
Concomitant Hereditary Spherocytosis and Pyruvate Kinase Deficiency in a Spanish Family with Chronic Hemolytic Anemia: Contribution of Laser Ektacytometry to Clinical Diagnosis

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tse, W.T.; Lux, S.E. Red blood cell membrane disorders. Br. J. Haematol. 1999, 104, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Vives Corrons, J.L. Anemias hemolíticas. Fisiopatología y diagnóstico. Anemias hemolíticas. congénitas. In Hematología Clínica, 5th ed.; Sans-Sabrafen, J., Besses Raebel, C., Vives Corrons, J.L., Eds.; Elsevier: Barcelona, Spain, 2006. [Google Scholar]
- Wu, Y.; Liao, L.; Lin, F. The diagnostic protocol for hereditary spherocytosis-2021 update. J. Clin. Lab. Anal. 2021, 35, e24034. [Google Scholar] [CrossRef] [PubMed]
- Llaudet-Planas, E.; Vives-Corrons, J.L.; Rizzuto, V.; Gómez-Ramírez, P.; Sevilla Navarro, J.; Coll Sibina, M.T.; García-Bernal, M.; Ruiz Llobet, A.; Badell, I.; Velasco-Puyó, P.; et al. Osmotic gradient ektacytometry: A valuable screening test for hereditary spherocytosis and other red blood cell membrane disorders. Int. J. Lab. Hematol. 2018, 40, 94–102. [Google Scholar] [CrossRef]
- Vives-Corrons, J.L.; Krishnevskaya, E.; Rodriguez, I.H.; Ancochea, A. Characterization of hereditary red blood cell membranopathies using combined targeted next-generation sequencing and osmotic gradient ektacytometry. Int. J. Hematol. 2021, 113, 163–174. [Google Scholar] [CrossRef]
- Yang, Y.M.; Donnell, C.; Wilborn, W.; Goodman, S.R.; Files, B.; Moore, R.B.; Mohandas, N.; Mankad, V.N. Splenic sequestration associated with sickle cell trait and hereditary spherocytosis. Am. J. Hematol. 1992, 40, 110–111. [Google Scholar] [CrossRef]
- Miraglia del Giudice, E.; Perrotta, S.; Nobili, B.; Pinto, L.; Cutillo, L.; Iolascon, A. Coexistence of hereditary spherocytosis (HS) due to band 3 deficiency and β-thalassaemia trait: Partial correction of HS phenotype. Br. J. Haematol. 1993, 85, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Alfinito, F.; Calabrò, V.; Cappellini, M.D.; Fiorelli, G.; Filosa, S.; Iolascon, A.; del Giudice, E.M.; Perrotta, S.; Migliorati, R.; Vallone, D.; et al. Glucose 6-phosphate dehydrogenase deficiency and red cell membrane defects: Additive or synergistic interaction in producing chronic haemolytic anaemia. Br. J. Haematol. 1994, 87, 148–152. [Google Scholar] [CrossRef]
- Brook, J.; Tanaka, K.R. Combination of pyruvate kinase (PK) deficiency and hereditary spherocytosis (HS). Clin. Res. 1970, 18, 176. [Google Scholar]
- Zarza, R.; Moscardó, M.; Alvarez, R.; García, J.; Morey, M.; Pujades, A.; Vives-Corrons, J.L. Co-existence of hereditary spherocytosis and a new red cell pyruvate kinase variant: PK Mallorca. Haematologica 2000, 85, 227–232. [Google Scholar]
- Vercellati, C.; Marcello, A.P.; Fermo, E.; Barcellini, W.; Zanella, A.; Bianchi, P. A case of hereditary spherocytosis misdiagnosed as pyruvate kinase deficient hemolytic anemia. Clin. Lab. 2013, 59, 421–424. [Google Scholar] [CrossRef]
- van Zwieten, R.; van Oirschot, B.A.; Veldthuis, M.; Dobbe, J.G.; Streekstra, G.J.; van Solinge, W.W.; Schutgens, R.E.; van Wijk, R. Partial pyruvate kinase deficiency aggravates the phenotypic expression of band 3 deficiency in a family with hereditary spherocytosis. Am. J. Hematol. 2015, 90, E35–E39. [Google Scholar] [CrossRef] [PubMed]
- Andres, O.; Loewecke, F.; Morbach, H.; Kraus, S.; Einsele, H.; Eber, S.; Speer, C.P. Hereditary spherocytosis is associated with decreased pyruvate kinase activity due to impaired structural integrity of the red blood cell membrane. Br. J. Haematol. 2019, 187, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Beutler, E.; Gelbart, T. Estimating the prevalence of pyruvate kinase deficiency from the gene frequency in the general white population. Blood 2000, 95, 3585–3588. [Google Scholar] [CrossRef] [PubMed]
- Secrest, M.H.; Storm, M.; Carrington, C.; Casso, D.; Gilroy, K.; Pladson, L.; Boscoe, A.N. Prevalence of pyruvate kinase deficiency: A systematic literature review. Eur. J. Haematol. 2020, 105, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Grace, R.F.; Bianchi, P.; van Beers, E.J.; Eber, S.W.; Glader, B.; Yaish, H.M.; Despotovic, J.M.; Rothman, J.A.; Sharma, M.; McNaull, M.M.; et al. Clinical spectrum of pyruvate kinase deficiency: Data from the pyruvate kinase deficiency natural history study. Blood 2018, 131, 2183–2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enegela, O.A.; Anjum, F. Pyruvate Kinase deficiency. In Stat Pearls; Stat Pearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Vives Corrons, J.L.; Marie, J.; Pujades, M.A.; Kahn, A. Hereditary erythrocyte pyruvate-kinase (PK) deficiency and chronic hemolytic anemia: Clinical, genetic and molecular studies in six new Spanish patients. Hum. Genet. 1980, 53, 401–408. [Google Scholar] [CrossRef]
- Zarza, R.; Alvarez, R.; Pujades, A.; Nomdedeu, B.; Carrera, A.; Estella, J.; Remacha, A.; Sánchez, J.M.; Morey, M.; Cortes, T.; et al. Molecular characterisation of PK-LR gene in pyruvate kinase-deficient patients. Br. J. Haematol. 1998, 103, 377–382. [Google Scholar] [CrossRef]
- Montllor, L.; Mañú-Pereira, M.D.; Llaudet-Planas, E.; Gómez Ramírez, P.; Sevilla Navarro, J.; Vives-Corrons, J.L. Red cell pyruvate kinase deficiency in Spain: A study of 15 cases. Med. Clin. 2017, 148, 23–27. [Google Scholar] [CrossRef]
- van Vuren, A.; van der Zwaag, B.; Huisjes, R.; Lak, N.; Bierings, M.; Gerritsen, E.; van Beers, E.; Bartels, M.; van Wijk, R. The Complexity of Genotype-Phenotype Correlations in Hereditary Spherocytosis: A Cohort of 95 Patients: Genotype-Phenotype Correlation in Hereditary Spherocytosis. Hemasphere 2019, 3, e276. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Vives Corrons, J.L.; Aguilar Bascompte, J.L. Manual of Hematology Laboratory Diagnostic Procedures, 4th ed.; Elsevier: Barcelona, Spain, 2014. (In Spanish) [Google Scholar]
- Beutler, E. Red Cell Metabolism: A Manual of Biochemical Methods; Grune & Stratton, Inc.: New York, NY, USA, 1984. [Google Scholar]
- King, M.J.; Behrens, J.; Rogers, C.; Flynn, C.; Greenwood, D.; Chambers, K. Rapid flow cytometric test for the diagnosis of membrane cytoskeleton-associated haemolytic anaemia. Br. J. Haematol. 2000, 111, 924–933. [Google Scholar] [PubMed]
- King, M.J.; Zanella, A. Hereditary red cell membrane disorders and laboratory diagnostic testing. Int. J. Lab. Hematol. 2013, 35, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Fermo, E.; Bianchi, P.; Chiarelli, L.R.; Cotton, F.; Vercellati, C.; Writzl, K.; Baker, K.; Hann, I.; Rodwell, R.; Valentini, G.; et al. Red cell pyruvate kinase deficiency: 17 new mutations of the PK-LR gene. Br. J. Haematol. 2005, 129, 839–846, Erratum in Br. J. Haematol. 2005, 130, 973. [Google Scholar] [CrossRef] [PubMed]
- Godal, H.C.; Heistø, H. High prevalence of increased osmotic fragility of red blood cells among Norwegian blood donors. Scand. J. Haematol. 1981, 27, 30–34. [Google Scholar] [CrossRef]
- Ricard, M.P.; Gilsanz, F.; Millan, I. Erythroid membrane protein defects in hereditary spherocytosis. A study of 62 Spanish cases. Haematologica 2000, 85, 994–995. [Google Scholar]
- Mariani, M.; Barcellini, W.; Vercellati, C.; Marcello, A.P.; Fermo, E.; Pedotti, P.; Boschetti, C.; Zanella, A. Clinical and hematologic features of 300 patients affected by hereditary spherocytosis grouped according to the type of the membrane protein defect. Haematologica 2008, 93, 1310–1317. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, P.; Fermo, E.; Vercellati, C.; Marcello, A.; Porretti, L.; Cortelezzi, A.; Barcellini, W.; Zanella, A. Diagnostic power of laboratory tests for hereditary spherocytosis: A comparison study in 150 patients grouped according to molecular and clinical characteristics. Haematologica 2012, 97, 516–523. [Google Scholar] [CrossRef]
- Perrotta, S.; Gallagher, P.G.; Mohandas, N. Hereditary spherocytosis. Lancet 2008, 18, 1411–1426. [Google Scholar] [CrossRef]
- Grace, R.F.; Zanella, A.; Neufeld, E.J.; Morton, D.H.; Eber, S.; Yaish, H.; Glader, B. Erythrocyte pyruvate kinase deficiency: 2015 status report. Am. J. Hematol. 2015, 90, 825–830. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, P.; Fermo, E. Molecular heterogeneity of pyruvate kinase deficiency. Haematologica 2020, 105, 2218–2228. [Google Scholar] [CrossRef]
- Fattizzo, B.; Giannotta, J.A.; Cecchi, N.; Barcellini, W. Confounding factors in the diagnosis and clinical course of rare congenital hemolytic anemias. Orphanet J. Rare Dis. 2021, 16, 415. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
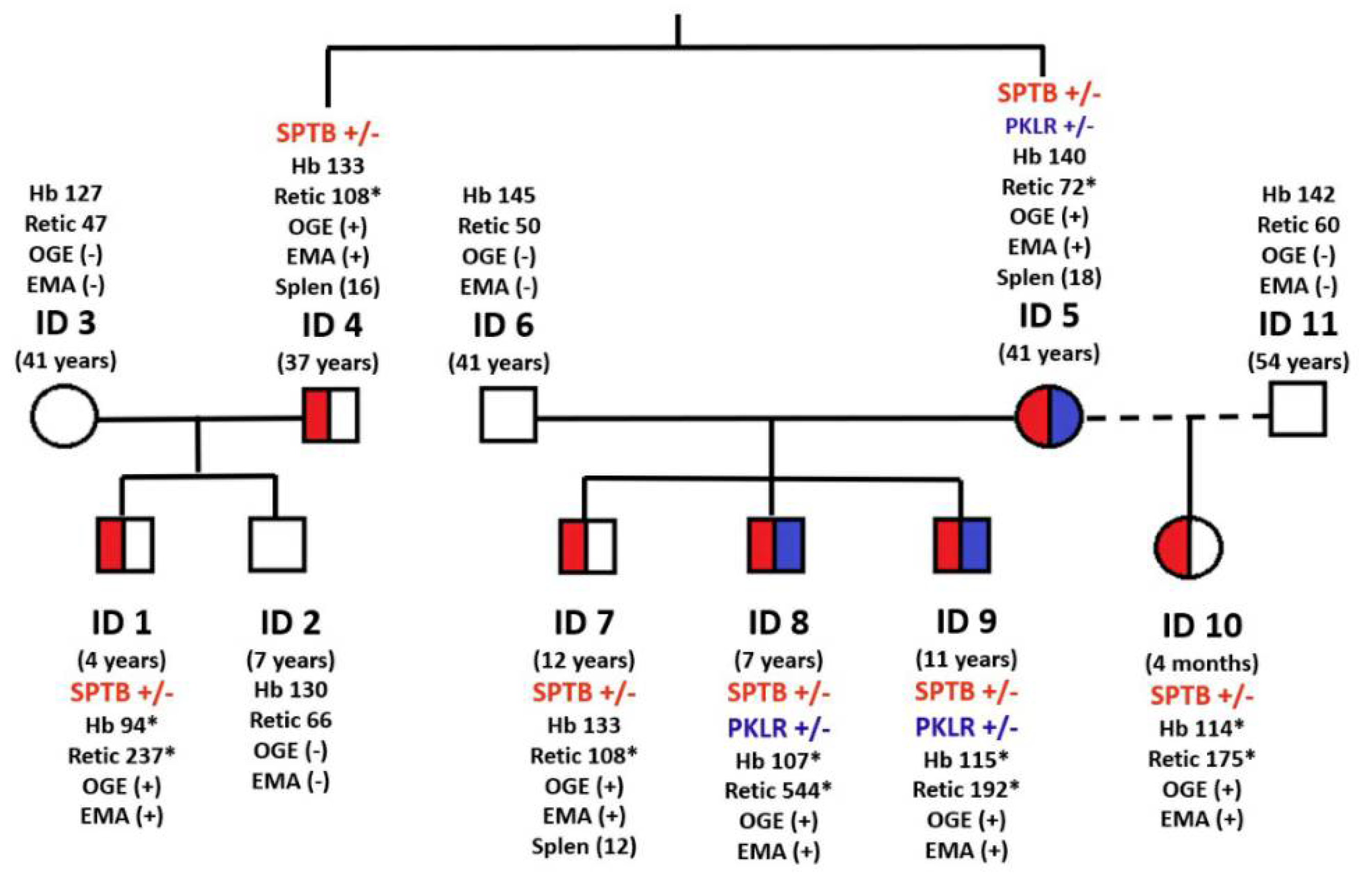
| ID1 | ID2 | ID3 | ID4 | ID5 | ID6 | ID7 | ID8 | ID9 | ID10 | ID11 | Reference Values | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age | 4 years | 7 years | 41 years | 37 years | 41 years | 41 years | 12 years | 7 years | 11 years | 4 months | 54 years | |
| Neonatal jaundice | Yes | No | No | No | No | No | Yes | No | No | Yes | No | |
| Splenomegaly | Yes | No | No | Yes | No | No | Yes | Yes | No | No | No | |
| Transfusion (No. of units) | No | No | No | No | No | No | 2 (6 and 8 years) | 1 (1 year) | No | No | No | |
| Hemoglobin (g/dL) * | 94 | 130 | 127 | 156 | 140 | 145 | 133 | 108 | 115 | 114 | 142 | 121–167 |
| MCV (fL) * | 76.1 | 81.6 | 89.9 | 85.8 | 86.9 | 93.3 | 83.5 | 87.1 | 81.5 | 83.3 | 93.4 | 80–94 |
| MCHC (g/L) * | 351 | 345 | 323 | 366 | 333 | 343 | 335 | 348 | 368 | 347 | 330 | 310–350 |
| Reticulocytes (109/L) * | 236.7 | 65.8 | 46.8 | 144.1 | 72.4 | 50 | 108.4 | 544 | 191.6 | 175.2 | 59.7 | 24–84 |
| Unconjugated bilirubin (mg/dL) | 2.6 | 0.1 | 0.2 | 0.61 | 0.60 | 0.56 | 2.04 | 0.98 | 0.6 | 5.94 | 0.54 | 0.2–0.8 |
| Haptoglobin (mg/dL) | <20 | 65 | 72 | 40 | 78 | 42 | 20 | 20 | 42 | <20 | 52 | 30–200 |
| Lactate Dehydrogenase (IU/L) | 767 | 250 | 380 | 520 | 266 | 318 | 280 | 582 | 310 | 393 | 183 | 230–460 |
| Direct antiglobulin test | (−) | (−) | (−) | (−) | (−) | (−) | (−) | (−) | (−) | (−) | (−) | (−) |
| Splenectomy (years) | No | No | No | Yes (16) | Yes (18) | No | Yes (12) | No | No | No | No | No |
| RBC OSMOTIC FRAGILITYY AND EMA--BINDING TEST | ||||||||||||
| OF in NaCl on fresh blood | Increased | Decreased | Normal | Normal | normal | normal | Increased | increased | increased | increased | normal | normal |
| OF in NaCl on incubated blood | Decreased | Decreased | Normal | Decreased | increased | normal | increased | increased | increased | increased | normal | normal |
| Acidified Glycerol Lysis (s) | 1800 | 1800 | 1800 | 1800 | <1800 | >1800 | >120 | <120 | <120 | >1800 | <120 | >1800 |
| EMA-binding test | –28.43 | –12.15 | –8.4 | –26.2 | –15.62 | –4.1 | –26.23 | –18.4 | –27.45 | –24.09 | –2.2 | >–11 |
| OSMOTIC GRADIENT EKTACYTOMETRY (OGE) | ||||||||||||
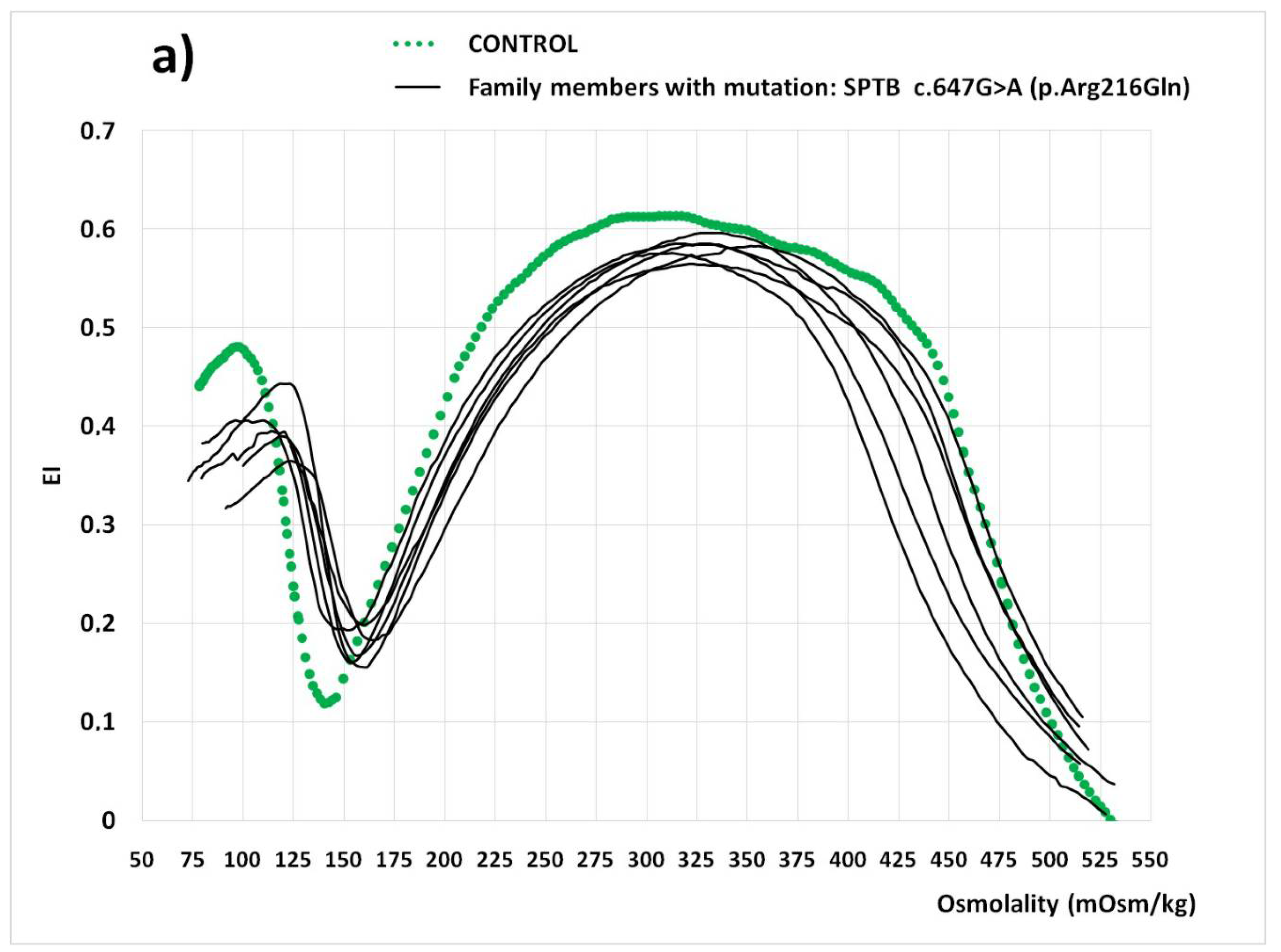
| EImax | 0.168 | 0.147 | 0.151 | 0.164 | 0.585 | 0.619 | 0.565 | 0.579 | 0.586 | 0.583 | 0.619 | 0.60–0.63 |
| Omin | 158 | 131 | 139 | 157 | 154 | 150 | 162 | 177 | 162 | 164 | 143 | 127–159 |
| Ohiper | 446 | 439 | 473 | 425 | 461 | 455 | 462 | 476 | 436 | 470 | 468 | 447–480 |
| AUC | 145.8 | 165.4 | 177.5 | 134.6 | 155.8 | 162.2 | 146.6 | 144.7 | 137.3 | 147.9 | 172.1 | 159–175 |
| ACTIVITY PYRUVATE KINASE (PK) | ||||||||||||
| PK (IU/gHb) | 11.5 | 12.31 | 14.9 | 8.59 | 7.95 | 9.17 | 9.8 | 9.61 | 9.61 | 14.6 | 8.73 | 8.4–15.2 |
| GENETIC DIAGNOSIS BY t-NGS, >500x | ||||||||||||
| PKLR c.1706G>A (p.Arg569Gln) | heterozygous | heterozygous | heterozygous | heterozygous | ||||||||
| SPTB c.647G>A (p.Arg216Gln) | heterozygous | heterozygous | heterozygous | heterozygous | heterozygous | heterozygous | heterozygous | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vives Corrons, J.-L.; Krishnevskaya, E.; Montllor, L.; Leguizamon, V.; Garcia Bernal, M. Concomitant Hereditary Spherocytosis and Pyruvate Kinase Deficiency in a Spanish Family with Chronic Hemolytic Anemia: Contribution of Laser Ektacytometry to Clinical Diagnosis. Cells 2022, 11, 1133. https://doi.org/10.3390/cells11071133
Vives Corrons J-L, Krishnevskaya E, Montllor L, Leguizamon V, Garcia Bernal M. Concomitant Hereditary Spherocytosis and Pyruvate Kinase Deficiency in a Spanish Family with Chronic Hemolytic Anemia: Contribution of Laser Ektacytometry to Clinical Diagnosis. Cells. 2022; 11(7):1133. https://doi.org/10.3390/cells11071133
Chicago/Turabian StyleVives Corrons, Joan-Lluis, Elena Krishnevskaya, Laura Montllor, Valentina Leguizamon, and Marta Garcia Bernal. 2022. "Concomitant Hereditary Spherocytosis and Pyruvate Kinase Deficiency in a Spanish Family with Chronic Hemolytic Anemia: Contribution of Laser Ektacytometry to Clinical Diagnosis" Cells 11, no. 7: 1133. https://doi.org/10.3390/cells11071133
APA StyleVives Corrons, J. -L., Krishnevskaya, E., Montllor, L., Leguizamon, V., & Garcia Bernal, M. (2022). Concomitant Hereditary Spherocytosis and Pyruvate Kinase Deficiency in a Spanish Family with Chronic Hemolytic Anemia: Contribution of Laser Ektacytometry to Clinical Diagnosis. Cells, 11(7), 1133. https://doi.org/10.3390/cells11071133