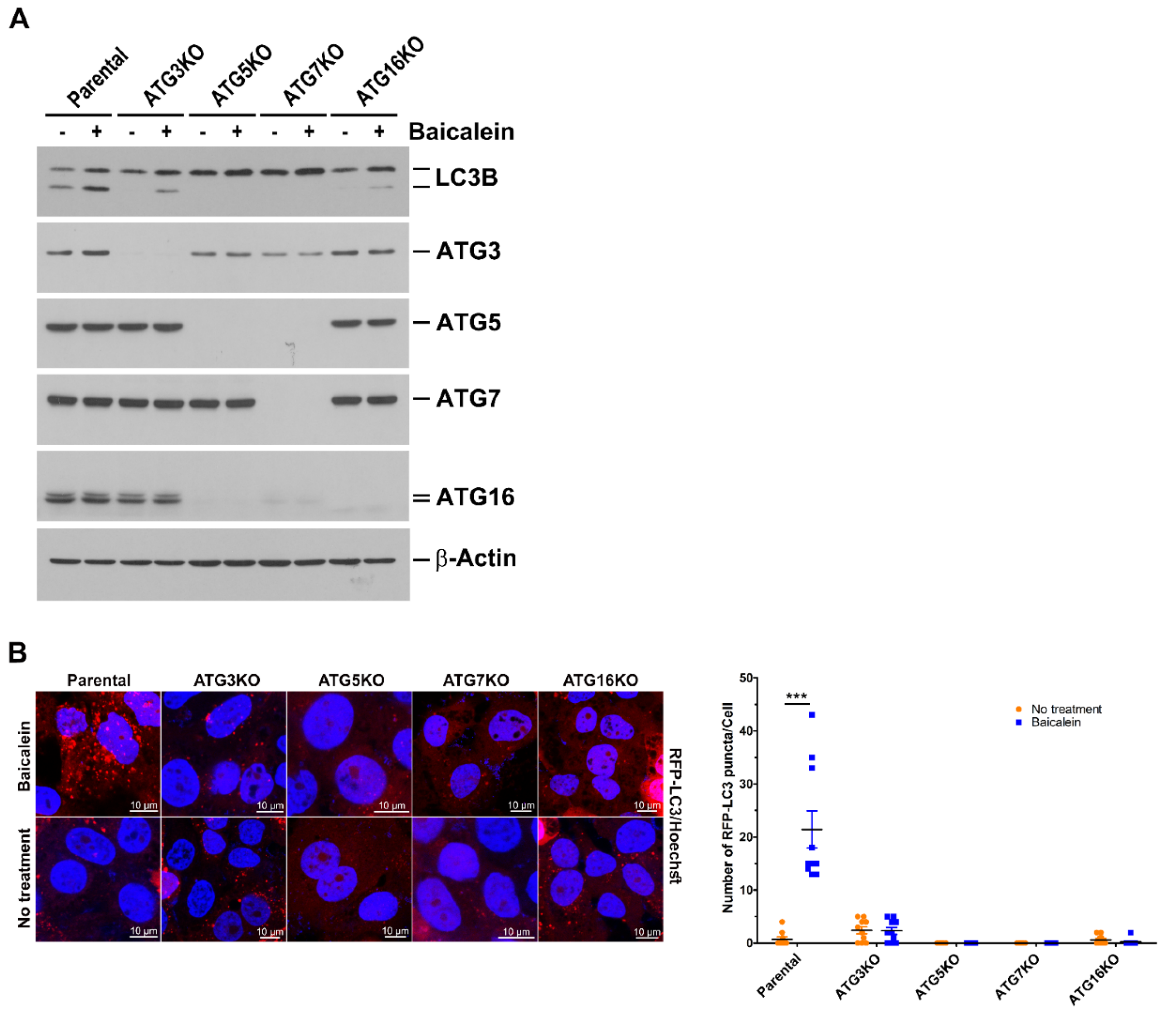
3.3. Baicalein Induces Autophagy through the Ubiquitin-Like (UBL) Conjugation System
3.4. Baicalein Activates the Autophagic Process to Engulf Mitochondria
3.5. Activation of Mitophagy by Baicalein
3.6. Baicalein Induces the Translocation of Parkin to Mitochondria, the Phosphorylation of Ubiquitin at Ser65, and the Stabilization of PINK1
3.7. Translocation of TBK1 to Mitochondria and Phosphorylation of TBK1 at Ser172 in Baicalein-Treated Cells
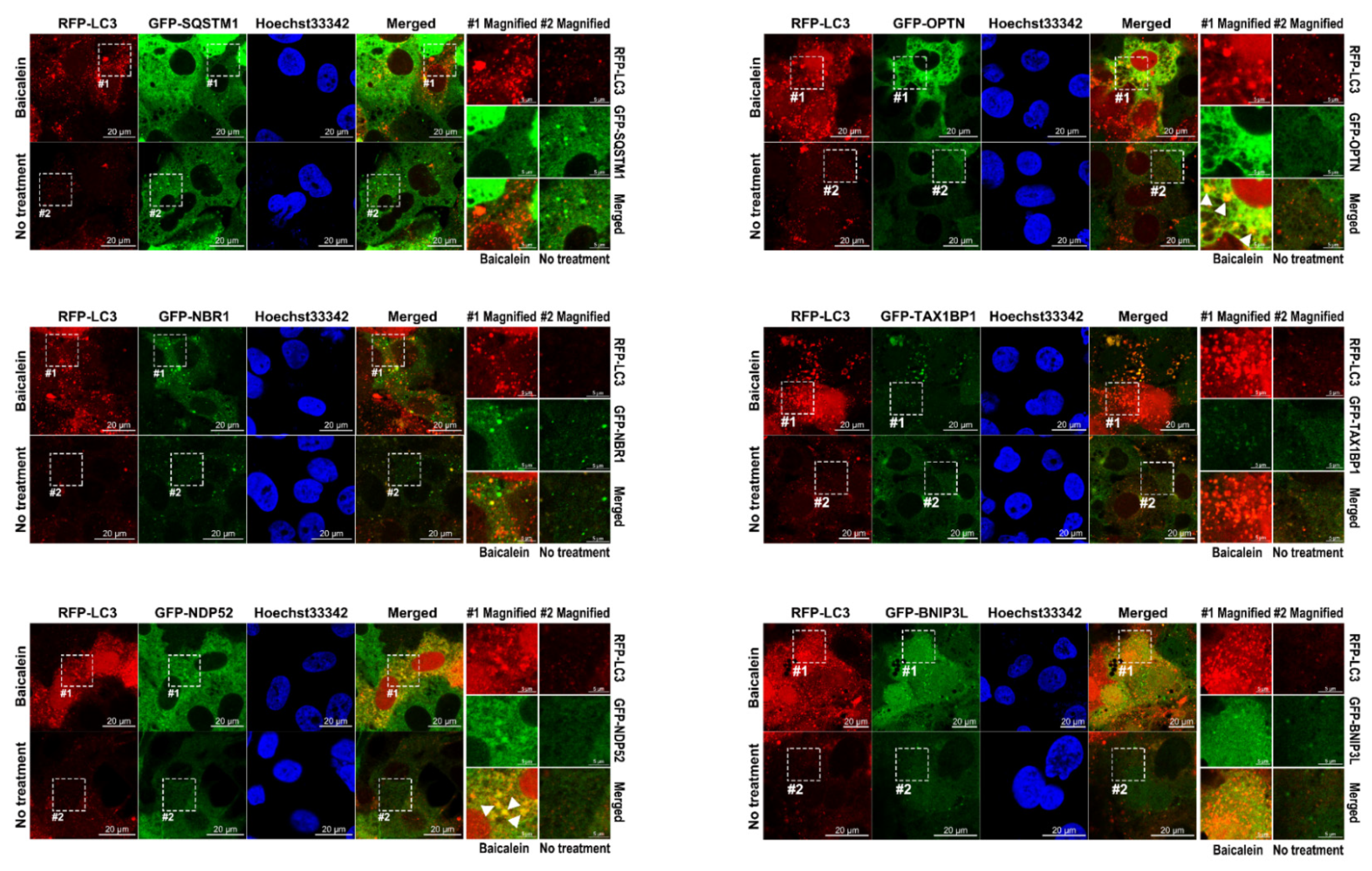
3.8. Baicalein Activates the Recruitment of NDP52 and OPTN in Mitophagy
3.9. Activation of Mitophagy Induced by NDP52 and OPTN in Baicalein-Treated Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Pedro, J.M.B.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B. Autophagy in Human Diseases. N. Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, T.; Yoshimori, T. Autophagosome biogenesis and human health. Cell Discov. 2020, 6, 1–14. [Google Scholar] [CrossRef]
- Zhao, Y.G.; Codogno, P.; Zhang, H. Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat. Rev. Mol. Cell Biol. 2021, 22, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N. The ATG conjugation systems in autophagy. Curr. Opin. Cell Biol. 2020, 63, 1–10. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Zaffagnini, G.; Martens, S. Mechanisms of Selective Autophagy. J. Mol. Biol. 2016, 428, 1714–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkin, V.; Rogov, V.V. A Diversity of Selective Autophagy Receptors Determines the Specificity of the Autophagy Pathway. Mol. Cell 2019, 76, 268–285. [Google Scholar] [CrossRef] [PubMed]
- Johansen, T.; Lamark, T. Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J. Mol. Biol. 2020, 432, 80–103. [Google Scholar] [CrossRef]
- Okamoto, K. Organellophagy: Eliminating cellular building blocks via selective autophagy. J. Cell Biol. 2014, 205, 435–445. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2012, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [Green Version]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, J.-M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, G.; Shimogori, T.; Hattori, N.; Nukina, N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum. Mol. Genet. 2015, 24, 4429–4442. [Google Scholar] [CrossRef] [Green Version]
- Moehlman, A.T.; Youle, R.J. Mitochondrial Quality Control and Restraining Innate Immunity. Annu. Rev. Cell Dev. Biol. 2020, 36, 265–289. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Ke, P.-Y. Mitophagy in the Pathogenesis of Liver Diseases. Cells 2020, 9, 831. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; McKeen, T.; Zhang, J.; Ding, W.-X. Role and Mechanisms of Mitophagy in Liver Diseases. Cells 2020, 9, 837. [Google Scholar] [CrossRef] [Green Version]
- Hazari, Y.; Bravo-San Pedro, J.M.; Hetz, C.; Galluzzi, L.; Kroemer, G. Autophagy in hepatic adaptation to stress. J. Hepatol. 2020, 72, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Allaire, M.; Rautou, P.-E.; Codogno, P.; Lotersztajn, S. Autophagy in liver diseases: Time for translation? J. Hepatol. 2019, 70, 985–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.-B.; Jiang, Y.; Chen, F. Separation methods used for Scutellaria baicalensis active components. J. Chromatogr. B 2004, 812, 277–290. [Google Scholar] [CrossRef]
- Li, H.-B.; Chen, F. Isolation and purification of baicalein, wogonin and oroxylin A from the medicinal plant Scutellaria baicalensis by high-speed counter-current chromatography. J. Chromatogr. A 2005, 1074, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Ku, S.-K.; Bae, J.-S. Anti-inflammatory Effects of Baicalin, Baicalein, and Wogonin In Vitro and In Vivo. Inflammation 2015, 38, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Zhang, R.; Piao, M.J.; Chae, S.; Kim, H.S.; Park, J.H.; Jung, K.S.; Hyun, J.W. Baicalein inhibits oxidative stress-induced cellular damage via antioxidant effects. Toxicol. Ind. Heal. 2011, 28, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-L. Hepatoprotective effects of baicalein against CCl4-induced acute liver injury in mice. World J. Gastroenterol. 2012, 18, 6605–6613. [Google Scholar] [CrossRef]
- Zhou, H.-C.; Wang, H.; Shi, K.; Li, J.-M.; Zong, Y.; Du, R. Hepatoprotective Effect of Baicalein Against Acetaminophen-Induced Acute Liver Injury in Mice. Molecules 2018, 24, 131. [Google Scholar] [CrossRef] [Green Version]
- Sowndhararajan, K.; Deepa, P.; Kim, M.; Park, S.J.; Kim, S. Baicalein as a potent neuroprotective agent: A review. Biomed. Pharmacother. 2017, 95, 1021–1032. [Google Scholar] [CrossRef]
- Liu, H.; Dong, Y.; Gao, Y.; Du, Z.; Wang, Y.; Cheng, P.; Chen, A.; Huang, H. The Fascinating Effects of Baicalein on Cancer: A Review. Int. J. Mol. Sci. 2016, 17, 1681. [Google Scholar] [CrossRef] [Green Version]
- Kaul, R.; Paul, P.; Kumar, S.; Büsselberg, D.; Dwivedi, V.D.; Chaari, A. Promising Antiviral Activities of Natural Flavonoids against SARS-CoV-2 Targets: Systematic Review. Int. J. Mol. Sci. 2021, 22, 11069. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.-L.; Hsieh, Y.-C.; Hsieh, W.-C.; Chou, C.-J. Baicalein induces a dual growth arrest by modulating multiple cell cycle regulatory molecules. Eur. J. Pharmacol. 2001, 425, 165–171. [Google Scholar] [CrossRef]
- Chao, J.-I.; Su, W.-C.; Liu, H.-F. Baicalein induces cancer cell death and proliferation retardation by the inhibition of CDC2 kinase and survivin associated with opposite role of p38 mitogen-activated protein kinase and AKT. Mol. Cancer Ther. 2007, 6, 3039–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Jiang, C.; Chen, W.; Zhang, G.; Luo, D.; Cao, Y.; Wu, J.; Ding, Y.; Liu, B. Baicalein Induces Apoptosis and Autophagy via Endoplasmic Reticulum Stress in Hepatocellular Carcinoma Cells. BioMed Res. Int. 2014, 2014, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Peng, B.; Nayak, Y.; Wang, C.; Si, F.; Liu, X.; Dou, J.; Xu, H.; Peng, G. Baicalein and Baicalin Promote Melanoma Apoptosis and Senescence via Metabolic Inhibition. Front. Cell Dev. Biol. 2020, 8, 836. [Google Scholar] [CrossRef]
- Lee, H.Z.; Leung, H.W.C.; Lai, M.Y.; Wu, C.H. Baicalein induced cell cycle arrest and apoptosis in human lung squamous carcinoma CH27 cells. Anticancer Res. 2005, 25, 959–964. [Google Scholar] [PubMed]
- Wu, R.; Murali, R.; Kabe, Y.; French, S.W.; Chiang, Y.; Liu, S.; Sher, L.; Wang, C.C.; Louie, S.; Tsukamoto, H. Baicalein Targets GTPase-Mediated Autophagy to Eliminate Liver Tumor–Initiating Stem Cell–Like Cells Resistant to mTORC1 Inhibition. Hepatol. 2018, 68, 1726–1740. [Google Scholar] [CrossRef] [PubMed]
- Üstüner, M.; Tanrikut, C.; Üstüner, D.; Kolaç, U.; Köroğlu, Z.Ö.; Burukoğlu, D.; Entok, E. The effect of baicalein on endoplasmic reticulum stress and autophagy on liver damage. Hum. Exp. Toxicol. 2021, 40, 1624–1633. [Google Scholar] [CrossRef]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [Green Version]
- Ke, P.-Y.; Chen, S.S.-L. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Investig. 2011, 121, 37–56. [Google Scholar] [CrossRef] [Green Version]
- Rosignol, I.; Villarejo-Zori, B.; Teresak, P.; Sierra-Filardi, E.; Pereiro, X.; Rodríguez-Muela, N.; Vecino, E.; Vieira, H.L.A.; Bell, K.; Boya, P. The mito-QC Reporter for Quantitative Mitophagy Assessment in Primary Retinal Ganglion Cells and Experimental Glaucoma Models. Int. J. Mol. Sci. 2020, 21, 1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, N.; Malide, D.; Liu, J.; I Rovira, I.; A Combs, C.; Finkel, T. A fluorescence-based imaging method to measure in vitro and in vivo mitophagy using mt-Keima. Nat. Protoc. 2017, 12, 1576–1587. [Google Scholar] [CrossRef] [PubMed]
- Razi, M.; Tooze, S.A. Chapter 17 Correlative Light and Electron Microscopy. Methods in Enzymology 2009, 452, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Reddick, L.E.; Alto, N.M. Correlative Light and Electron Microscopy (CLEM) as a Tool to Visualize Microinjected Molecules and their Eukaryotic Sub-cellular Targets. J. Vis. Exp. 2012, e3650, e3650. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, S.; Kahlhofer, J.; Baylac, N.; Kallio, K.; Eskelinen, E.L. Correlative Light and Electron Microscopy of Autophagosomes. Methods Mol. Biol. 2019, 1880, 199–209. [Google Scholar] [PubMed]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Birsoy, K.; Hughes, N.W.; Krupczak, K.M.; Post, Y.; Wei, J.J.; Lander, E.S.; Sabatini, D.M. Identification and characterization of essential genes in the human genome. Science 2015, 350, 1096–1101. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the Autophagosome Maturation Process by a Novel Reporter Protein, Tandem Fluorescent-Tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef] [Green Version]
- Eskelinen, E.-L. Maturation of Autophagic Vacuoles in Mammalian Cells. Autophagy 2005, 1, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in Mammalian Autophagy Research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, A.; Tagawa, Y.; Yoshimori, T.; Moriyama, Y.; Masaki, R.; Tashiro, Y. Bafilomycin A1 Prevents Maturation of Autophagic Vacuoles by Inhibiting Fusion between Autophagosomes and Lysosomes in Rat Hepatoma Cell Line, H-4-II-E Cells. Cell Struct. Funct. 1998, 23, 33–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, G.F.G.; Toth, R.; James, J.; Ganley, I. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 2013, 14, 1127–1135. [Google Scholar] [CrossRef]
- McWilliams, T.; Prescott, A.R.; Allen, G.F.; Tamjar, J.; Munson, M.J.; Thomson, C.; Muqit, M.M.; Ganley, I.G. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 2016, 214, 333–345. [Google Scholar] [CrossRef]
- Liu, Y.-T.; Sliter, D.A.; Shammas, M.K.; Huang, X.; Wang, C.; Calvelli, H.; Maric, D.S.; Narendra, D.P. Mt-Keima detects PINK1-PRKN mitophagy in vivo with greater sensitivity than mito-QC. Autophagy 2021, 17, 3753–3762. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLOS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [Green Version]
- McLelland, G.-L.; Goiran, T.; Yi, W.; Dorval, G.; Chen, C.X.; Lauinger, N.D.; I Krahn, A.; Valimehr, S.; Rakovic, A.; Rouiller, I.; et al. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. eLife 2018, 7, e32866. [Google Scholar] [CrossRef]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.-F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G. Autophagy: A druggable process that is deregulated in aging and human disease. J. Clin. Investig. 2015, 125, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Kroemer, G. Therapeutic modulation of autophagy: Which disease comes first? Cell Death Differ. 2019, 26, 680–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisner, V.; Picard, M.; Hajnóczky, G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 2018, 20, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, T.M.K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef]
- Ichimura, Y.; Waguri, S.; Sou, Y.-S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 Activates the Keap1-Nrf2 Pathway during Selective Autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.A.; Ni, H.-M.; Ding, Y.; Ding, W.-X. Parkin regulates mitophagy and mitochondrial function to protect against alcohol-induced liver injury and steatosis in mice. Am. J. Physiol. Liver Physiol. 2015, 309, G324–G340. [Google Scholar] [CrossRef] [Green Version]
- Eid, N.; Ito, Y.; Otsuki, Y. Mitophagy in steatotic hepatocytes of ethanol-treated wild-type and Parkin knockout mice. Am. J. Physiol. Liver Physiol. 2015, 309, G513–G514. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-F.; Li, T.; Tang, Z.-H.; Chang, L.-L.; Zhu, H.; Chen, X.-P.; Wang, Y.-T.; Lu, J.-J. Baicalein Triggers Autophagy and Inhibits the Protein Kinase B/Mammalian Target of Rapamycin Pathway in Hepatocellular Carcinoma HepG2 Cells. Phytotherapy Res. 2015, 29, 674–679. [Google Scholar] [CrossRef]
- Aryal, P.; Kim, K.; Park, P.-H.; Ham, S.; Cho, J.; Song, K. Baicalein induces autophagic cell death through AMPK/ULK1 activation and downregulation of mTORC1 complex components in human cancer cells. FEBS J. 2014, 281, 4644–4658. [Google Scholar] [CrossRef]
- Zachari, M.; Gudmundsson, S.R.; Li, Z.; Manifava, M.; Cugliandolo, F.; Shah, R.; Smith, M.; Stronge, J.; Karanasios, E.; Piunti, C.; et al. Selective Autophagy of Mitochondria on a Ubiquitin-Endoplasmic-Reticulum Platform. Dev. Cell 2019, 50, 627–643.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, J.N.S.; Wang, C.; Bunker, E.; Hao, L.; Maric, D.; Schiavo, G.; Randow, F.; Youle, R.J. Spatiotemporal Control of ULK1 Activation by NDP52 and TBK1 during Selective Autophagy. Mol. Cell 2019, 74, 347–362.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, R.; Kumar, M.; Katyal, A. 12/15–Lipoxygenase debilitates mitochondrial health in intermittent hypobaric hypoxia induced neuronal damage: An in vivo study. Redox Biol. 2021, 49, 102228. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yuan, Y.; Gong, X.; Zhang, L.; Zhou, Q.; Wu, S.; Zhang, X.; Hu, J.; Kuang, G.; Yin, X.; et al. Baicalin and its nanoliposomes ameliorates nonalcoholic fatty liver disease via suppression of TLR4 signaling cascade in mice. Int. Immunopharmacol. 2020, 80, 106208. [Google Scholar] [CrossRef]
- Sun, W.; Liu, P.; Wang, T.; Wang, X.; Zheng, W.; Li, J. Baicalein reduces hepatic fat accumulation by activating AMPK in oleic acid-induced HepG2 cells and high-fat diet-induced non-insulin-resistant mice. Food Funct. 2020, 11, 711–721. [Google Scholar] [CrossRef]
- Liu, K.; Lee, J.; Kim, J.Y.; Wang, L.; Tian, Y.; Chan, S.T.; Cho, C.; Machida, K.; Chen, D.; Ou, J.-H.J. Mitophagy Controls the Activities of Tumor Suppressor p53 to Regulate Hepatic Cancer Stem Cells. Mol. Cell 2017, 68, 281–292.e5. [Google Scholar] [CrossRef] [Green Version]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ke, P.-Y.; Chang, C.-W.; Hsiao, Y.-C. Baicalein Activates Parkin-Dependent Mitophagy through NDP52 and OPTN. Cells 2022, 11, 1132. https://doi.org/10.3390/cells11071132
Ke P-Y, Chang C-W, Hsiao Y-C. Baicalein Activates Parkin-Dependent Mitophagy through NDP52 and OPTN. Cells. 2022; 11(7):1132. https://doi.org/10.3390/cells11071132
Chicago/Turabian StyleKe, Po-Yuan, Chih-Wei Chang, and Yuan-Chao Hsiao. 2022. "Baicalein Activates Parkin-Dependent Mitophagy through NDP52 and OPTN" Cells 11, no. 7: 1132. https://doi.org/10.3390/cells11071132
APA StyleKe, P. -Y., Chang, C. -W., & Hsiao, Y. -C. (2022). Baicalein Activates Parkin-Dependent Mitophagy through NDP52 and OPTN. Cells, 11(7), 1132. https://doi.org/10.3390/cells11071132