
Early Protective Role of Inflammation in Cardiac Remodeling and Heart Failure: Focus on TNFα and Resident Macrophages



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction to Short-Term Adaptive Cardiac Remodeling and Transition to Heart Failure
2. Cardiac Remodeling, HF, and Inflammation
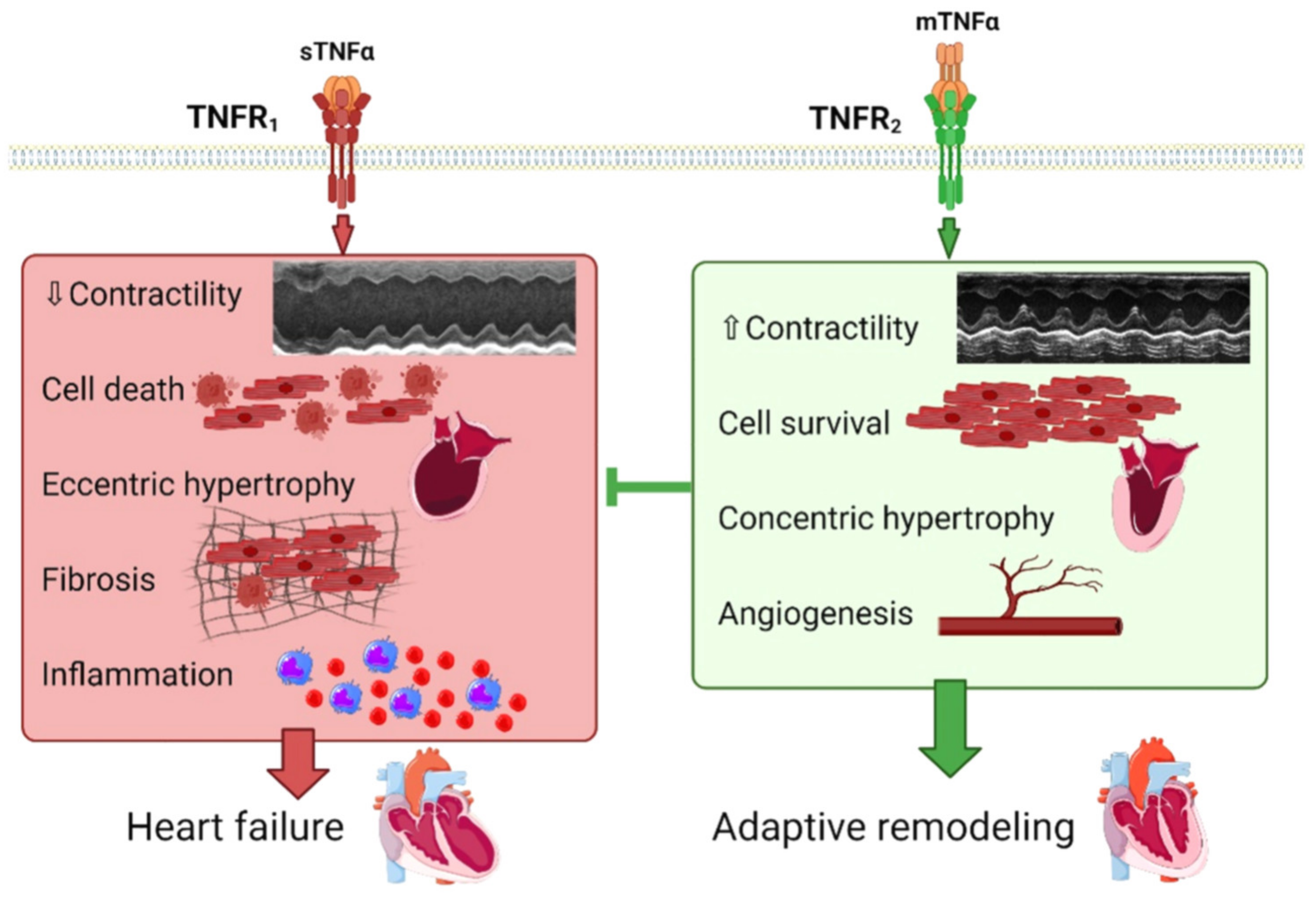
2.1. TNFα, Early Adaptive Remodeling and HF
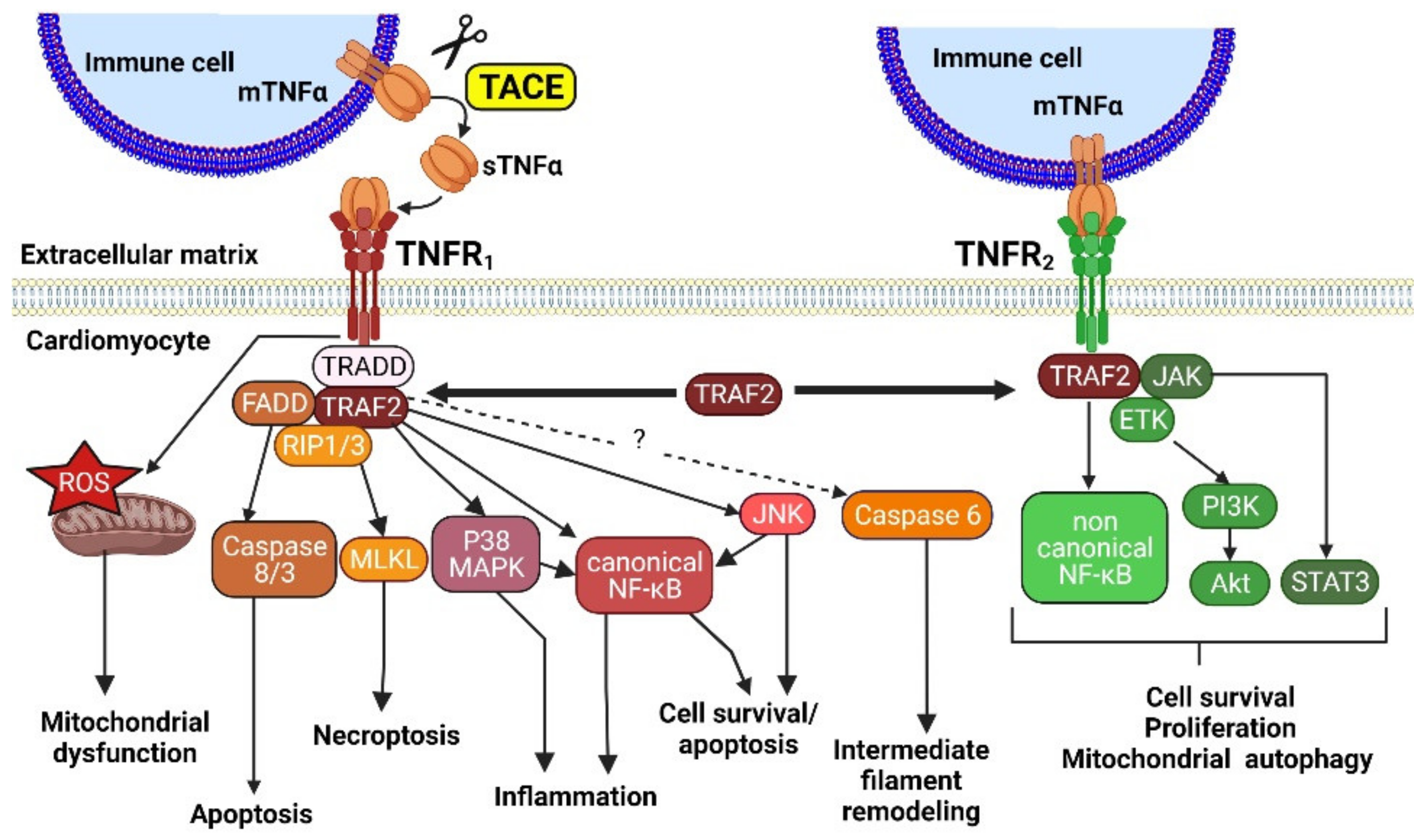
2.2. TNFR and NF-κB Signaling, Cell Survival and HF
2.3. TNFα, Contractile Function and HF
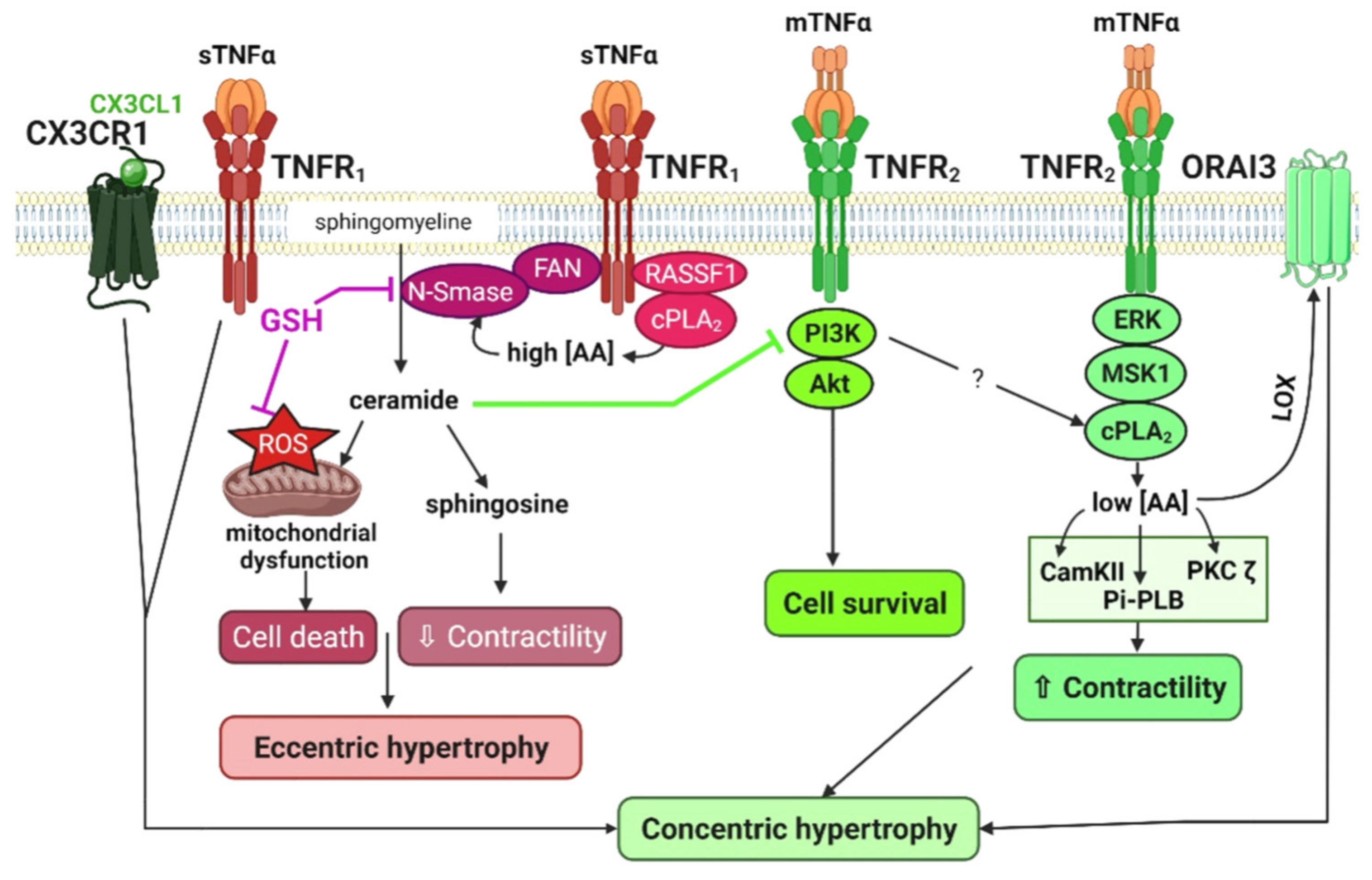
2.3.1. The Neutral Sphingomyelinase, a Determinant of TNFR1 Deleterious Signaling
2.3.2. The Cardiac cPLA2, a Determinant TNFR2 Protective Signaling Pathway: Involvement in β2-Adrenergic Signaling and Relationship with PI3Kinase Activity
2.4. Combined Signaling of TNFα with the CX3CL1 Chemokine
3. Innate Immunity, Cardiac Remodeling and HF
4. Macrophages Subsets and Cardiac Remodeling
4.1. Resident Mφ Are Requisite for the Adaptive Response to Pressure Overload or Hypertension
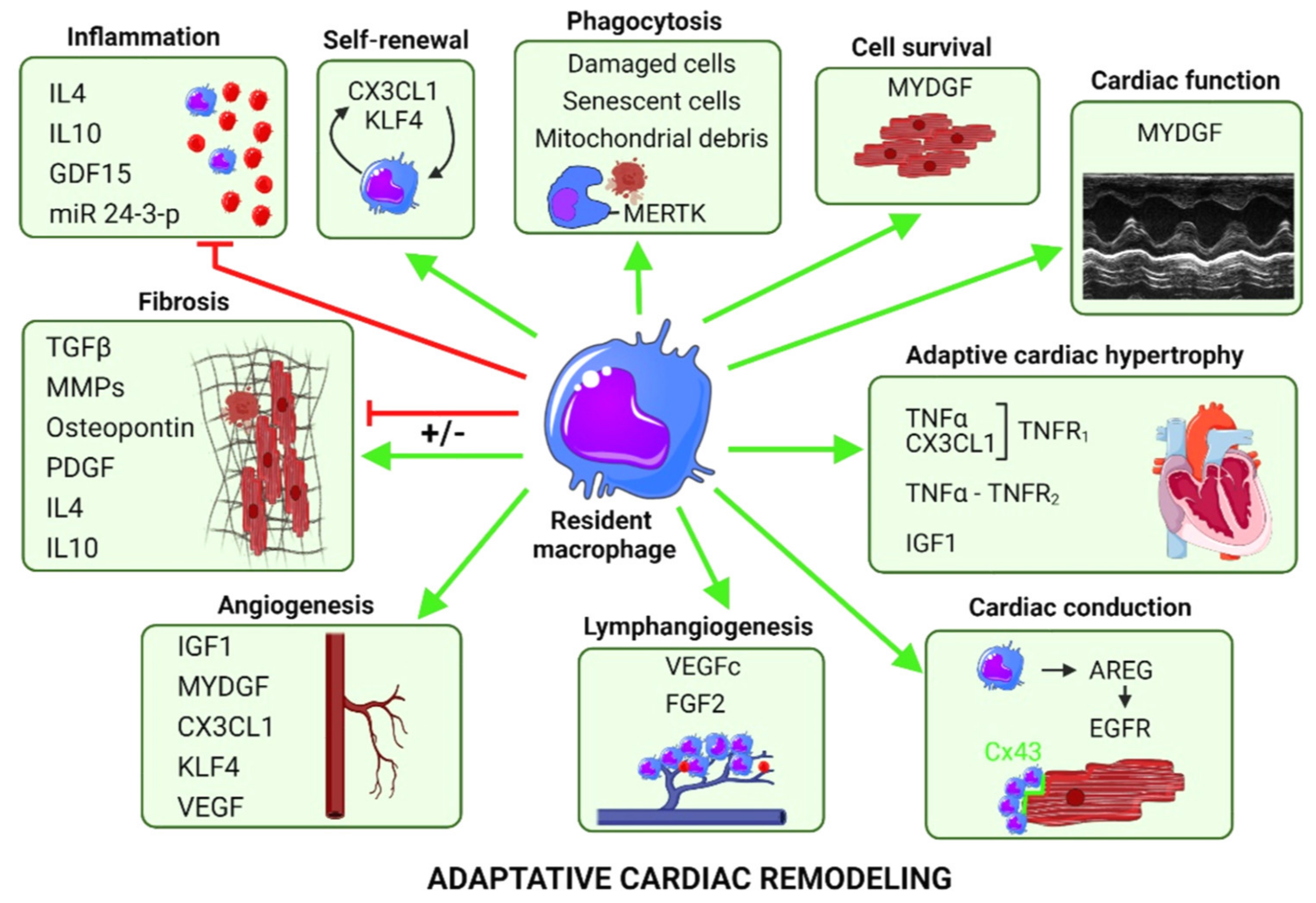
4.1.1. Protective Growth Factor Secretion by Resident Mφ
IGF1
AREG/EGFR
MYDGF
GDF15
VEGFc-d and FGF2
4.2. Protective Phagocytic Activity of Cardiac Mφ
4.3. Protective Signals Favoring Proliferation of Resident Mφ
4.4. Exosomes, Mir and Cardiac Mφ
4.5. Immune Response and Fibrosis in Aging and Myocardial Diseases
5. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Verma, A.; Meris, A.; Skali, H.; Ghali, J.K.; Arnold, J.M.O.; Bourgoun, M.; Velazquez, E.J.; McMurray, J.J.V.; Kober, L.; Pfeffer, M.A.; et al. Prognostic Implications of Left Ventricular Mass and Geometry Following Myocardial Infarction: The VALIANT (VALsartan in Acute Myocardial INfarcTion) Echocardiographic Study. JACC Cardiovas. Imaging 2008, 1, 582–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, D.L. Stress-Activated Cytokines and the Heart: From Adaptation to Maladaptation. Annu. Rev. Physiol. 2003, 65, 81–101. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Mechanisms of Physiological and Pathological Cardiac Hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef] [PubMed]
- Sasayama, S.; Ross, J.; Franklin, D.; Bloor, C.M.; Bishop, S.; Dilley, R.B. Adaptations of the Left Ventricle to Chronic Pressure Overload. Circ. Res. 1976, 38, 172–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flamant, M.; Mougenot, N.; Balse, E.; Le Fèvre, L.; Atassi, F.; Gautier, E.L.; Le Goff, W.; Keck, M.; Nadaud, S.; Combadière, C.; et al. Early Activation of the Cardiac CX3CL1/CX3CR1 Axis Delays β-Adrenergic-Induced Heart Failure. Sci. Rep. 2021, 11, 17982. [Google Scholar] [CrossRef] [PubMed]
- Lieb, W.; Gona, P.; Larson, M.G.; Aragam, J.; Zile, M.R.; Cheng, S.; Benjamin, E.J.; Vasan, R.S. The Natural History of Left Ventricular Geometry in the Community: Clinical Correlates and Prognostic Significance of Change in LV Geometric Pattern. JACC Cardiovasc. Imaging 2014, 7, 870–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, R.V.; Ahmed, M.I.; Marjan, M.; Aban, I.B.; Zile, M.R.; Ahmed, A. Natural History of Concentric Left Ventricular Geometry in Community-Dwelling Older Adults without Heart Failure during Seven Years of Follow-Up. Am. J. Cardiol. 2011, 107, 321–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maillet, M.; van Berlo, J.H.; Molkentin, J.D. Molecular Basis of Physiological Heart Growth: Fundamental Concepts and New Players. Nat. Rev. Mol. Cell Biol. 2013, 14, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Haider, A.W.; Larson, M.G.; Benjamin, E.J.; Levy, D. Increased Left Ventricular Mass and Hypertrophy Are Associated with Increased Risk for Sudden Death. J. Am. Coll. Cardiol. 1998, 32, 1454–1459. [Google Scholar] [CrossRef] [Green Version]
- Katz, A.M. Maladaptive Growth in the Failing Heart: The Cardiomyopathy of Overload. Cardiovasc. Drugs Ther. 2002, 16, 245–249. [Google Scholar] [CrossRef]
- Heineke, J.; Molkentin, J.D. Regulation of Cardiac Hypertrophy by Intracellular Signalling Pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600. [Google Scholar] [CrossRef]
- Meng, G.; Liu, J.; Liu, S.; Song, Q.; Liu, L.; Xie, L.; Han, Y.; Ji, Y. Hydrogen Sulfide Pretreatment Improves Mitochondrial Function in Myocardial Hypertrophy via a SIRT3-Dependent Manner. Br. J. Pharmacol. 2018, 175, 1126–1145. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, H.; Nishida, K.; Otsu, K. Macromolecular Degradation Systems and Cardiovascular Aging. Circ. Res. 2016, 118, 1577–1592. [Google Scholar] [CrossRef] [Green Version]
- Van de Veire, N.R.; De Backer, J.; Ascoop, A.-K.; Middernacht, B.; Velghe, A.; Sutter, J.D. Echocardiographically Estimated Left Ventricular End-Diastolic and Right Ventricular Systolic Pressure in Normotensive Healthy Individuals. Int. J. Cardiovasc. Imaging 2006, 22, 633–641. [Google Scholar] [CrossRef]
- Garciarena, C.D.; Pinilla, O.A.; Nolly, M.B.; Laguens, R.P.; Escudero, E.M.; Cingolani, H.E.; Ennis, I.L. Endurance Training in the Spontaneously Hypertensive Rat: Conversion of Pathological into Physiological Cardiac Hypertrophy. Hypertension 2009, 53, 708–714. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Li, G.; An, J.; Liu, C.; Zhu, X.; Xu, Y.; Gao, Y.; Li, J.; Liu, J.; Yan, J.; et al. Exercise Training Protects Against Heart Failure Via Expansion of Myeloid-Derived Suppressor Cells Through Regulating IL-10/STAT3/S100A9 Pathway. Circ. Heart Fail. 2021, 15, e008550. [Google Scholar] [CrossRef]
- Sharma, K.; Kass, D.A. Heart Failure with Preserved Ejection Fraction: Mechanisms, Clinical Features, and Therapies. Circ. Res. 2014, 115, 79–96. [Google Scholar] [CrossRef] [Green Version]
- Keller, K.M.; Howlett, S.E. Sex Differences in the Biology and Pathology of the Aging Heart. Can. J. Cardiol. 2016, 32, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- Hanna, A.; Frangogiannis, N.G. Inflammatory Cytokines and Chemokines as Therapeutic Targets in Heart Failure. Cardiovasc. Drugs Ther. 2020, 34, 849–863. [Google Scholar] [CrossRef]
- Dick, S.A.; Epelman, S. Chronic Heart Failure and Inflammation: What Do We Really Know? Circ. Res. 2016, 119, 159–176. [Google Scholar] [CrossRef] [Green Version]
- Frangogiannis, N.G. The Immune System and Cardiac Repair. Pharmacol. Res. 2008, 58, 88–111. [Google Scholar] [CrossRef] [Green Version]
- Hein, S.; Arnon, E.; Kostin, S.; Schönburg, M.; Elsässer, A.; Polyakova, V.; Bauer, E.P.; Klövekorn, W.-P.; Schaper, J. Progression from Compensated Hypertrophy to Failure in the Pressure-Overloaded Human Heart: Structural Deterioration and Compensatory Mechanisms. Circulation 2003, 107, 984–991. [Google Scholar] [CrossRef] [Green Version]
- Balakumar, P.; Jagadeesh, G. Multifarious Molecular Signaling Cascades of Cardiac Hypertrophy: Can the Muddy Waters Be Cleared? Pharmacol. Res. 2010, 62, 365–383. [Google Scholar] [CrossRef]
- Crozatier, B.; Ventura-Clapier, R. Inhibition of Hypertrophy, per Se, May Not Be a Good Therapeutic Strategy in Ventricular Pressure Overload: Other Approaches Could Be More Beneficial. Circulation 2015, 131, 1448–1457. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.J.; Borlaug, B.A.; Kitzman, D.W.; McCulloch, A.D.; Blaxall, B.C.; Agarwal, R.; Chirinos, J.A.; Collins, S.; Deo, R.C.; Gladwin, M.T.; et al. Research Priorities for Heart Failure with Preserved Ejection Fraction: National Heart, Lung, and Blood Institute Working Group Summary. Circulation 2020, 141, 1001–1026. [Google Scholar] [CrossRef]
- Levine, B.; Kalman, J.; Mayer, L.; Fillit, H.M.; Packer, M. Elevated Circulating Levels of Tumor Necrosis Factor in Severe Chronic Heart Failure. N. Engl. J. Med. 1990, 323, 236–241. [Google Scholar] [CrossRef]
- Mann, D.L. Innate Immunity and the Failing Heart: The Cytokine Hypothesis Revisited. Circ. Res. 2015, 116, 1254–1268. [Google Scholar] [CrossRef] [Green Version]
- Suetomi, T.; Willeford, A.; Brand, C.S.; Cho, Y.; Ross, R.S.; Miyamoto, S.; Brown, J.H. Inflammation and NLRP3 Inflammasome Activation Initiated in Response to Pressure Overload by Ca2+/Calmodulin-Dependent Protein Kinase II δ Signaling in Cardiomyocytes Are Essential for Adverse Cardiac Remodeling. Circulation 2018, 138, 2530–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Zhang, J.; Xu, Y.; Liu, J.; Ye, J.; Wang, Z.; Ye, D.; Feng, Y.; Xu, S.; Pan, W.; et al. Selective Inhibition of NLRP3 Inflammasome Reverses Pressure Overload-Induced Pathological Cardiac Remodeling by Attenuating Hypertrophy, Fibrosis, and Inflammation. Int. Immunopharmacol. 2021, 99, 108046. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Hastings, M.H.; Rhee, J.; Trager, L.E.; Roh, J.D.; Rosenzweig, A. Targeting Age-Related Pathways in Heart Failure. Circ. Res. 2020, 126, 533–551. [Google Scholar] [CrossRef] [PubMed]
- Meschiari, C.A.; Ero, O.K.; Pan, H.; Finkel, T.; Lindsey, M.L. The Impact of Aging on Cardiac Extracellular Matrix. Geroscience 2017, 39, 7–18. [Google Scholar] [CrossRef]
- Wu, J.; Xia, S.; Kalionis, B.; Wan, W.; Sun, T. The Role of Oxidative Stress and Inflammation in Cardiovascular Aging. BioMed Res. Int. 2014, 2014, 615312. [Google Scholar] [CrossRef]
- Izzo, C.; Vitillo, P.; Di Pietro, P.; Visco, V.; Strianese, A.; Virtuoso, N.; Ciccarelli, M.; Galasso, G.; Carrizzo, A.; Vecchione, C. The Role of Oxidative Stress in Cardiovascular Aging and Cardiovascular Diseases. Life 2021, 11, 60. [Google Scholar] [CrossRef]
- Kubota, T.; McTiernan, C.F.; Frye, C.S.; Slawson, S.E.; Lemster, B.H.; Koretsky, A.P.; Demetris, A.J.; Feldman, A.M. Dilated Cardiomyopathy in Transgenic Mice with Cardiac-Specific Overexpression of Tumor Necrosis Factor-Alpha. Circ. Res. 1997, 81, 627–635. [Google Scholar] [CrossRef]
- Bryant, D.; Becker, L.; Richardson, J.; Shelton, J.; Franco, F.; Peshock, R.; Thompson, M.; Giroir, B. Cardiac Failure in Transgenic Mice with Myocardial Expression of Tumor Necrosis Factor-Alpha. Circulation 1998, 97, 1375–1381. [Google Scholar] [CrossRef] [Green Version]
- Sivasubramanian, N.; Coker, M.L.; Kurrelmeyer, K.M.; MacLellan, W.R.; DeMayo, F.J.; Spinale, F.G.; Mann, D.L. Left Ventricular Remodeling in Transgenic Mice with Cardiac Restricted Overexpression of Tumor Necrosis Factor. Circulation 2001, 104, 826–831. [Google Scholar] [CrossRef] [Green Version]
- Franco, F.; Thomas, G.D.; Giroir, B.; Bryant, D.; Bullock, M.C.; Chwialkowski, M.C.; Victor, R.G.; Peshock, R.M. Magnetic Resonance Imaging and Invasive Evaluation of Development of Heart Failure in Transgenic Mice with Myocardial Expression of Tumor Necrosis Factor-Alpha. Circulation 1999, 99, 448–454. [Google Scholar] [CrossRef] [Green Version]
- Machida, Y.; Kubota, T.; Kawamura, N.; Funakoshi, H.; Ide, T.; Utsumi, H.; Li, Y.Y.; Feldman, A.M.; Tsutsui, H.; Shimokawa, H.; et al. Overexpression of Tumor Necrosis Factor-Alpha Increases Production of Hydroxyl Radical in Murine Myocardium. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H449–H455. [Google Scholar] [CrossRef] [Green Version]
- Engel, D.; Peshock, R.; Armstong, R.C.; Sivasubramanian, N.; Mann, D.L. Cardiac Myocyte Apoptosis Provokes Adverse Cardiac Remodeling in Transgenic Mice with Targeted TNF Overexpression. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1303–H1311. [Google Scholar] [CrossRef] [Green Version]
- Meldrum, D.R. Tumor Necrosis Factor in the Heart. Am. J. Physiol. 1998, 274, R577–R595. [Google Scholar] [CrossRef]
- Dutka, D.P.; Elborn, J.S.; Delamere, F.; Shale, D.J.; Morris, G.K. Tumour Necrosis Factor Alpha in Severe Congestive Cardiac Failure. Br. Heart J. 1993, 70, 141–143. [Google Scholar] [CrossRef] [Green Version]
- Matsumori, A.; Yamada, T.; Suzuki, H.; Matoba, Y.; Sasayama, S. Increased Circulating Cytokines in Patients with Myocarditis and Cardiomyopathy. Br. Heart J. 1994, 72, 561–566. [Google Scholar] [CrossRef]
- Torre-Amione, G.; Kapadia, S.; Benedict, C.; Oral, H.; Young, J.B.; Mann, D.L. Proinflammatory Cytokine Levels in Patients with Depressed Left Ventricular Ejection Fraction: A Report from the Studies of Left Ventricular Dysfunction (SOLVD). J. Am. Coll. Cardiol. 1996, 27, 1201–1206. [Google Scholar] [CrossRef] [Green Version]
- Seta, Y.; Shan, K.; Bozkurt, B.; Oral, H.; Mann, D.L. Basic Mechanisms in Heart Failure: The Cytokine Hypothesis. J. Card. Fail. 1996, 2, 243–249. [Google Scholar] [CrossRef]
- Sun, M.; Chen, M.; Dawood, F.; Zurawska, U.; Li, J.Y.; Parker, T.; Kassiri, Z.; Kirshenbaum, L.A.; Arnold, M.; Khokha, R.; et al. Tumor Necrosis Factor-Alpha Mediates Cardiac Remodeling and Ventricular Dysfunction after Pressure Overload State. Circulation 2007, 115, 1398–1407. [Google Scholar] [CrossRef] [Green Version]
- Mann, D.L.; McMurray, J.J.V.; Packer, M.; Swedberg, K.; Borer, J.S.; Colucci, W.S.; Djian, J.; Drexler, H.; Feldman, A.; Kober, L.; et al. Targeted Anticytokine Therapy in Patients with Chronic Heart Failure: Results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation 2004, 109, 1594–1602. [Google Scholar] [CrossRef] [Green Version]
- Chung, E.S.; Packer, M.; Lo, K.H.; Fasanmade, A.A.; Willerson, J.T. Anti-TNF Therapy against Congestive Heart Failure Investigators Randomized, Double-Blind, Placebo-Controlled, Pilot Trial of Infliximab, a Chimeric Monoclonal Antibody to Tumor Necrosis Factor-Alpha, in Patients with Moderate-to-Severe Heart Failure: Results of the Anti-TNF Therapy against Congestive Heart Failure (ATTACH) Trial. Circulation 2003, 107, 3133–3140. [Google Scholar] [CrossRef] [Green Version]
- Anker, S.D.; Coats, A.J.S. How to RECOVER from RENAISSANCE? The Significance of the Results of RECOVER, RENAISSANCE, RENEWAL and ATTACH. Int. J. Cardiol. 2002, 86, 123–130. [Google Scholar] [CrossRef]
- Mann, D.L. Inflammatory Mediators and the Failing Heart: Past, Present, and the Foreseeable Future. Circ. Res. 2002, 91, 988–998. [Google Scholar] [CrossRef]
- Mann, D.L. The Effect of Tumor Necrosis Factor-Alpha on Cardiac Structure and Function: A Tale of Two Cytokines. J. Card. Fail. 1996, 2, S165–S172. [Google Scholar] [CrossRef]
- Sack, M. Tumor Necrosis Factor-Alpha in Cardiovascular Biology and the Potential Role for Anti-Tumor Necrosis Factor-Alpha Therapy in Heart Disease. Pharmacol. Ther. 2002, 94, 123–135. [Google Scholar] [CrossRef]
- Hajjar, R.J.; Leopold, J.A. Inflammation and Heart Failure: Friend or Foe? Circulation 2021, 144, 1241–1243. [Google Scholar] [CrossRef] [PubMed]
- Adamo, L.; Rocha-Resende, C.; Prabhu, S.D.; Mann, D.L. Reappraising the Role of Inflammation in Heart Failure. Nat. Rev. Cardiol. 2020, 17, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Vanderheyden, M.; Paulus, W.J.; Voss, M.; Knuefermann, P.; Sivasubramanian, N.; Mann, D.; Baumgarten, G. Myocardial Cytokine Gene Expression Is Higher in Aortic Stenosis than in Idiopathic Dilated Cardiomyopathy. Heart 2005, 91, 926–931. [Google Scholar] [CrossRef]
- Norton, G.R.; Peterson, V.R.; Robinson, C.; Norman, G.; Libhaber, C.D.; Libhaber, E.; Gomes, M.; Sareli, P.; Woodiwiss, A.J. Independent of Left Ventricular Mass, Circulating Inflammatory Markers Rather than Pressure Load Are Associated with Concentric Left Ventricular Remodelling. Int. J. Cardiol. 2019, 274, 342–347. [Google Scholar] [CrossRef]
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An Endotoxin-Induced Serum Factor That Causes Necrosis of Tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666–3670. [Google Scholar] [CrossRef] [Green Version]
- Torre-Amione, G.; Kapadia, S.; Lee, J.; Bies, R.D.; Lebovitz, R.; Mann, D.L. Expression and Functional Significance of Tumor Necrosis Factor Receptors in Human Myocardium. Circulation 1995, 92, 1487–1493. [Google Scholar] [CrossRef]
- Grell, M.; Douni, E.; Wajant, H.; Löhden, M.; Clauss, M.; Maxeiner, B.; Georgopoulos, S.; Lesslauer, W.; Kollias, G.; Pfizenmaier, K.; et al. The Transmembrane Form of Tumor Necrosis Factor Is the Prime Activating Ligand of the 80 KDa Tumor Necrosis Factor Receptor. Cell 1995, 83, 793–802. [Google Scholar] [CrossRef] [Green Version]
- Borghi, A.; Verstrepen, L.; Beyaert, R. TRAF2 Multitasking in TNF Receptor-Induced Signaling to NF-ΚB, MAP Kinases and Cell Death. Biochem. Pharmacol. 2016, 116, 9. [Google Scholar] [CrossRef]
- Gough, P.; Myles, I.A. Tumor Necrosis Factor Receptors: Pleiotropic Signaling Complexes and Their Differential Effects. Front. Immunol. 2020, 11, 585880. [Google Scholar] [CrossRef]
- Schumacher, S.M.; Naga Prasad, S.V. Tumor Necrosis Factor-α in Heart Failure: An Updated Review. Curr. Cardiol. Rep. 2018, 20, 117. [Google Scholar] [CrossRef]
- Zee, K.J.V.; Kohno, T.; Fischer, E.; Rock, C.S.; Moldawer, L.L.; Lowry, S.F. Tumor Necrosis Factor Soluble Receptors Circulate during Experimental and Clinical Inflammation and Can Protect against Excessive Tumor Necrosis Factor Alpha In Vitro and In Vivo. Proc. Natl. Acad. Sci. USA 1992, 89, 4845–4849. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, T.; Nakano, M.; Bednarczyk, J.L.; McIntyre, B.W.; Entman, M.; Mann, D.L. Tumor Necrosis Factor-Alpha Provokes a Hypertrophic Growth Response in Adult Cardiac Myocytes. Circulation 1997, 95, 1247–1252. [Google Scholar] [CrossRef]
- Sekiguchi, K.; Li, X.; Coker, M.; Flesch, M.; Barger, P.M.; Sivasubramanian, N.; Mann, D.L. Cross-Regulation between the Renin-Angiotensin System and Inflammatory Mediators in Cardiac Hypertrophy and Failure. Cardiovasc. Res. 2004, 63, 433–442. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, Y.; Otsu, K.; Nishida, K.; Hirotani, S.; Nakayama, H.; Yamaguchi, O.; Matsumura, Y.; Ueno, H.; Tada, M.; Hori, M. Involvement of Reactive Oxygen Species-Mediated NF- κ B Activation in TNF- α -Induced Cardiomyocyte Hypertrophy. J. Mol. Cell. Cardiol. 2002, 34, 233–240. [Google Scholar] [CrossRef]
- Nakamura, K.; Fushimi, K.; Kouchi, H.; Mihara, K.; Miyazaki, M.; Ohe, T.; Namba, M. Inhibitory Effects of Antioxidants on Neonatal Rat Cardiac Myocyte Hypertrophy Induced by Tumor Necrosis Factor-Alpha and Angiotensin II. Circulation 1998, 98, 794–799. [Google Scholar] [CrossRef] [Green Version]
- Condorelli, G.; Morisco, C.; Latronico, M.V.G.; Claudio, P.P.; Dent, P.; Tsichlis, P.; Condorelli, G.; Frati, G.; Drusco, A.; Croce, C.M.; et al. TNF-Alpha Signal Transduction in Rat Neonatal Cardiac Myocytes: Definition of Pathways Generating from the TNF-Alpha Receptor. FASEB J. 2002, 16, 1732–1737. [Google Scholar] [CrossRef]
- Higuchi, Y.; McTiernan, C.F.; Frye, C.B.; McGowan, B.S.; Chan, T.O.; Feldman, A.M. Tumor Necrosis Factor Receptors 1 and 2 Differentially Regulate Survival, Cardiac Dysfunction, and Remodeling in Transgenic Mice with Tumor Necrosis Factor-Alpha-Induced Cardiomyopathy. Circulation 2004, 109, 1892–1897. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.Y.; Feng, Y.Q.; Kadokami, T.; McTiernan, C.F.; Draviam, R.; Watkins, S.C.; Feldman, A.M. Myocardial Extracellular Matrix Remodeling in Transgenic Mice Overexpressing Tumor Necrosis Factor Alpha Can Be Modulated by Anti-Tumor Necrosis Factor Alpha Therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 12746–12751. [Google Scholar] [CrossRef] [Green Version]
- Dibbs, Z.I.; Diwan, A.; Nemoto, S.; DeFreitas, G.; Abdellatif, M.; Carabello, B.A.; Spinale, F.G.; Feuerstein, G.; Sivasubramanian, N.; Mann, D.L. Targeted Overexpression of Transmembrane Tumor Necrosis Factor Provokes a Concentric Cardiac Hypertrophic Phenotype. Circulation 2003, 108, 1002–1008. [Google Scholar] [CrossRef] [Green Version]
- Diwan, A.; Dibbs, Z.; Nemoto, S.; DeFreitas, G.; Carabello, B.A.; Sivasubramanian, N.; Wilson, E.M.; Spinale, F.G.; Mann, D.L. Targeted Overexpression of Noncleavable and Secreted Forms of Tumor Necrosis Factor Provokes Disparate Cardiac Phenotypes. Circulation 2004, 109, 262–268. [Google Scholar] [CrossRef] [Green Version]
- Miao, K.; Zhou, L.; Ba, H.; Li, C.; Gu, H.; Yin, B.; Wang, J.; Yang, X.-P.; Li, Z.; Wang, D.W. Transmembrane Tumor Necrosis Factor Alpha Attenuates Pressure-Overload Cardiac Hypertrophy via Tumor Necrosis Factor Receptor 2. PLoS Biol. 2020, 18, e3000967. [Google Scholar] [CrossRef]
- Dittrich, G.M.; Heineke, J. TNF-α Signaling: TACE Inhibition to Put out the Burning Heart. PLoS Biol. 2020, 18, e3001037. [Google Scholar] [CrossRef]
- Garlie, J.B.; Hamid, T.; Gu, Y.; Ismahil, M.A.; Chandrasekar, B.; Prabhu, S.D. Tumor Necrosis Factor Receptor 2 Signaling Limits Beta-Adrenergic Receptor-Mediated Cardiac Hypertrophy In Vivo. Basic Res. Cardiol. 2011, 106, 1193–1205. [Google Scholar] [CrossRef]
- Chan, T.O.; Tsichlis, P.N. PDK2: A Complex Tail in One Akt. Sci. STKE 2001, 2001, pe1. [Google Scholar] [CrossRef]
- Capetanaki, Y.; Papathanasiou, S.; Diokmetzidou, A.; Vatsellas, G.; Tsikitis, M. Desmin Related Disease: A Matter of Cell Survival Failure. Curr. Opin. Cell Biol. 2015, 32, 113–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panagopoulou, P.; Davos, C.H.; Milner, D.J.; Varela, E.; Cameron, J.; Mann, D.L.; Capetanaki, Y. Desmin Mediates TNF-Alpha-Induced Aggregate Formation and Intercalated Disk Reorganization in Heart Failure. J. Cell Biol. 2008, 181, 761–775. [Google Scholar] [CrossRef] [PubMed]
- Papathanasiou, S.; Rickelt, S.; Soriano, M.E.; Schips, T.G.; Maier, H.J.; Davos, C.H.; Varela, A.; Kaklamanis, L.; Mann, D.L.; Capetanaki, Y. Tumor Necrosis Factor-α Confers Cardioprotection through Ectopic Expression of Keratins K8 and K18. Nat. Med. 2015, 21, 1076–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartekova, M.; Radosinska, J.; Jelemensky, M.; Dhalla, N.S. Role of Cytokines and Inflammation in Heart Function during Health and Disease. Heart Fail. Rev. 2018, 23, 733–758. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.W.; Shaw, J.A.; Kirshenbaum, L.A. Multiple Facets of NF-ΚB in the Heart. Circ. Res. 2011, 108, 1122–1132. [Google Scholar] [CrossRef] [Green Version]
- Regula, K.M.; Baetz, D.; Kirshenbaum, L.A. Nuclear Factor-KappaB Represses Hypoxia-Induced Mitochondrial Defects and Cell Death of Ventricular Myocytes. Circulation 2004, 110, 3795–3802. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Joseph, L.C.; Gurin, M.I.; Thorp, E.B.; Morrow, J.P. Extracellular Signal-Regulated Kinase Activation during Cardiac Hypertrophy Reduces Sarcoplasmic/Endoplasmic Reticulum Calcium ATPase 2 (SERCA2) Transcription. J. Mol. Cell. Cardiol. 2014, 75, 58–63. [Google Scholar] [CrossRef] [Green Version]
- Forman, K.; Vara, E.; García, C.; Kireev, R.; Cuesta, S.; Acuña-Castroviejo, D.; Tresguerres, J.A.F. Influence of Aging and Growth Hormone on Different Members of the NFkB Family and IkB Expression in the Heart from a Murine Model of Senescence-Accelerated Aging. Exp. Gerontol. 2016, 73, 114–120. [Google Scholar] [CrossRef]
- Helenius, M.; Hänninen, M.; Lehtinen, S.K.; Salminen, A. Aging-Induced up-Regulation of Nuclear Binding Activities of Oxidative Stress Responsive NF-KB Transcription Factor in Mouse Cardiac Muscle. J. Mol. Cell. Cardiol. 1996, 28, 487–498. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Ong, H.; Tan, T.; Park, K.H.; Bian, Z.; Zou, X.; Haggard, E.; Janssen, P.M.; Merritt, R.E.; et al. MG53 Suppresses NF-ΚB Activation to Mitigate Age-Related Heart Failure. JCI Insight 2021, 6, 148375. [Google Scholar] [CrossRef]
- Qu, Y.-C.; Du, Y.-M.; Wu, S.-L.; Chen, Q.-X.; Wu, H.-L.; Zhou, S.-F. Activated Nuclear Factor-KappaB and Increased Tumor Necrosis Factor-Alpha in Atrial Tissue of Atrial Fibrillation. Scand. Cardiovasc. J. 2009, 43, 292–297. [Google Scholar] [CrossRef]
- Mishra, A.; Srivastava, A.; Mittal, T.; Garg, N.; Mittal, B. Role of Inflammatory Gene Polymorphisms in Left Ventricular Dysfunction (LVD) Susceptibility in Coronary Artery Disease (CAD) Patients. Cytokine 2013, 61, 856–861. [Google Scholar] [CrossRef]
- Santos, D.G.B.; Resende, M.F.; Mill, J.G.; Mansur, A.J.; Krieger, J.E.; Pereira, A.C. Nuclear Factor (NF) KappaB Polymorphism Is Associated with Heart Function in Patients with Heart Failure. BMC Med. Genet. 2010, 11, 89. [Google Scholar] [CrossRef]
- Chandrasekar, B.; Colston, J.T.; de la Rosa, S.D.; Rao, P.P.; Freeman, G.L. TNF-Alpha and H2O2 Induce IL-18 and IL-18R Beta Expression in Cardiomyocytes via NF-Kappa B Activation. Biochem. Biophys. Res. Commun. 2003, 303, 1152–1158. [Google Scholar] [CrossRef]
- Lecour, S.; James, R.W. When Are Pro-Inflammatory Cytokines SAFE in Heart Failure? Eur. Heart J. 2011, 32, 680–685. [Google Scholar] [CrossRef] [Green Version]
- Evans, S.; Tzeng, H.-P.; Veis, D.J.; Matkovich, S.; Weinheimer, C.; Kovacs, A.; Barger, P.M.; Mann, D.L. TNF Receptor–Activated Factor 2 Mediates Cardiac Protection through Noncanonical NF-ΚB Signaling. JCI Insight 2018, 3, e98278. [Google Scholar] [CrossRef]
- Amgalan, D.; Chen, Y.; Kitsis, R.N. Death Receptor Signaling in the Heart: Cell Survival, Apoptosis, and Necroptosis. Circulation 2017, 136, 743–746. [Google Scholar] [CrossRef]
- Guo, X.; Yin, H.; Li, L.; Chen, Y.; Li, J.; Doan, J.; Steinmetz, R.; Liu, Q. Cardioprotective Role of Tumor Necrosis Factor Receptor-Associated Factor 2 by Suppressing Apoptosis and Necroptosis. Circulation 2017, 136, 729–742. [Google Scholar] [CrossRef]
- Yang, K.-C.; Ma, X.; Liu, H.; Murphy, J.; Barger, P.M.; Mann, D.L.; Diwan, A. Tumor Necrosis Factor Receptor-Associated Factor 2 Mediates Mitochondrial Autophagy. Circ. Heart Fail. 2015, 8, 175–187. [Google Scholar] [CrossRef] [Green Version]
- Mehra, V.C.; Ramgolam, V.S.; Bender, J.R. Cytokines and Cardiovascular Disease. J. Leukoc. Biol. 2005, 78, 805–818. [Google Scholar] [CrossRef] [Green Version]
- Prabhu, S.D. Cytokine-Induced Modulation of Cardiac Function. Circ. Res. 2004, 95, 1140–1153. [Google Scholar] [CrossRef]
- Yokoyama, T.; Vaca, L.; Rossen, R.D.; Durante, W.; Hazarika, P.; Mann, D.L. Cellular Basis for the Negative Inotropic Effects of Tumor Necrosis Factor-Alpha in the Adult Mammalian Heart. J. Clin. Investig. 1993, 92, 2303–2312. [Google Scholar] [CrossRef] [Green Version]
- Suematsu, N.; Tsutsui, H.; Wen, J.; Kang, D.; Ikeuchi, M.; Ide, T.; Hayashidani, S.; Shiomi, T.; Kubota, T.; Hamasaki, N.; et al. Oxidative Stress Mediates Tumor Necrosis Factor-Alpha-Induced Mitochondrial DNA Damage and Dysfunction in Cardiac Myocytes. Circulation 2003, 107, 1418–1423. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Andrieu-Abadie, N.; Levade, T.; Zhang, P.; Obeid, L.M.; Hannun, Y.A. Glutathione Regulation of Neutral Sphingomyelinase in Tumor Necrosis Factor-Alpha-Induced Cell Death. J. Biol. Chem. 1998, 273, 11313–11320. [Google Scholar] [CrossRef] [Green Version]
- Oral, H.; Dorn, G.W.; Mann, D.L. Sphingosine Mediates the Immediate Negative Inotropic Effects of Tumor Necrosis Factor-Alpha in the Adult Mammalian Cardiac Myocyte. J. Biol. Chem. 1997, 272, 4836–4842. [Google Scholar] [CrossRef] [Green Version]
- Defer, N.; Azroyan, A.; Pecker, F.; Pavoine, C. TNFR1 and TNFR2 Signaling Interplay in Cardiac Myocytes. J. Biol. Chem. 2007, 282, 35564–35573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cailleret, M.; Amadou, A.; Andrieu-Abadie, N.; Nawrocki, A.; Adamy, C.; Ait-Mamar, B.; Rocaries, F.; Best-Belpomme, M.; Levade, T.; Pavoine, C.; et al. N-Acetylcysteine Prevents the Deleterious Effect of Tumor Necrosis Factor-(Alpha) on Calcium Transients and Contraction in Adult Rat Cardiomyocytes. Circulation 2004, 109, 406–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, T.M.A.; Zi, M.; Prehar, S.; Maqsood, A.; Abou-Leisa, R.; Nguyen, L.; Pfeifer, G.P.; Cartwright, E.J.; Neyses, L.; Oceandy, D. The Tumour Suppressor Ras-Association Domain Family Protein 1A (RASSF1A) Regulates TNF-α Signalling in Cardiomyocytes. Cardiovasc. Res. 2014, 103, 47–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, D.R.; Freeman, G.L. Tumor Necrosis Factor-Alpha Induces a Biphasic Effect on Myocardial Contractility in Conscious Dogs. Circ. Res. 1996, 78, 154–160. [Google Scholar] [CrossRef]
- Pagani, F.D.; Baker, L.S.; Hsi, C.; Knox, M.; Fink, M.P.; Visner, M.S. Left Ventricular Systolic and Diastolic Dysfunction after Infusion of Tumor Necrosis Factor-Alpha in Conscious Dogs. J. Clin. Investig. 1992, 90, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Krown, K.A.; Yasui, K.; Brooker, M.J.; Dubin, A.E.; Nguyen, C.; Harris, G.L.; McDonough, P.M.; Glembotski, C.C.; Palade, P.T.; Sabbadini, R.A. TNF Alpha Receptor Expression in Rat Cardiac Myocytes: TNF Alpha Inhibition of L-Type Ca2+ Current and Ca2+ Transients. FEBS Lett. 1995, 376, 24–30. [Google Scholar] [CrossRef] [Green Version]
- Amadou, A.; Nawrocki, A.; Best-Belpomme, M.; Pavoine, C.; Pecker, F. Arachidonic Acid Mediates Dual Effect of TNF-Alpha on Ca2+ Transients and Contraction of Adult Rat Cardiomyocytes. Am. J. Physiol. Cell Physiol. 2002, 282, C1339–C1347. [Google Scholar] [CrossRef] [Green Version]
- Bourraindeloup, M.; Adamy, C.; Candiani, G.; Cailleret, M.; Bourin, M.-C.; Badoual, T.; Su, J.B.; Adubeiro, S.; Roudot-Thoraval, F.; Dubois-Rande, J.-L.; et al. N-Acetylcysteine Treatment Normalizes Serum Tumor Necrosis Factor-Alpha Level and Hinders the Progression of Cardiac Injury in Hypertensive Rats. Circulation 2004, 110, 2003–2009. [Google Scholar] [CrossRef] [Green Version]
- Adamy, C.; Le Corvoisier, P.; Candiani, G.; Kirsch, M.; Pavoine, C.; Defer, N.; Bourin, M.C.; Su, J.B.; Vermes, E.; Hittinger, L.; et al. Tumor Necrosis Factor Alpha and Glutathione Interplay in Chronic Heart Failure. Arch. Malad. Coeur Vaiss. 2005, 98, 906–912. [Google Scholar]
- Leslie, C.C. Cytosolic Phospholipase A2: Physiological Function and Role in Disease. J. Lipid Res. 2015, 56, 1386–1402. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, M.; Tucker, D.E.; Burchett, S.A.; Leslie, C.C. Properties of the Group IV Phospholipase A2 Family. Prog. Lipid Res. 2006, 45, 487–510. [Google Scholar] [CrossRef]
- Jayadev, S.; Linardic, C.M.; Hannun, Y.A. Identification of Arachidonic Acid as a Mediator of Sphingomyelin Hydrolysis in Response to Tumor Necrosis Factor Alpha. J. Biol. Chem. 1994, 269, 5757–5763. [Google Scholar] [CrossRef]
- Jupp, O.J.; Vandenabeele, P.; MacEwan, D.J. Distinct Regulation of Cytosolic Phospholipase A2 Phosphorylation, Translocation, Proteolysis and Activation by Tumour Necrosis Factor-Receptor Subtypes. Biochem. J. 2003, 374, 453–461. [Google Scholar] [CrossRef]
- Murray, D.R.; Prabhu, S.D.; Chandrasekar, B. Chronic Beta-Adrenergic Stimulation Induces Myocardial Proinflammatory Cytokine Expression. Circulation 2000, 101, 2338–2341. [Google Scholar] [CrossRef] [Green Version]
- Brodde, O.E.; Michel, M.C. Adrenergic and Muscarinic Receptors in the Human Heart. Pharmacol. Rev. 1999, 51, 651–690. [Google Scholar]
- Ait-Mamar, B.; Cailleret, M.; Rucker-Martin, C.; Bouabdallah, A.; Candiani, G.; Adamy, C.; Duvaldestin, P.; Pecker, F.; Defer, N.; Pavoine, C. The Cytosolic Phospholipase A2 Pathway, a Safeguard of Beta2-Adrenergic Cardiac Effects in Rat. J. Biol. Chem. 2005, 280, 18881–18890. [Google Scholar] [CrossRef] [Green Version]
- Pavoine, C.; Magne, S.; Sauvadet, A.; Pecker, F. Evidence for a Beta2-Adrenergic/Arachidonic Acid Pathway in Ventricular Cardiomyocytes. Regulation by the Beta1-Adrenergic/Camp Pathway. J. Biol. Chem. 1999, 274, 628–637. [Google Scholar] [CrossRef] [Green Version]
- Pavoine, C.; Defer, N. The Cardiac Beta2-Adrenergic Signalling a New Role for the CPLA2. Cell. Signal. 2005, 17, 141–152. [Google Scholar] [CrossRef]
- Pavoine, C.; Behforouz, N.; Gauthier, C.; Le Gouvello, S.; Roudot-Thoraval, F.; Martin, C.R.; Pawlak, A.; Feral, C.; Defer, N.; Houel, R.; et al. Beta2-Adrenergic Signaling in Human Heart: Shift from the Cyclic AMP to the Arachidonic Acid Pathway. Mol. Pharmacol. 2003, 64, 1117–1125. [Google Scholar] [CrossRef] [Green Version]
- Magne, S.; Couchie, D.; Pecker, F.; Pavoine, C. Beta(2)-Adrenergic Receptor Agonists Increase Intracellular Free Ca(2+) Concentration Cycling in Ventricular Cardiomyocytes through P38 and P42/44 MAPK-Mediated Cytosolic Phospholipase A(2) Activation. J. Biol. Chem. 2001, 276, 39539–39548. [Google Scholar] [CrossRef] [Green Version]
- Pabbidi, M.R.; Ji, X.; Maxwell, J.T.; Mignery, G.A.; Samarel, A.M.; Lipsius, S.L. Inhibition of CAMP-Dependent PKA Activates Β2-Adrenergic Receptor Stimulation of Cytosolic Phospholipase A2 via Raf-1/MEK/ERK and IP3-Dependent Ca2+ Signaling in Atrial Myocytes. PLoS ONE 2016, 11, e0168505. [Google Scholar] [CrossRef]
- Jo, S.-H.; Leblais, V.; Wang, P.H.; Crow, M.T.; Xiao, R.-P. Phosphatidylinositol 3-Kinase Functionally Compartmentalizes the Concurrent G(s) Signaling during Beta2-Adrenergic Stimulation. Circ. Res. 2002, 91, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Chesley, A.; Lundberg, M.S.; Asai, T.; Xiao, R.P.; Ohtani, S.; Lakatta, E.G.; Crow, M.T. The Beta(2)-Adrenergic Receptor Delivers an Antiapoptotic Signal to Cardiac Myocytes through G(i)-Dependent Coupling to Phosphatidylinositol 3’-Kinase. Circ. Res. 2000, 87, 1172–1179. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.Z.; Zheng, M.; Koch, W.J.; Lefkowitz, R.J.; Kobilka, B.K.; Xiao, R.P. Dual Modulation of Cell Survival and Cell Death by Beta(2)-Adrenergic Signaling in Adult Mouse Cardiac Myocytes. Proc. Natl. Acad. Sci. USA 2001, 98, 1607–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Wang, J.; Brand, D.D.; Zheng, S.G. Role of TNF–TNF Receptor 2 Signal in Regulatory T Cells and Its Therapeutic Implications. Front. Immunol. 2018, 9, 784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haq, S.; Kilter, H.; Michael, A.; Tao, J.; O’Leary, E.; Sun, X.M.; Walters, B.; Bhattacharya, K.; Chen, X.; Cui, L.; et al. Deletion of Cytosolic Phospholipase A2 Promotes Striated Muscle Growth. Nat. Med. 2003, 9, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.F. Cardiac Actions of Protein Kinase C Isoforms. Physiology 2012, 27, 130–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunapuli, P.; Lawson, J.A.; Rokach, J.A.; Meinkoth, J.L.; FitzGerald, G.A. Prostaglandin F2alpha (PGF2alpha) and the Isoprostane, 8, 12-Iso-Isoprostane F2alpha-III, Induce Cardiomyocyte Hypertrophy. Differential Activation of Downstream Signaling Pathways. J. Biol. Chem. 1998, 273, 22442–22452. [Google Scholar] [CrossRef] [Green Version]
- Lai, J.; Jin, H.; Yang, R.; Winer, J.; Li, W.; Yen, R.; King, K.L.; Zeigler, F.; Ko, A.; Cheng, J.; et al. Prostaglandin F2 Alpha Induces Cardiac Myocyte Hypertrophy In Vitro and Cardiac Growth In Vivo. Am. J. Physiol. 1996, 271, H2197–H2208. [Google Scholar] [CrossRef]
- Saliba, Y.; Keck, M.; Marchand, A.; Atassi, F.; Ouille, A.; Cazorla, O.; Trebak, M.; Pavoine, C.; Lacampagne, A.; Hulot, J.S.; et al. Emergence of Orai3 Activity during Cardiac Hypertrophy. Cardiovasc. Res. 2015, 105, 248–259. [Google Scholar] [CrossRef] [Green Version]
- Keck, M.; Flamant, M.; Mougenot, N.; Favier, S.; Atassi, F.; Barbier, C.; Nadaud, S.; Lompré, A.-M.; Hulot, J.-S.; Pavoine, C. Cardiac Inflammatory CD11b/c Cells Exert a Protective Role in Hypertrophied Cardiomyocyte by Promoting TNFR 2—and Orai3- Dependent Signaling. Sci. Rep. 2019, 9, 6047. [Google Scholar] [CrossRef] [Green Version]
- Shuttleworth, T.J. Selective Activation of Distinct Orai Channels by STIM1. Cell Calcium 2017, 63, 40–42. [Google Scholar] [CrossRef] [Green Version]
- Trebak, M.; Putney, J.W. ORAI Calcium Channels. Physiology 2017, 32, 332–342. [Google Scholar] [CrossRef]
- Skoda, M.; Stangret, A.; Szukiewicz, D. Fractalkine and Placental Growth Factor: A Duet of Inflammation and Angiogenesis in Cardiovascular Disorders. Cytokine Growth Factor Rev. 2018, 39, 116–123. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, R.; Chakrabarti, S.; Su, Z. Resident Macrophages as Potential Therapeutic Targets for Cardiac Ageing and Injury. Clin. Transl. Immunol. 2020, 9, e1167. [Google Scholar] [CrossRef]
- Alvarez-Argote, S.; O’Meara, C.C. The Evolving Roles of Cardiac Macrophages in Homeostasis, Regeneration, and Repair. Int. J. Mol. Sci. 2021, 22, 7923. [Google Scholar] [CrossRef]
- Fujiu, K.; Wang, J.; Nagai, R. Cardioprotective Function of Cardiac Macrophages. Cardiovasc. Res. 2014, 102, 232–239. [Google Scholar] [CrossRef]
- Pinto, A.R.; Godwin, J.W.; Rosenthal, N.A. Macrophages in Cardiac Homeostasis, Injury Responses and Progenitor Cell Mobilisation. Stem Cell Res. 2014, 13, 705–714. [Google Scholar] [CrossRef] [Green Version]
- Frantz, S.; Nahrendorf, M. Cardiac Macrophages and Their Role in Ischaemic Heart Disease. Cardiovasc. Res. 2014, 102, 240–248. [Google Scholar] [CrossRef]
- DeBerge, M.; Shah, S.J.; Wilsbacher, L.; Thorp, E.B. Macrophages in Heart Failure with Reduced versus Preserved Ejection Fraction. Trends Mol. Med. 2019, 25, 328–340. [Google Scholar] [CrossRef]
- Nadaud, S.; Flamant, M.; Le Goff, W.; Balse, E.; Pavoine, C. Transcriptomic and Lipidomic Mapping of Macrophages in the Hub of Chronic Beta-Adrenergic-Stimulation Unravels Hypertrophy-, Proliferation-, and Lipid Metabolism-Related Genes as Novel Potential Markers of Early Hypertrophy or Heart Failure. Biomedicines 2022, 10, 221. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Horng, T. Lipid Metabolism in Regulation of Macrophage Functions. Trends Cell Biol. 2020, 30, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Cabrera-Fuentes, H.A.; Devaux, Y.; Frangogiannis, N.G.; Frantz, S.; Guzik, T.; Liehn, E.A.; Gomes, C.P.C.; Schulz, R.; Hausenloy, D.J. Immune Cells as Targets for Cardioprotection: New Players and Novel Therapeutic Opportunities. Cardiovasc. Res. 2019, 115, 1117–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, X.; Lee, K.; Li, N.; Corbett, D.; Mendoza, L.; Frangogiannis, N.G. Characterization of the Inflammatory and Fibrotic Response in a Mouse Model of Cardiac Pressure Overload. Histochem. Cell Biol. 2009, 131, 471. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Kusakari, Y.; Xiao, C.-Y.; Kinsella, S.D.; Rosenberg, M.A.; Scherrer-Crosbie, M.; Hara, K.; Rosenzweig, A.; Matsui, T. MTOR Attenuates the Inflammatory Response in Cardiomyocytes and Prevents Cardiac Dysfunction in Pathological Hypertrophy. Am. J. Physiol. Cell Physiol. 2010, 299, C1256–C1266. [Google Scholar] [CrossRef] [Green Version]
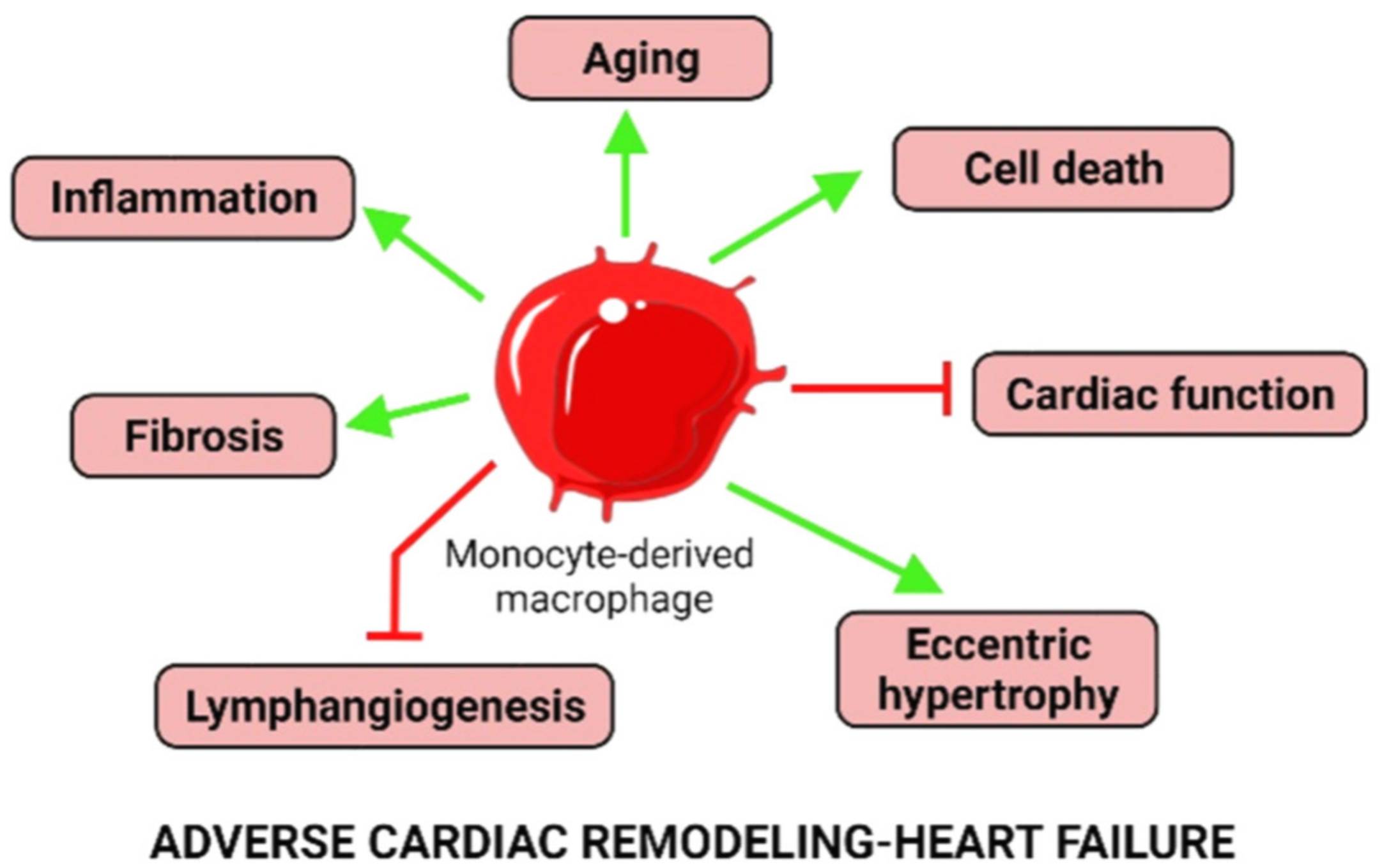
- Chen, B.; Frangogiannis, N.G. The Role of Macrophages in Nonischemic Heart Failure. JACC Basic Transl. Sci. 2018, 3, 245–248. [Google Scholar] [CrossRef]
- Patel, B.; Bansal, S.S.; Ismahil, M.A.; Hamid, T.; Rokosh, G.; Mack, M.; Prabhu, S.D. CCR2+ Monocyte-Derived Infiltrating Macrophages Are Required for Adverse Cardiac Remodeling During Pressure Overload. JACC Basic Transl. Sci. 2018, 3, 230–244. [Google Scholar] [CrossRef]
- Damilano, F.; Franco, I.; Perrino, C.; Schaefer, K.; Azzolino, O.; Carnevale, D.; Cifelli, G.; Carullo, P.; Ragona, R.; Ghigo, A.; et al. Distinct Effects of Leukocyte and Cardiac Phosphoinositide 3-Kinase γ Activity in Pressure Overload–Induced Cardiac Failure. Circulation 2011, 123, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Weisheit, C.; Zhang, Y.; Faron, A.; Köpke, O.; Weisheit, G.; Steinsträsser, A.; Frede, S.; Meyer, R.; Boehm, O.; Hoeft, A.; et al. Ly6C(Low) and Not Ly6C(High) Macrophages Accumulate First in the Heart in a Model of Murine Pressure-Overload. PLoS ONE 2014, 9, e112710. [Google Scholar] [CrossRef]
- Tian, Y.; Luo, J.; Xu, Q.; Liu, Y.; Cai, R.; Zhou, M.-S. Macrophage Depletion Protects against Endothelial Dysfunction and Cardiac Remodeling in Angiotensin II Hypertensive Mice. Clin. Exp. Hypertens. 2021, 43, 699–706. [Google Scholar] [CrossRef]
- Lavine, K.J.; Pinto, A.R.; Epelman, S.; Kopecky, B.J.; Clemente-Casares, X.; Godwin, J.; Rosenthal, N.; Kovacic, J.C. The Macrophage in Cardiac Homeostasis and Disease: JACC Macrophage in CVD Series (Part 4). J. Am. Coll. Cardiol. 2018, 72, 2213–2230. [Google Scholar] [CrossRef]
- Tauber, A.I. Metchnikoff and the Phagocytosis Theory. Nat. Rev. Mol. Cell Biol. 2003, 4, 897–901. [Google Scholar] [CrossRef]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Ramos, G.C.; van den Berg, A.; Nunes-Silva, V.; Weirather, J.; Peters, L.; Burkard, M.; Friedrich, M.; Pinnecker, J.; Abeßer, M.; Heinze, K.G.; et al. Myocardial Aging as a T-Cell-Mediated Phenomenon. Proc. Natl. Acad. Sci. USA 2017, 114, E2420–E2429. [Google Scholar] [CrossRef] [Green Version]
- Psarras, S.; Beis, D.; Nikouli, S.; Tsikitis, M.; Capetanaki, Y. Three in a Box: Understanding Cardiomyocyte, Fibroblast, and Innate Immune Cell Interactions to Orchestrate Cardiac Repair Processes. Front. Cardiovasc. Med. 2019, 6, 32. [Google Scholar] [CrossRef]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; Althagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-Renewing Resident Cardiac Macrophages Limit Adverse Remodeling Following Myocardial Infarction. Nat. Immunol. 2019, 20, 29–39. [Google Scholar] [CrossRef]
- Van Furth, R.; Cohn, Z.A. The Origin and Kinetics of Mononuclear Phagocytes. J. Exp. Med. 1968, 128, 415–435. [Google Scholar] [CrossRef]
- Davies, L.C.; Taylor, P.R. Tissue-Resident Macrophages: Then and Now. Immunology 2015, 144, 541–548. [Google Scholar] [CrossRef] [Green Version]
- Davies, L.C.; Rosas, M.; Jenkins, S.J.; Liao, C.-T.; Scurr, M.J.; Brombacher, F.; Fraser, D.J.; Allen, J.E.; Jones, S.A.; Taylor, P.R. Distinct Bone Marrow-Derived and Tissue-Resident Macrophage Lineages Proliferate at Key Stages during Inflammation. Nat. Commun. 2013, 4, 1886. [Google Scholar] [CrossRef] [Green Version]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and Adult-Derived Resident Cardiac Macrophages Are Maintained through Distinct Mechanisms at Steady State and during Inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Bajpai, G.; Schneider, C.; Wong, N.; Bredemeyer, A.; Hulsmans, M.; Nahrendorf, M.; Epelman, S.; Kreisel, D.; Liu, Y.; Itoh, A.; et al. The Human Heart Contains Distinct Macrophage Subsets with Divergent Origins and Functions. Nat. Med. 2018, 24, 1234–1245. [Google Scholar] [CrossRef]
- Ma, Y.; Mouton, A.J.; Lindsey, M.L. Cardiac Macrophage Biology in the Steady-State Heart, the Aging Heart, and Following Myocardial Infarction. Transl. Res. 2018, 191, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Bizou, M.; Itier, R.; Majdoubi, M.; Abbadi, D.; Pichery, E.; Dutaur, M.; Marsal, D.; Calise, D.; Garmy-Susini, B.; Douin-Echinard, V.; et al. Cardiac Macrophage Subsets Differentially Regulate Lymphatic Network Remodeling during Pressure Overload. Sci. Rep. 2021, 11, 16801. [Google Scholar] [CrossRef]
- Fujiu, K.; Shibata, M.; Nakayama, Y.; Ogata, F.; Matsumoto, S.; Noshita, K.; Iwami, S.; Nakae, S.; Komuro, I.; Nagai, R.; et al. A Heart-Brain-Kidney Network Controls Adaptation to Cardiac Stress through Tissue Macrophage Activation. Nat. Med. 2017, 23, 611–622. [Google Scholar] [CrossRef]
- Liao, X.; Shen, Y.; Zhang, R.; Sugi, K.; Vasudevan, N.T.; Alaiti, M.A.; Sweet, D.R.; Zhou, L.; Qing, Y.; Gerson, S.L.; et al. Distinct Roles of Resident and Nonresident Macrophages in Nonischemic Cardiomyopathy. Proc. Natl. Acad. Sci. USA 2018, 115, E4661–E4669. [Google Scholar] [CrossRef] [Green Version]
- Sadoshima, J. MRTF-A in Macrophages Mediates Pathological Hypertrophy. Cardiovasc. Res. 2022, 118, cvab376. [Google Scholar] [CrossRef]
- Kain, D.; Amit, U.; Yagil, C.; Landa, N.; Naftali-Shani, N.; Molotski, N.; Aviv, V.; Feinberg, M.S.; Goitein, O.; Kushnir, T.; et al. Macrophages Dictate the Progression and Manifestation of Hypertensive Heart Disease. Int. J. Cardiol. 2016, 203, 381–395. [Google Scholar] [CrossRef]
- Zandbergen, H.R.; Sharma, U.C.; Gupta, S.; Verjans, J.W.H.; van den Borne, S.; Pokharel, S.; van Brakel, T.; Duijvestijn, A.; van Rooijen, N.; Maessen, J.G.; et al. Macrophage Depletion in Hypertensive Rats Accelerates Development of Cardiomyopathy. J. Cardiovasc. Pharmacol. Ther. 2009, 14, 68–75. [Google Scholar] [CrossRef]
- Zaman, R.; Hamidzada, H.; Kantores, C.; Wong, A.; Dick, S.A.; Wang, Y.; Momen, A.; Aronoff, L.; Lin, J.; Razani, B.; et al. Selective Loss of Resident Macrophage-Derived Insulin-like Growth Factor-1 Abolishes Adaptive Cardiac Growth to Stress. Immunity 2021, 54, 2057–2071.e6. [Google Scholar] [CrossRef]
- Ishibashi, M.; Hiasa, K.; Zhao, Q.; Inoue, S.; Ohtani, K.; Kitamoto, S.; Tsuchihashi, M.; Sugaya, T.; Charo, I.F.; Kura, S.; et al. Critical Role of Monocyte Chemoattractant Protein-1 Receptor CCR2 on Monocytes in Hypertension-Induced Vascular Inflammation and Remodeling. Circ. Res. 2004, 94, 1203–1210. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, F.; Kai, H.; Tokuda, K.; Takeya, M.; Takeshita, A.; Egashira, K.; Imaizumi, T. Hypertensive Myocardial Fibrosis and Diastolic Dysfunction: Another Model of Inflammation? Hypertension 2004, 43, 739–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revelo, X.S.; Parthiban, P.; Chen, C.; Barrow, F.; Fredrickson, G.; Wang, H.; Yücel, D.; Herman, A.; van Berlo, J.H. Cardiac Resident Macrophages Prevent Fibrosis and Stimulate Angiogenesis. Circ. Res. 2021, 129, 1086–1101. [Google Scholar] [CrossRef] [PubMed]
- Ismahil, M.A.; Prabhu, S.D. Resident Macrophages in the Heart: Cardioprotective Under Pressure. Circ. Res. 2021, 129, 1102–1104. [Google Scholar] [CrossRef] [PubMed]
- Leid, J.; Carrelha, J.; Boukarabila, H.; Epelman, S.; Jacobsen, S.E.W.; Lavine, K.J. Primitive Embryonic Macrophages Are Required for Coronary Development and Maturation. Circ. Res. 2016, 118, 1498–1511. [Google Scholar] [CrossRef]
- Wong, N.R.; Mohan, J.; Kopecky, B.J.; Guo, S.; Du, L.; Leid, J.; Feng, G.; Lokshina, I.; Dmytrenko, O.; Luehmann, H.; et al. Resident Cardiac Macrophages Mediate Adaptive Myocardial Remodeling. Immunity 2021, 54, 2072–2088.e7. [Google Scholar] [CrossRef]
- Sugita, J.; Fujiu, K.; Nakayama, Y.; Matsubara, T.; Matsuda, J.; Oshima, T.; Liu, Y.; Maru, Y.; Hasumi, E.; Kojima, T.; et al. Cardiac Macrophages Prevent Sudden Death during Heart Stress. Nat. Commun. 2021, 12, 1910. [Google Scholar] [CrossRef]
- Son, G.H.; Park, S.H.; Kim, Y.; Kim, J.Y.; Kim, J.W.; Chung, S.; Kim, Y.-H.; Kim, H.; Hwang, J.-J.; Seo, J.-S. Postmortem MRNA Expression Patterns in Left Ventricular Myocardial Tissues and Their Implications for Forensic Diagnosis of Sudden Cardiac Death. Mol. Cells 2014, 37, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wülfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 2017, 169, 510–522.e20. [Google Scholar] [CrossRef] [Green Version]
- Harari, E.; Guo, L.; Smith, S.L.; Braumann, R.E.; Virmani, R.; Finn, A.V. Heart-Resident Macrophages: Are They Involved in the Rhythm of Every Beat? J. Thorac. Dis. 2017, 9, 2264–2267. [Google Scholar] [CrossRef] [Green Version]
- Munshi, N.V. Resident Macrophages: Near and Dear to Your Heart. Cell 2017, 169, 376–377. [Google Scholar] [CrossRef] [Green Version]
- Bu, J.; Huang, S.; Wang, J.; Xia, T.; Liu, H.; You, Y.; Wang, Z.; Liu, K. The GABAA Receptor Influences Pressure Overload-Induced Heart Failure by Modulating Macrophages in Mice. Front. Immunol. 2021, 12, 670153. [Google Scholar] [CrossRef]
- Korf-Klingebiel, M.; Reboll, M.R.; Polten, F.; Weber, N.; Jäckle, F.; Wu, X.; Kallikourdis, M.; Kunderfranco, P.; Condorelli, G.; Giannitsis, E.; et al. Myeloid-Derived Growth Factor Protects against Pressure Overload-Induced Heart Failure by Preserving Sarco/Endoplasmic Reticulum Ca2+-ATPase Expression in Cardiomyocytes. Circulation 2021, 144, 1227–1240. [Google Scholar] [CrossRef]
- Martini, E.; Kunderfranco, P.; Peano, C.; Carullo, P.; Cremonesi, M.; Schorn, T.; Carriero, R.; Termanini, A.; Colombo, F.S.; Jachetti, E.; et al. Single-Cell Sequencing of Mouse Heart Immune Infiltrate in Pressure Overload-Driven Heart Failure Reveals Extent of Immune Activation. Circulation 2019, 140, 2089–2107. [Google Scholar] [CrossRef]
- Korf-Klingebiel, M.; Reboll, M.R.; Klede, S.; Brod, T.; Pich, A.; Polten, F.; Napp, L.C.; Bauersachs, J.; Ganser, A.; Brinkmann, E.; et al. Myeloid-Derived Growth Factor (C19orf10) Mediates Cardiac Repair Following Myocardial Infarction. Nat. Med. 2015, 21, 140–149. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Feng, J.; Liu, W.; Li, Y.; Liu, J.; Yin, Q.; Lian, H.; Liu, L.; Nie, Y. Mydgf Promotes Cardiomyocyte Proliferation and Neonatal Heart Regeneration. Theranostics 2020, 10, 9100–9112. [Google Scholar] [CrossRef]
- Di Candia, A.M.; de Avila, D.X.; Moreira, G.R.; Villacorta, H.; Maisel, A.S. Growth Differentiation Factor-15, a Novel Systemic Biomarker of Oxidative Stress, Inflammation, and Cellular Aging: Potential Role in Cardiovascular Diseases. Am. Heart J. Plus Cardiol. Res. Prac. 2021, 9, 100046. [Google Scholar] [CrossRef]
- Rochette, L.; Dogon, G.; Zeller, M.; Cottin, Y.; Vergely, C. GDF15 and Cardiac Cells: Current Concepts and New Insights. Int. J. Mol. Sci. 2021, 22, 8889. [Google Scholar] [CrossRef]
- Unsicker, K.; Spittau, B.; Krieglstein, K. The Multiple Facets of the TGF-β Family Cytokine Growth/Differentiation Factor-15/Macrophage Inhibitory Cytokine-1. Cytokine Growth Factor Rev. 2013, 24, 373–384. [Google Scholar] [CrossRef]
- Xu, J.; Kimball, T.R.; Lorenz, J.N.; Brown, D.A.; Bauskin, A.R.; Klevitsky, R.; Hewett, T.E.; Breit, S.N.; Molkentin, J.D. GDF15/MIC-1 Functions as a Protective and Antihypertrophic Factor Released from the Myocardium in Association with SMAD Protein Activation. Circ. Res. 2006, 98, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Kempf, T.; Zarbock, A.; Widera, C.; Butz, S.; Stadtmann, A.; Rossaint, J.; Bolomini-Vittori, M.; Korf-Klingebiel, M.; Napp, L.C.; Hansen, B.; et al. GDF-15 Is an Inhibitor of Leukocyte Integrin Activation Required for Survival after Myocardial Infarction in Mice. Nat. Med. 2011, 17, 581–588. [Google Scholar] [CrossRef]
- Jung, S.B.; Choi, M.J.; Ryu, D.; Yi, H.S.; Lee, S.E.; Chang, J.Y.; Chung, H.K.; Kim, Y.K.; Kang, S.G.; Lee, J.H.; et al. Reduced Oxidative Capacity in Macrophages Results in Systemic Insulin Resistance. Nat. Commun. 2018, 9, 1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez Fernandez, A.B.; Ferrero-Gregori, A.; Garcia-Osuna, A.; Mirabet-Perez, S.; Pirla-Buxo, M.J.; Cinca-Cuscullola, J.; Ordonez-Llanos, J.; Roig Minguell, E. Growth Differentiation Factor 15 as Mortality Predictor in Heart Failure Patients with Non-Reduced Ejection Fraction. ESC Heart Fail. 2020, 7, 2223–2229. [Google Scholar] [CrossRef] [PubMed]
- Brakenhielm, E.; González, A.; Díez, J. Role of Cardiac Lymphatics in Myocardial Edema and Fibrosis: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2020, 76, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Brakenhielm, E.; Alitalo, K. Cardiac Lymphatics in Health and Disease. Nat. Rev. Cardiol. 2019, 16, 56–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira, J.M.; Norman, S.; del Campo, C.V.; Cahill, T.J.; Barnette, D.N.; Gunadasa-Rohling, M.; Johnson, L.A.; Greaves, D.R.; Carr, C.A.; Jackson, D.G.; et al. The Cardiac Lymphatic System Stimulates Resolution of Inflammation Following Myocardial Infarction. J. Clin. Investig 2018, 8, 3402–3412. [Google Scholar] [CrossRef]
- Stables, M.J.; Shah, S.; Camon, E.B.; Lovering, R.C.; Newson, J.; Bystrom, J.; Farrow, S.; Gilroy, D.W. Transcriptomic Analyses of Murine Resolution-Phase Macrophages. Blood 2011, 118, e192–e208. [Google Scholar] [CrossRef] [Green Version]
- Ren Zongna; Yu Peng; Li Dandan; Li Zheng; Liao Yingnan; Wang Yin; Zhou Bingying; Wang Li Single-Cell Reconstruction of Progression Trajectory Reveals Intervention Principles in Pathological Cardiac Hypertrophy. Circulation 2020, 141, 1704–1719. [CrossRef]
- Wang, J.J.-C.; Rau, C.; Avetisyan, R.; Ren, S.; Romay, M.C.; Stolin, G.; Gong, K.W.; Wang, Y.; Lusis, A.J. Genetic Dissection of Cardiac Remodeling in an Isoproterenol-Induced Heart Failure Mouse Model. PLOS Genet. 2016, 12, e1006038. [Google Scholar] [CrossRef]
- Lau, E.; Cao, Q.; Lam, M.P.Y.; Wang, J.; Ng, D.C.M.; Bleakley, B.J.; Lee, J.M.; Liem, D.A.; Wang, D.; Hermjakob, H.; et al. Integrated Omics Dissection of Proteome Dynamics during Cardiac Remodeling. Nat. Commun. 2018, 9, 120. [Google Scholar] [CrossRef]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.F.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert Consensus Document: Mitochondrial Function as a Therapeutic Target in Heart Failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef]
- Nicolás-Ávila, J.A.; Lechuga-Vieco, A.V.; Esteban-Martínez, L.; Sánchez-Díaz, M.; Díaz-García, E.; Santiago, D.J.; Rubio-Ponce, A.; Li, J.L.; Balachander, A.; Quintana, J.A.; et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 2020, 183, 94–109.e23. [Google Scholar] [CrossRef]
- Tang, X.; Li, P.-H.; Chen, H.-Z. Cardiomyocyte Senescence and Cellular Communications Within Myocardial Microenvironments. Front. Endocrinol. 2020, 11, 280. [Google Scholar] [CrossRef]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018, 9, 586. [Google Scholar] [CrossRef]
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-Independent Telomere Damage Drives Post-Mitotic Cardiomyocyte Senescence. EMBO J. 2019, 38, e100492. [Google Scholar] [CrossRef]
- Banerjee, P.; Kotla, S.; Reddy Velatooru, L.; Abe, R.J.; Davis, E.A.; Cooke, J.P.; Schadler, K.; Deswal, A.; Herrmann, J.; Lin, S.H.; et al. Senescence-Associated Secretory Phenotype as a Hinge Between Cardiovascular Diseases and Cancer. Front. Cardiovasc. Med. 2021, 8, 763930. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, C.; Liu, L.; Xi, A.; Chen, B.; Li, Y.; Du, J. Macrophage-Derived Mir-155-Containing Exosomes Suppress Fibroblast Proliferation and Promote Fibroblast Inflammation during Cardiac Injury. Mol. Ther. 2017, 25, 192–204. [Google Scholar] [CrossRef] [Green Version]
- Heymans, S.; Corsten, M.F.; Verhesen, W.; Carai, P.; van Leeuwen, R.E.W.; Custers, K.; Peters, T.; Hazebroek, M.; Stöger, L.; Wijnands, E.; et al. Macrophage MicroRNA-155 Promotes Cardiac Hypertrophy and Failure. Circulation 2013, 128, 1420–1432. [Google Scholar] [CrossRef] [Green Version]
- Ramanujam, D.; Schön, A.P.; Beck, C.; Vaccarello, P.; Felician, G.; Dueck, A.; Esfandyari, D.; Meister, G.; Meitinger, T.; Schulz, C.; et al. MicroRNA-21-Dependent Macrophage-to-Fibroblast Signaling Determines the Cardiac Response to Pressure Overload. Circulation 2021, 143, 1513–1525. [Google Scholar] [CrossRef]
- Sun, X.; Liu, Y.; Wang, J.; Zhang, M.; Wang, M. Cardioprotection of M2 Macrophages-Derived Exosomal MicroRNA-24-3p/Tnfsf10 Axis against Myocardial Injury after Sepsis. Mol. Immunol. 2022, 141, 309–317. [Google Scholar] [CrossRef]
- Usuelli, V.; Ben Nasr, M.; D’Addio, F.; Liu, K.; Vergani, A.; El Essawy, B.; Yang, J.; Assi, E.; Uehara, M.; Rossi, C.; et al. MiR-21 Antagonism Reprograms Macrophage Metabolism and Abrogates Chronic Allograft Vasculopathy. Am. J. Transpl. 2021, 21, 3280–3295. [Google Scholar] [CrossRef]
- Mirna, M.; Paar, V.; Topf, A.; Kraus, T.; Sotlar, K.; Aigner, A.; Ewe, A.; Watzinger, S.; Podesser, B.K.; Hackl, M.; et al. A New Player in the Game: Treatment with AntagomiR-21a-5p Significantly Attenuates Histological and Echocardiographic Effects of Experimental Autoimmune Myocarditis. Cardiovasc. Res. 2022, 118, 556–572. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac Fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef]
- Huet, E.; Gabison, E.; Vallee, B.; Mougenot, N.; Linguet, G.; Riou, B.; Jarosz, C.; Menashi, S.; Besse, S. Deletion of Extracellular Matrix Metalloproteinase Inducer/CD147 Induces Altered Cardiac Extracellular Matrix Remodeling in Aging Mice. J. Physiol. Pharmacol. 2015, 66, 355–366. [Google Scholar]
- Loredo-Mendoza, M.L.; Ramirez-Sanchez, I.; Bustamante-Pozo, M.M.; Ayala, M.; Navarrete, V.; Garate-Carrillo, A.; Ito, B.R.; Ceballos, G.; Omens, J.; Villarreal, F. The Role of Inflammation in Driving Left Ventricular Remodeling in a Pre-HFpEF Model. Exp. Biol. Med. 2020, 245, 748–757. [Google Scholar] [CrossRef]
- Chiao, Y.A.; Dai, Q.; Zhang, J.; Lin, J.; Lopez, E.F.; Ahuja, S.S.; Chou, Y.-M.; Lindsey, M.L.; Jin, Y.-F. Multi-Analyte Profiling Reveals Matrix Metalloproteinase-9 and Monocyte Chemotactic Protein-1 as Plasma Biomarkers of Cardiac Aging. Circ. Cardiovasc. Genet. 2011, 4, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Wagner, J.U.G.; Dimmeler, S. Cellular Cross-Talks in the Diseased and Aging Heart. J. Mol. Cell. Cardiol. 2020, 138, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Chiao, Y.A.; Ramirez, T.A.; Zamilpa, R.; Okoronkwo, S.M.; Dai, Q.; Zhang, J.; Jin, Y.-F.; Lindsey, M.L. Matrix Metalloproteinase-9 Deletion Attenuates Myocardial Fibrosis and Diastolic Dysfunction in Ageing Mice. Cardiovasc. Res. 2012, 96, 444–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aujla, P.K.; Kassiri, Z. Diverse Origins and Activation of Fibroblasts in Cardiac Fibrosis. Cell Signal. 2021, 78, 109869. [Google Scholar] [CrossRef]
- Kanellakis, P.; Ditiatkovski, M.; Kostolias, G.; Bobik, A. A Pro-Fibrotic Role for Interleukin-4 in Cardiac Pressure Overload. Cardiovasc. Res. 2012, 95, 77–85. [Google Scholar] [CrossRef]
- Peng, H.; Sarwar, Z.; Yang, X.-P.; Peterson, E.L.; Xu, J.; Janic, B.; Rhaleb, N.; Carretero, O.A.; Rhaleb, N.-E. Profibrotic Role for Interleukin-4 in Cardiac Remodeling and Dysfunction. Hypertension 2015, 66, 582–589. [Google Scholar] [CrossRef] [Green Version]
- Deniset, J.F.; Belke, D.; Lee, W.-Y.; Jorch, S.K.; Deppermann, C.; Hassanabad, A.F.; Turnbull, J.D.; Teng, G.; Rozich, I.; Hudspeth, K.; et al. Gata6+ Pericardial Cavity Macrophages Relocate to the Injured Heart and Prevent Cardiac Fibrosis. Immunity 2019, 51, 131–140.e5. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Dong, E.; Zhang, Y.; Xiao, H. The Role of the Inflammasome in Heart Failure. Front. Physiol. 2021, 12, 709703. [Google Scholar] [CrossRef] [PubMed]
- Everett, B.M.; Cornel, J.H.; Lainscak, M.; Anker, S.D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Anti-Inflammatory Therapy with Canakinumab for the Prevention of Hospitalization for Heart Failure. Circulation 2019, 139, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Rolski, F.; Błyszczuk, P. Complexity of TNF-α Signaling in Heart Disease. J. Clin. Med. 2020, 9, 3267. [Google Scholar] [CrossRef]
- Fischer, R.; Kontermann, R.E.; Pfizenmaier, K. Selective Targeting of TNF Receptors as a Novel Therapeutic Approach. Front. Cell Dev. Biol. 2020, 8, 401. [Google Scholar] [CrossRef]
- Adamy, C.; Mulder, P.; Khouzami, L.; Andrieu-abadie, N.; Defer, N.; Candiani, G.; Pavoine, C.; Caramelle, P.; Souktani, R.; Le Corvoisier, P.; et al. Neutral Sphingomyelinase Inhibition Participates to the Benefits of N-Acetylcysteine Treatment in Post-Myocardial Infarction Failing Heart Rats. J. Mol. Cell. Cardiol. 2007, 43, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Kologrivova, I.; Shtatolkina, M.; Suslova, T.; Ryabov, V. Cells of the Immune System in Cardiac Remodeling: Main Players in Resolution of Inflammation and Repair After Myocardial Infarction. Front. Immunol. 2021, 12, 664457. [Google Scholar] [CrossRef]
- Chaudhry, F.; Isherwood, J.; Bawa, T.; Patel, D.; Gurdziel, K.; Lanfear, D.E.; Ruden, D.M.; Levy, P.D. Single-Cell RNA Sequencing of the Cardiovascular System: New Looks for Old Diseases. Front. Cardiovasc. Med. 2019, 6, 173. [Google Scholar] [CrossRef]
- Gladka, M.M. Single-Cell RNA Sequencing of the Adult Mammalian Heart-State-of-the-Art and Future Perspectives. Curr. Heart Fail. Rep. 2021, 18, 64–70. [Google Scholar] [CrossRef]
- Vannella, K.M.; Wynn, T.A. Mechanisms of Organ Injury and Repair by Macrophages. Annu. Rev. Physiol. 2017, 79, 593–617. [Google Scholar] [CrossRef]
- Walter, W.; Alonso-Herranz, L.; Trappetti, V.; Crespo, I.; Ibberson, M.; Cedenilla, M.; Karaszewska, A.; Núñez, V.; Xenarios, I.; Arroyo, A.G.; et al. Deciphering the Dynamic Transcriptional and Post-Transcriptional Networks of Macrophages in the Healthy Heart and after Myocardial Injury. Cell Rep. 2018, 23, 622–636. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.J.; Pearce, E.J. Immunometabolism Governs Dendritic Cell and Macrophage Function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef]
- Bertero, E.; Maack, C. Metabolic Remodelling in Heart Failure. Nat. Rev. Cardiol. 2018, 15, 457–470. [Google Scholar] [CrossRef]
- Batista-Gonzalez, A.; Vidal, R.; Criollo, A.; Carreño, L.J. New Insights on the Role of Lipid Metabolism in the Metabolic Reprogramming of Macrophages. Front. Immunol. 2019, 10, 2993. [Google Scholar] [CrossRef]
- Ardura, J.A.; Rackov, G.; Izquierdo, E.; Alonso, V.; Gortazar, A.R.; Escribese, M.M. Targeting Macrophages: Friends or Foes in Disease? Front. Pharmacol. 2019, 10, 1255. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Besse, S.; Nadaud, S.; Balse, E.; Pavoine, C. Early Protective Role of Inflammation in Cardiac Remodeling and Heart Failure: Focus on TNFα and Resident Macrophages. Cells 2022, 11, 1249. https://doi.org/10.3390/cells11071249
Besse S, Nadaud S, Balse E, Pavoine C. Early Protective Role of Inflammation in Cardiac Remodeling and Heart Failure: Focus on TNFα and Resident Macrophages. Cells. 2022; 11(7):1249. https://doi.org/10.3390/cells11071249
Chicago/Turabian StyleBesse, Sophie, Sophie Nadaud, Elise Balse, and Catherine Pavoine. 2022. "Early Protective Role of Inflammation in Cardiac Remodeling and Heart Failure: Focus on TNFα and Resident Macrophages" Cells 11, no. 7: 1249. https://doi.org/10.3390/cells11071249
APA StyleBesse, S., Nadaud, S., Balse, E., & Pavoine, C. (2022). Early Protective Role of Inflammation in Cardiac Remodeling and Heart Failure: Focus on TNFα and Resident Macrophages. Cells, 11(7), 1249. https://doi.org/10.3390/cells11071249