
Dance of The Golgi: Understanding Golgi Dynamics in Cancer Metastasis


{kind=link}
{kind=link}
Abstract
:1. Introduction
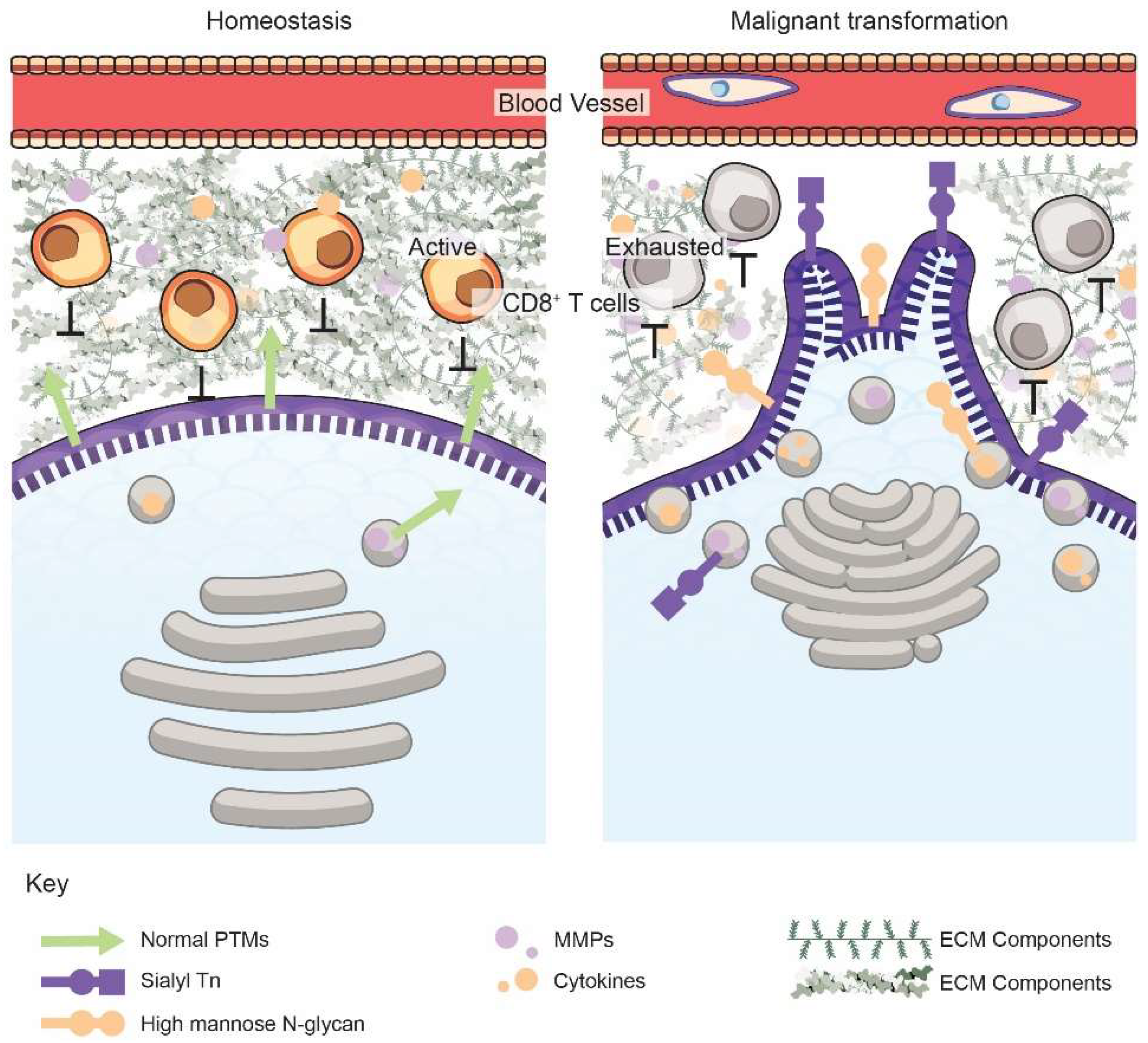
2. The Golgi Alters the Cancer Cell Secretome, the Tumor ECM, and Immune Surveillance
3. Golgi Dynamics in Cancer Metastasis
3.1. Golgi-Mediated Vesicular Trafficking and Exocytosis
3.2. Golgi Orientation Governs Cell Polarity and Directional Migration
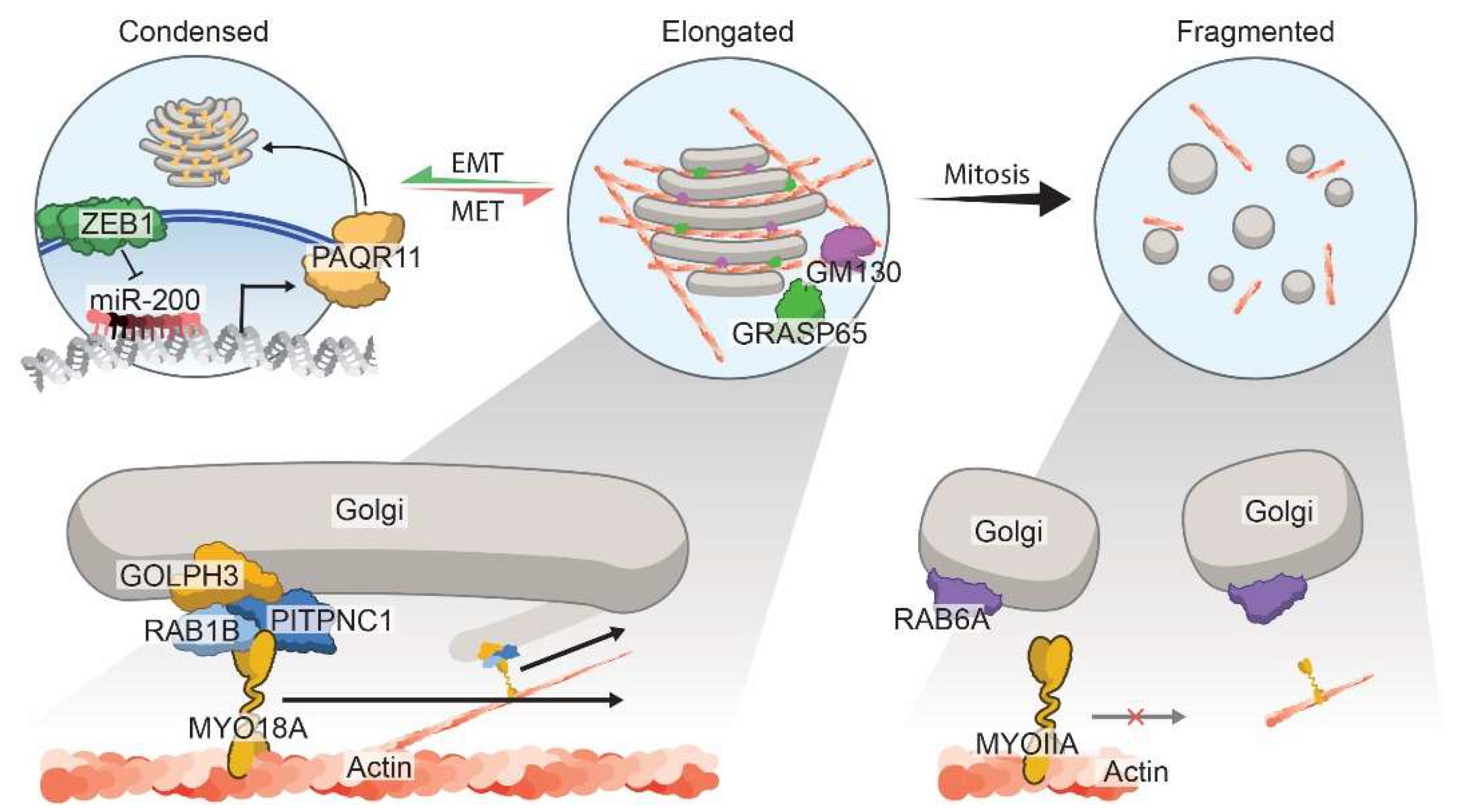
3.3. Morphology of the Golgi Apparatus
4. Golgi-Mediated Post-Translational Modifications (PTMs)
5. Therapeutic Targeting of Tumor Secretion
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ER | Endoplasmic reticulum |
| PM | Plasma membrane |
| EC | Extracellular |
| CPS | Conventional protein secretion |
| UPS | Unconventional protein secretion |
| PTM | Post-translational modification |
| ECM | Extracellular matrix |
| KDELR | KDEL receptor |
| NSCLC | Non-small cell lung cancer |
| BM | Basement membrane |
| MMPs | Matrix metalloproteases |
| TME | Tumor microenvironment |
| GEFs | Guanine exchange factors |
| GAPs | GTPase activating proteins |
| ARF | ADP-ribosylation factor |
| IMPAD1 | Inositol monophosphatase domain containing 1 |
| NO | Nitric oxide |
| UPR | Unfolded protein response |
| EMT | Epithelial-to-mesenchymal transition |
| miR-200 | miRNA-200 family |
| PAQR11 | Progestin and adipoQ receptor family member 11 |
| TIME | Tumor immune microenvironment |
| GAGs | Glycosaminoglycans |
| GPI | Glycosylphosphatidylinositol |
| GSL | Glycosphingolipids |
| CS | Chondroitin sulfate |
| HS | Heparan sulfate |
| BFA | Brefeldin-A |
References
- Viotti, C. ER to Golgi-Dependent Protein Secretion: The Conventional Pathway. Methods Mol. Biol. 2016, 1459, 3–29. [Google Scholar] [PubMed]
- Palade, G. Intracellular Aspects of the Process of Protein Synthesis. Science 1975, 189, 347–358. [Google Scholar] [CrossRef]
- Paltridge, J.L.; Belle, L.; Khew-Goodall, Y. The secretome in cancer progression. Biochim. Biophys Acta 2013, 1834, 2233–2241. [Google Scholar] [CrossRef] [PubMed]
- Rabouille, C. Pathways of Unconventional Protein Secretion. Trends Cell Biol. 2017, 27, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Rabouille, C.; Malhotra, V.; Nickel, W. Diversity in unconventional protein secretion. J. Cell Sci. 2012, 125 Pt 22, 5251–5255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickel, W.; Rabouille, C. Mechanisms of regulated unconventional protein secretion. Nat. Rev. Mol. Cell Biol. 2009, 10, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Bentivoglio, M.; Jones, E.G.; Mazzarello, P.; Ribak, C.E.; Shepherd, G.M.; Swanson, L.W. Camillo Golgi and modern neuroscience. Brain Res. Rev. 2011, 66, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GM, C. The Endoplasmic Reticulum. In The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2000. [Google Scholar]
- Cooper, G. The Golgi Apparatus. In The Cell: A Molecular Approach; Sinauer Associates: Sunderland, MA, USA, 2000. [Google Scholar]
- Lodish, H.B.A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Overview of the Secretory Pathway. In Molecular Cell Biology, 4th ed.; W. H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Lodish, H.B.A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Protein Glycosylation in the ER and Golgi Complex. In Molecular Cell Biology; W. H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Wiersma, V.R.; Michalak, M.; Abdullah, T.M.; Bremer, E.; Eggleton, P. Mechanisms of Translocation of ER Chaperones to the Cell Surface and Immunomodulatory Roles in Cancer and Autoimmunity. Front. Oncol. 2015, 5, 7. [Google Scholar] [CrossRef]
- Tan, X.; Banerjee, P.; Pham, E.A.; Rutaganira, F.U.N.; Basu, K.; Bota-Rabassedas, N.; Guo, H.F.; Grzeskowiak, C.L.; Liu, X.; Yu, J.; et al. PI4KIIIβ is a therapeutic target in chromosome 1q-amplified lung adenocarcinoma. Sci. Transl. Med. 2020, 12, eaax3772. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Halberg, N.; Sengelaub, C.A.; Navrazhina, K.; Molina, H.; Uryu, K.; Tavazoie, S.F. PITPNC1 Recruits RAB1B to the Golgi Network to Drive Malignant Secretion. Cancer Cell 2016, 29, 339–353. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, G.K.; Jwa, N.S.; Lebrun, M.H.; Job, D.; Rakwal, R. Plant secretome: Unlocking secrets of the secreted proteins. Proteomics 2010, 10, 799–827. [Google Scholar] [CrossRef]
- Han, H.M.; Bouchet-Marquis, C.; Huebinger, J.; Grabenbauer, M. Golgi apparatus analyzed by cryo-electron microscopy. Histochem. Cell Biol. 2013, 140, 369–381. [Google Scholar] [CrossRef] [Green Version]
- Kurokawa, K.; Ishii, M.; Suda, Y.; Ichihara, A.; Nakano, A. Live cell visualization of Golgi membrane dynamics by super-resolution confocal live imaging microscopy. Methods Cell Biol. 2013, 118, 235–242. [Google Scholar]
- Snapp, E.L. Photobleaching methods to study Golgi complex dynamics in living cells. Methods Cell Biol. 2013, 118, 195–216. [Google Scholar]
- Tannous, B.A. Gaussia luciferase reporter assay for monitoring biological processes in culture and in vivo. Nat. Protoc. 2009, 4, 582–591. [Google Scholar] [CrossRef]
- Egea, G.; Franci, C.; Gambus, G.; Lesuffleur, T.; Zweibaum, A.; Real, F.X. cis-Golgi resident proteins and O-glycans are abnormally compartmentalized in the RER of colon cancer cells. J. Cell Sci. 1993, 105 Pt 3, 819–830. [Google Scholar] [CrossRef]
- Kellokumpu, S.; Sormunen, R.; Kellokumpu, I. Abnormal glycosylation and altered Golgi structure in colorectal cancer: Dependence on intra-Golgi pH. Febs. Lett. 2002, 516, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Albacete-Albacete, L.; Sanchez-Alvarez, M.; Del Pozo, M.A. Extracellular Vesicles: An Emerging Mechanism Governing the Secretion and Biological Roles of Tenascin-C. Front. Immunol. 2021, 12, 671485. [Google Scholar] [CrossRef]
- Duffy, M.J.; Maguire, T.M.; Hill, A.; McDermott, E.; O’Higgins, N. Metalloproteinases: Role in breast carcinogenesis, invasion and metastasis. Breast Cancer Res. 2000, 2, 252. [Google Scholar] [CrossRef] [Green Version]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Stephens, D.C.; Harris, D.A. Organizing ‘Elements’: Facilitating Exocytosis and Promoting Metastasis. Trends Cancer 2020, 6, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Sbai, O.; Ferhat, L.; Bernard, A.; Gueye, Y.; Ould-Yahoui, A.; Thiolloy, S.; Charrat, E.; Charton, G.; Tremblay, E.; Risso, J.; et al. vesicular trafficking and secretion of matrix metalloproteinases-2, -9 and tissue inhibitor of metalloproteinases-1 in neuronal cells. Mol. Cell. Neurosci. 2008, 39, 549–568. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, T.; Hasegawa, K.; Aoki, Y.; Watanabe, T.; Otagiri, Y.; Arasaki, K.; Wakana, Y.; Asano, K.; Tanaka, M.; Yamaguchi, H.; et al. MT1-MMP recruits the ER-Golgi SNARE Bet1 for efficient MT1-MMP transport to the plasma membrane. Cancer Cell Biol. 2019, 218, 3355–3371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleiner, D.E.; Stetler-Stevenson, W.G. Matrix metalloproteinases and metastasis. Cancer Chemother. Pharmacol. 1999, 43, S42–S51. [Google Scholar] [CrossRef]
- Jin, D.; Tao, J.; Li, D.; Wang, Y.; Li, L.; Hu, Z.; Zhou, Z.; Chang, X.; Qu, C.; Zhang, H. Golgi protein 73 activation of MMP-13 promotes hepatocellular carcinoma cell invasion. Oncotarget 2015, 6, 33523–33533. [Google Scholar] [CrossRef] [Green Version]
- Condamine, T.; Ramachandran, I.; Youn, J.I.; Gabrilovich, D.I. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu. Rev. Med. 2015, 66, 97–110. [Google Scholar] [CrossRef] [Green Version]
- Song, J.W.; Zhu, J.; Wu, X.X.; Tu, T.; Huang, J.Q.; Chen, G.Z.; Liang, L.Y.; Zhou, C.H.; Xu, X.; Gong, L.Y. GOLPH3/CKAP4 promotes metastasis and tumorigenicity by enhancing the secretion of exosomal WNT3A in non-small-cell lung cancer. Cell Death Dis. 2021, 12, 976. [Google Scholar] [CrossRef]
- Schinzari, V.; Timperi, E.; Pecora, G.; Palmucci, F.; Gallerano, D.; Grimaldi, A.; Covino, D.A.; Guglielmo, N.; Melandro, F.; Manzi, E.; et al. Wnt3a/β-Catenin Signaling Conditions Differentiation of Partially Exhausted T-effector Cells in Human Cancers. Cancer Immunol. Res. 2018, 6, 941–952. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, G.S.; Pavlou, M.P.; Diamandis, E.P. Cancer secretomics reveal pathophysiological pathways in cancer molecular oncology. Mol. Oncol. 2010, 4, 496–510. [Google Scholar] [CrossRef] [Green Version]
- Galdiero, M.R.; Marone, G.; Mantovani, A. Cancer Inflammation and Cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, a028662. [Google Scholar] [CrossRef] [Green Version]
- Lieu, Z.Z.; Lock, J.G.; Hammond, L.A.; La Gruta, N.L.; Stow, J.L.; Gleeson, P.A. A trans-Golgi network golgin is required for the regulated secretion of TNF in activated macrophages in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 3351–3356. [Google Scholar] [CrossRef] [Green Version]
- Capece, D.; Verzella, D.; Tessitore, A.; Alesse, E.; Capalbo, C.; Zazzeroni, F. Cancer secretome and inflammation: The bright and the dark sides of NF-κB. Semin. Cell Dev. Biol. 2018, 78, 51–61. [Google Scholar] [CrossRef]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef]
- Sehgal, P.B. Interleukin-6 at the Host-Tumor Interface: STAT3 in Biomolecular Condensates in Cancer Cells. Cells 2022, 11, 1164. [Google Scholar] [CrossRef]
- Goldenring, J.R. A central role for vesicle trafficking in epithelial neoplasia: Intracellular highways to carcinogenesis. Nat. Reviews. Cancer 2013, 13, 813–820. [Google Scholar] [CrossRef]
- Bhuin, T.; Roy, J.K. Rab proteins: The key regulators of intracellular vesicle transport. Exp. Cell Res. 2014, 328, 1–19. [Google Scholar] [CrossRef]
- Mughees, M.; Chugh, H.; Wajid, S. Vesicular trafficking-related proteins as the potential therapeutic target for breast cancer. Protoplasma 2020, 257, 345–352. [Google Scholar] [CrossRef]
- Tzeng, H.-T.; Wang, Y.-C. Rab-mediated vesicle trafficking in cancer. J. Biomed. Sci. 2016, 23, 70. [Google Scholar] [CrossRef] [Green Version]
- Acob, A.; Jing, J.; Lee, J.; Schedin, P.; Gilbert, S.M.; Peden, A.A.; Junutula, J.R.; Prekeris, R. Rab40b regulates trafficking of MMP2 and MMP9 during invadopodia formation and invasion of breast cancer cells. J. Cell Sci. 2013, 126 Pt 20, 4647–4658. [Google Scholar]
- Hendrix, A.; Maynard, D.; Pauwels, P.; Braems, G.; Denys, H.; Van den Broecke, R.; Lambert, J.; Van Belle, S.; Cocquyt, V.; Gespach, C.; et al. Effect of the secretory small GTPase Rab27B on breast cancer growth, invasion, and metastasis. J. Natl. Cancer Inst. 2010, 102, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, L.A.; Kumar, R.; Hales, C.M.; Navarre, J.; Bhartur, S.G.; Burnette, J.O.; Provance, D.W., Jr.; Mercer, J.A.; Bähler, M.; Goldenring, J.R. Myosin vb is associated with plasma membrane recycling systems. Mol. Biol. Cell 2001, 12, 1843–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welz, T.; Kerkhoff, E. Exploring the iceberg: Prospects of coordinated myosin V and actin assembly functions in transport processes. Small GTPases 2019, 10, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Howe, E.N.; Burnette, M.D.; Justice, M.E.; Schnepp, P.M.; Hedrick, V.; Clancy, J.W.; Guldner, I.H.; Lamere, A.T.; Li, J.; Aryal, U.K.; et al. Rab11b-mediated integrin recycling promotes brain metastatic adaptation and outgrowth. Nat. Commun. 2020, 11, 3017. [Google Scholar] [CrossRef] [PubMed]
- Ferro, E.; Bosia, C.; Campa, C.C. RAB11-mediated trafficking and human cancers: An updated review. Biology 2020, 10, 26. [Google Scholar] [CrossRef]
- Miserey-Lenkei, S.; Bousquet, H.; Pylypenko, O.; Bardin, S.; Dimitrov, A.; Bressanelli, G.; Bonifay, R.; Fraisier, V.; Guillou, C.; Bougeret, C.; et al. Coupling fission and exit of RAB6 vesicles at Golgi hotspots through kinesin-myosin interactions. Nat. Commun. 2017, 8, 1254. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, M.S.; McPherson, P.S. Regulation of Cancer Cell Behavior by the Small GTPase Rab13. J. Biol. Chem. 2016, 291, 9929–9937. [Google Scholar] [CrossRef] [Green Version]
- Neary, C.L.; Nesterova, M.; Cho, Y.S.; Cheadle, C.; Becker, K.G.; Cho-Chung, Y.S. Protein kinase A isozyme switching: Eliciting differential cAMP signaling and tumor reversion. Oncogene 2004, 23, 8847–8856. [Google Scholar] [CrossRef]
- Casalou, C.; Faustino, A.; Barral, D.C. Arf proteins in cancer cell migration. Small GTPases 2016, 7, 270–282. [Google Scholar] [CrossRef]
- Casalou, C.; Ferreira, A.; Barral, D.C. The Role of ARF Family Proteins and Their Regulators and Effectors in Cancer Progression: A Therapeutic Perspective. Front. Cell Dev. Biol. 2020, 8, 217. [Google Scholar] [CrossRef] [Green Version]
- Schlienger, S.; Campbell, S.; Claing, A. ARF1 regulates the Rho/MLC pathway to control EGF-dependent breast cancer cell invasion. Mol. Biol. Cell 2014, 25, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Tang, S.; Cai, Y.; Pi, W.; Deng, L.; Wu, G.; Chavanieu, A.; Teng, Y. Suppression of breast cancer metastasis through the inactivation of ADP-ribosylation factor 1. Oncotarget 2016, 7, 58111–58120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howley, B.V.; Link, L.A.; Grelet, S.; El-Sabban, M.; Howe, P.H. A CREB3-regulated ER-Golgi trafficking signature promotes metastatic progression in breast cancer. Oncogene 2018, 37, 1308–1325. [Google Scholar] [CrossRef] [PubMed]
- Howley, B.V.; Howe, P.H. Metastasis-associated upregulation of ER-Golgi trafficking kinetics: Regulation of cancer progression via the Golgi apparatus. Oncoscience 2018, 5, 142–143. [Google Scholar] [CrossRef]
- Ruggiero, C.; Grossi, M.; Fragassi, G.; Di Campli, A.; Di Ilio, C.; Luini, A.; Sallese, M. The KDEL receptor signalling cascade targets focal adhesion kinase on focal adhesions and invadopodia. Oncotarget 2018, 9, 10228–10246. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, R.; Kundu, S.T.; Grzeskowiak, C.L.; Fradette, J.J.; Scott, K.L.; Creighton, C.J.; Gibbons, D.L. IMPAD1 and KDELR2 drive invasion and metastasis by enhancing Golgi-mediated secretion. Oncogene 2020, 39, 5979–5994. [Google Scholar] [CrossRef]
- Malsam, J.; Söllner, T.H. Organization of SNAREs within the Golgi stack. Cold Spring Harb Perspect. Biol. 2011, 3, a005249. [Google Scholar] [CrossRef] [Green Version]
- Sehgal, P.B.; Mukhopadhyay, S.; Xu, F.; Patel, K.; Shah, M. Dysfunction of Golgi tethers, SNAREs, and SNAPs in monocrotaline-induced pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L1526–L1542. [Google Scholar] [CrossRef]
- Steffen, A.; Le Dez, G.; Poincloux, R.; Recchi, C.; Nassoy, P.; Rottner, K.; Galli, T.; Chavrier, P. MT1-MMP-Dependent Invasion Is Regulated by TI-VAMP/VAMP7. Curr. Biol. 2008, 18, 926–931. [Google Scholar] [CrossRef]
- Meng, J.; Wang, J. Role of SNARE proteins in tumourigenesis and their potential as targets for novel anti-cancer therapeutics. Biochim. Et Biophys. Acta (BBA) Rev. Cancer 2015, 1856, 1–12. [Google Scholar] [CrossRef]
- Peak, T.C.; Su, Y.; Chapple, A.G.; Chyr, J.; Deep, G. Syntaxin 6: A novel predictive and prognostic biomarker in papillary renal cell carcinoma. Sci. Rep. 2019, 9, 3146. [Google Scholar] [CrossRef]
- Du, Y.; Shen, J.; Hsu, J.L.; Han, Z.; Hsu, M.C.; Yang, C.C.; Kuo, H.P.; Wang, Y.N.; Yamaguchi, H.; Miller, S.A.; et al. Syntaxin 6-mediated Golgi translocation plays an important role in nuclear functions of EGFR through microtubule-dependent trafficking. Oncogene 2014, 33, 756–770. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Smith, M.; Halder, J.B.; Meltzer, P.S.; Gonda, T.A.; Mangala, L.S.; Rupaimoole, R.; Lu, C.; Nagaraja, A.S.; Gharpure, K.M.; Kang, Y.; et al. ATP11B mediates platinum resistance in ovarian cancer. J. Clin. Investig. 2013, 123, 2119–2130. [Google Scholar] [CrossRef]
- Boddul, S.V.; Meng, J.; Dolly, J.O.; Wang, J. SNAP-23 and VAMP-3 contribute to the release of IL-6 and TNFα from a human synovial sarcoma cell line. FEBS J. 2014, 281, 750–765. [Google Scholar] [CrossRef]
- Wang, L.; Brautigan, D.L. α-SNAP inhibits AMPK signaling to reduce mitochondrial biogenesis and dephosphorylates Thr172 in AMPKα in vitro. Nat. Commun. 2013, 4, 1559. [Google Scholar] [CrossRef] [Green Version]
- Naydenov, N.G.; Brown, B.; Harris, G.; Dohn, M.R.; Morales, V.M.; Baranwal, S.; Reynolds, A.B.; Ivanov, A.I. A membrane fusion protein αSNAP is a novel regulator of epithelial apical junctions. PLoS ONE 2012, 7, e34320. [Google Scholar] [CrossRef] [Green Version]
- Ravichandran, Y.; Goud, B.; Manneville, J.B. The Golgi apparatus and cell polarity: Roles of the cytoskeleton, the Golgi matrix, and Golgi membranes. Curr. Opin. Cell Biol. 2020, 62, 104–113. [Google Scholar] [CrossRef]
- Rizzo, R.; Russo, D.; Kurokawa, K.; Sahu, P.; Lombardi, B.; Supino, D.; Zhukovsky, M.A.; Vocat, A.; Pothukuchi, P.; Kunnathully, V.; et al. Golgi maturation-dependent glycoenzyme recycling controls glycosphingolipid biosynthesis and cell growth via GOLPH3. EMBO J. 2021, 40, e107238. [Google Scholar] [CrossRef]
- Kellokumpu, S. Golgi pH, Ion and Redox Homeostasis: How Much Do They Really Matter? Front. Cell Dev. Biol. 2019, 7, 93. [Google Scholar] [CrossRef] [Green Version]
- Mennerich, D.; Kellokumpu, S.; Kietzmann, T. Hypoxia and Reactive Oxygen Species as Modulators of Endoplasmic Reticulum and Golgi Homeostasis. Antioxid. Redox Signal. 2019, 30, 113–137. [Google Scholar] [CrossRef]
- Rankin, E.B.; Nam, J.M.; Giaccia, A.J. Hypoxia: Signaling the Metastatic Cascade. Trends Cancer 2016, 2, 295–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arsenault, D.; Lucien, F.; Dubois, C.M. Hypoxia enhances cancer cell invasion through relocalization of the proprotein convertase furin from the trans-Golgi network to the cell surface. J. Cell. Physiol. 2012, 227, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Bensellam, M.; Maxwell, E.L.; Chan, J.Y.; Luzuriaga, J.; West, P.K.; Jonas, J.-C.; Gunton, J.E.; Laybutt, D.R. Hypoxia reduces ER-to-Golgi protein trafficking and increases cell death by inhibiting the adaptive unfolded protein response in mouse beta cells. Diabetologia 2016, 59, 1492–1502. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Wang, L.; Mollica, M.; Re, A.T.; Wu, S.; Zuo, L. Nitric oxide in cancer metastasis. Cancer lett. 2014, 353, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Iwakiri, Y.; Satoh, A.; Chatterjee, S.; Toomre, D.K.; Chalouni, C.M.; Fulton, D.; Groszmann, R.J.; Shah, V.H.; Sessa, W.C. Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc. Natl. Acad. Sci. USA 2006, 103, 19777–19782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, B.J.; Allen, J.L.; Caswell, P.T. Vesicle trafficking pathways that direct cell migration in 3D matrices and in vivo. Traffic 2018, 19, 899–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kupfer, A.; Louvard, D.; Singer, S.J. Polarization of the Golgi apparatus and the microtubule-organizing center in cultured fibroblasts at the edge of an experimental wound. Proc. Natl. Acad. Sci. USA 1982, 79, 2603–2607. [Google Scholar] [CrossRef] [Green Version]
- Mu, G.; Ding, Q.; Li, H.; Zhang, L.; Zhang, L.; He, K.; Wu, L.; Deng, Y.; Yang, D.; Wu, L.; et al. Gastrin stimulates pancreatic cancer cell directional migration by activating the Gz12/13-RhoA-ROCK signaling pathway. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Zhang, S.; Schafer-Hales, K.; Khuri, F.R.; Zhou, W.; Vertino, P.M.; Marcus, A.I. The tumor suppressor LKB1 regulates lung cancer cell polarity by mediating cdc42 recruitment and activity. Cell Tumor Stem. Cell Biol. 2008, 68, 740–748. [Google Scholar] [CrossRef] [Green Version]
- Xing, M.; Peterman, M.C.; Davis, R.L.; Oegema, K.; Shiau, A.K.; Field, S.J. GOLPH3 drives cell migration by promoting Golgi reorientation and directional trafficking to the leading edge. Mol. Biol. Cell 2016, 27, 3828–3840. [Google Scholar] [CrossRef]
- Baschieri, F.; Confalonieri, S.; Bertalot, G.; Di Fiore, P.P.; Dietmaier, W.; Leist, M.; Crespo, P.; Macara, I.G.; Farhan, H. Spatial control of Cdc42 signalling by a GM130-RasGRF complex regulates polarity and tumorigenesis. Nat. Commun. 2014, 5, 4839. [Google Scholar] [CrossRef]
- Baschieri, F.; Farhan, H. Endomembrane control of cell polarity: Relevance to cancer. Small GTPases 2015, 6, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Baschieri, F.; Uetz-von Allmen, E.; Legler, D.F.; Farhan, H. Loss of GM130 in breast cancer cells and its effects on cell migration, invasion and polarity. Cell Cycle 2015, 14, 1139–1147. [Google Scholar] [CrossRef]
- Dubois, F.; Alpha, K.; Turner, C.E. Paxillin regulates cell polarization and anterograde vesicle trafficking during cell migration. Mol. Biol. Cell 2017, 28, 3815–3831. [Google Scholar] [CrossRef]
- Mousson, A.; Legrand, M.; Steffan, T.; Vauchelles, R.; Carl, P.; Gies, J.P.; Lehmann, M.; Zuber, G.; De Mey, J.; Dujardin, D.; et al. Inhibiting FAK-Paxillin Interaction Reduces Migration and Invadopodia-Mediated Matrix Degradation in Metastatic Melanoma Cells. Cancers 2021, 13, 1871. [Google Scholar] [CrossRef]
- Yadav, S.; Puri, S.; Linstedt, A.D. A primary role for Golgi positioning in directed secretion, cell polarity and wound healing. Mol. Biol. Cell 2009, 20, 1728–1736. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Banerjee, P.; Guo, H.F.; Ireland, S.; Pankova, D.; Ahn, Y.H.; Nikolaidis, I.M.; Liu, X.; Zhao, Y.; Xue, Y.; et al. Epithelial-to-mesenchymal transition drives a pro-metastatic Golgi compaction process through scaffolding protein PAQR11. J. Clin. investig. 2017, 127, 117–131. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Peng, D.H.; Ungewiss, C.; Tong, P.; Byers, L.A.; Wang, J.; Canales, J.R.; Villalobos, P.A.; Uraoka, N.; Mino, B.; Behrens, C.; et al. ZEB1 induces LOXL2-mediated collagen stabilization and deposition in the extracellular matrix to drive lung cancer invasion and metastasis. Oncogene 2017, 36, 1925–1938. [Google Scholar] [CrossRef] [Green Version]
- Gibbons, D.L.; Lin, W.; Creighton, C.J.; Rizvi, Z.H.; Gregory, P.A.; Goodall, G.J.; Thilaganathan, N.; Du, L.; Zhang, Y.; Pertsemlidis, A.; et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009, 23, 2140–2151. [Google Scholar] [CrossRef] [Green Version]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Ungewiss, C.; Rizvi, Z.H.; Roybal, J.D.; Peng, D.H.; Gold, K.A.; Shin, D.-H.; Creighton, C.J.; Gibbons, D.L. The microRNA-200/Zeb1 axis regulates ECM-dependent β1-integrin/FAK signaling, cancer cell invasion and metastasis through CRKL. Sci. Rep. 2016, 6, 18652. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.-F.; Bota-Rabassedas, N.; Terajima, M.; Leticia Rodriguez, B.; Gibbons, D.L.; Chen, Y.; Banerjee, P.; Tsai, C.-L.; Tan, X.; Liu, X.; et al. A collagen glucosyltransferase drives lung adenocarcinoma progression in mice. Commun. Biol. 2021, 4, 482. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.H.; Rodriguez, B.L.; Diao, L.; Chen, L.; Wang, J.; Byers, L.A.; Wei, Y.; Chapman, H.A.; Yamauchi, M.; Behrens, C.; et al. Collagen promotes anti-PD-1/PD-L1 resistance in cancer through LAIR1-dependent CD8(+) T cell exhaustion. Nat. Commun. 2020, 11, 4520. [Google Scholar] [CrossRef]
- Sechi, S.; Frappaolo, A.; Karimpour-Ghahnavieh, A.; Piergentili, R.; Giansanti, M.G. Oncogenic Roles of GOLPH3 in the Physiopathology of Cancer. Int. J. Mol. Sci. 2020, 21, 933. [Google Scholar] [CrossRef] [Green Version]
- Rahajeng, J.; Kuna, R.S.; Makowski, S.L.; Tran, T.T.T.; Buschman, M.D.; Li, S.; Cheng, N.; Ng, M.M.; Field, S.J. Efficient Golgi Forward Trafficking Requires GOLPH3-Driven, PI4P-Dependent Membrane Curvature. Dev. Cell 2019, 50, 573–585.e575. [Google Scholar] [CrossRef]
- Scott, K.L.; Kabbarah, O.; Liang, M.C.; Ivanova, E.; Anagnostou, V.; Wu, J.; Dhakal, S.; Wu, M.; Chen, S.; Feinberg, T.; et al. GOLPH3 modulates mTOR signalling and rapamycin sensitivity in cancer. Nature 2009, 459, 1085–1090. [Google Scholar] [CrossRef] [Green Version]
- Kuna, R.S.; Field, S.J. GOLPH3: A Golgi phosphatidylinositol(4)phosphate effector that directs vesicle trafficking and drives cancer. J. Lipid Res. 2019, 60, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Makhoul, C.; Gosavi, P.; Gleeson, P.A. Golgi Dynamics: The Morphology of the Mammalian Golgi Apparatus in Health and Disease. Front. Cell Dev. Biol. 2019, 7, 112. [Google Scholar] [CrossRef] [Green Version]
- Saraste, J.; Prydz, K. A New Look at the Functional Organization of the Golgi Ribbon. Front. Cell Dev. Biol. 2019, 7, 171. [Google Scholar] [CrossRef]
- Stow, J.L.; Murray, R.Z. Intracellular trafficking and secretion of inflammatory cytokines. Cytokine Growth Factor Rev. 2013, 24, 227–239. [Google Scholar] [CrossRef]
- Robbins, E.; Gonatas, N.K. The Ultrastructure of a Mammalian Cell during the Mitotic Cycle. J. Cell Biol. 1964, 21, 429–463. [Google Scholar] [CrossRef]
- Valente, C.; Colanzi, A. Mechanisms and Regulation of the Mitotic Inheritance of the Golgi Complex. Front. Cell Dev. Biol. 2015, 3, 79. [Google Scholar] [CrossRef] [Green Version]
- Petrosyan, A. Unlocking Golgi: Why Does Morphology Matter? Biochemistry 2019, 84, 1490–1501. [Google Scholar] [CrossRef]
- Petrosyan, A. Onco-Golgi: Is Fragmentation a Gate to Cancer Progression? Biochem. Mol. Biol. J. 2015, 1, 16. [Google Scholar] [CrossRef] [Green Version]
- Chia, J.; Goh, G.; Racine, V.; Ng, S.; Kumar, P.; Bard, F. RNAi screening reveals a large signaling network controlling the Golgi apparatus in human cells. Mol. Syst. Biol. 2012, 8, 629. [Google Scholar] [CrossRef]
- Lin, S.C.; Chien, C.W.; Lee, J.C.; Yeh, Y.C.; Hsu, K.F.; Lai, Y.Y.; Lin, S.C.; Tsai, S.J. Suppression of dual-specificity phosphatase-2 by hypoxia increases chemoresistance and malignancy in human cancer cells. J. Clin. Investig. 2011, 121, 1905–1916. [Google Scholar] [CrossRef]
- Lee, J.E.; Patel, K.; Almodóvar, S.; Tuder, R.M.; Flores, S.C.; Sehgal, P.B. Dependence of Golgi apparatus integrity on nitric oxide in vascular cells: Implications in pulmonary arterial hypertension. Am. J. Physiology. Heart Circ. Physiol. 2011, 300, H1141–H1158. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.E.; Yuan, H.; Liang, F.X.; Sehgal, P.B. Nitric oxide scavenging causes remodeling of the endoplasmic reticulum, Golgi apparatus and mitochondria in pulmonary arterial endothelial cells. Nitric Oxide Biol. Chem. 2013, 33, 64–73. [Google Scholar] [CrossRef] [Green Version]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Reviews. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Ferrer, L.; Legler, K.; Milde-Langosch, K. Role of protein glycosylation in cancer metastasis. Semin. Cancer Biol. 2017, 44, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Hakomori, S. Aberrant glycosylation in tumors and tumor-associated carbohydrate antigens. Adv. Cancer Res. 1989, 52, 257–331. [Google Scholar]
- Hakomori, S.-I.; Cummings, R.D. Glycosylation effects on cancer development. Glycoconj. J. 2012, 29, 565–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakomori, S. Glycosylation defining cancer malignancy: New wine in an old bottle. Proc. Natl. Acad. Sci. USA 2002, 99, 10231–10233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, M.; Nishikawa, A.; Ihara, Y.; Taniguchi, S.; Taniguchi, N. Suppression of lung metastasis of B16 mouse melanoma by N-acetylglucosaminyltransferase III gene transfection. Proc. Natl. Acad. Sci. USA 1995, 92, 8754–8758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, M.; Ihara, Y.; Matsuzawa, Y.; Taniguchi, N. Aberrant glycosylation of E-cadherin enhances cell-cell binding to suppress metastasis. J. Biol. Chem. 1996, 271, 13811–13815. [Google Scholar] [CrossRef] [Green Version]
- Kakugawa, Y.; Wada, T.; Yamaguchi, K.; Yamanami, H.; Ouchi, K.; Sato, I.; Miyagi, T. Up-regulation of plasma membrane-associated ganglioside sialidase (Neu3) in human colon cancer and its involvement in apoptosis suppression. Proc. Natl. Acad. Sci. USA 2002, 99, 10718–10723. [Google Scholar] [CrossRef] [Green Version]
- Rivinoja, A.; Pujol, F.M.; Hassinen, A.; Kellokumpu, S. Golgi pH, its regulation and roles in human disease. Ann. Med. 2010, 44, 542–554. [Google Scholar] [CrossRef]
- Julien, S.; Picco, G.; Sewell, R.; Vercoutter-Edouart, A.S.; Tarp, M.; Miles, D.; Clausen, H.; Taylor-Papadimitriou, J.; Burchell, J.M. Sialyl-Tn vaccine induces antibody-mediated tumour protection in a relevant murine model. Br. J. Cancer 2009, 100, 1746–1754. [Google Scholar] [CrossRef] [Green Version]
- Belo, A.I.; van Vliet, S.J.; Maus, A.; Laan, L.C.; Nauta, T.D.; Koolwijk, P.; Tefsen, B.; van Die, I. Hypoxia inducible factor 1α down regulates cell surface expression of α1,2-fucosylated glycans in human pancreatic adenocarcinoma cells. FEBS Lett. 2015, 589, 2359–2366. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.H.; Lin, S.Y.; Weng, R.R.; Juan, Y.H.; Chen, Y.W.; Hou, H.H.; Hung, Z.C.; Oswita, G.A.; Huang, Y.J.; Guu, S.Y.; et al. Fucosyltransferase 4 shapes oncogenic glycoproteome to drive metastasis of lung adenocarcinoma. EBioMedicine 2020, 57, 102846. [Google Scholar] [CrossRef]
- Pothukuchi, P.; Agliarulo, I.; Pirozzi, M.; Rizzo, R.; Russo, D.; Turacchio, G.; Nüchel, J.; Yang, J.-S.; Gehin, C.; Capolupo, L.; et al. GRASP55 regulates intra-Golgi localization of glycosylation enzymes to control glycoshingolipid biosynthesis. EMBO J. 2021, 40, e107766. [Google Scholar] [CrossRef]
- Stevenson, N.L.; Bergen, D.J.M.; Skinner, R.E.H.; Kague, E.; Martin-Silverstone, E.; Brown, K.A.R.; Hammond, C.L.; Stephens, D.J. Giantin-knockout models reveal a feedback loop between Golgi function and glycosyltrasferase expression. J. Cell Sci. 2017, 130, 4132–4143. [Google Scholar]
- Conroy, L.R.; Stanback, A.E.; Young, L.E.A.; Clarke, H.A.; Austin, G.L.; Liu, J.; Allison, D.B.; Sun, R.C. In Situ analysis of N-linked glycans as potential biomarkers of clinical course in human prostate cancer. Mol. Cancer Res. 2021, 19, 1727–1738. [Google Scholar] [CrossRef]
- Langford, J.K.; Stanley, M.J.; Cao, D.; Sanderson, R.D. Multiple heparan sulfate chains are required for optimal syndecan-1 function. J. Biol. Chem. 1998, 273, 29965–29971. [Google Scholar] [CrossRef] [Green Version]
- Vicente, C.M.; da Silva, D.A.; Sartorio, P.V.; Silva, T.D.; Saad, S.S.; Nader, H.B.; Forones, N.M.; Toma, L. Heparan Sulfate Proteoglycans in Human Colorectal Cancer. Anal. Cell. Pathol. 2018, 2018, 8389595. [Google Scholar] [CrossRef] [Green Version]
- Pudełko, A.; Wisowski, G.; Olczyk, K.; Koźma, E.M. The dual role of the glycosaminoglycan chondroitin-6-sulfate in the development, progression and metastasis of cancer. FEBS J. 2019, 286, 1815–1837. [Google Scholar] [CrossRef]
- Jin, H.; Zangar, R.C. Protein modifications as potential biomarkers in breast cancer. Biomark. Insights 2009, 4, S2557. [Google Scholar] [CrossRef]
- Dela Cruz, C.S.; Lee, Y.; Viswanathan, S.R.; El-Guindy, A.S.; Gerlach, J.; Nikiforow, S.; Shedd, D.; Gradoville, L.; Miller, G. N-linked glycosylation is required for optimal function of Kaposi’s sarcoma herpesvirus-encoded, but not cellular, interleukin 6. J. Exp. Med. 2004, 199, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Zappa, F.; Failli, M.; De Matteis, M.A. The Golgi complex in disease and therapy. Curr. Opin. Cell Biol. 2018, 50, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Shi, L.; Banerjee, P.; Liu, X.; Guo, H.; Yu, J.; Bota-Rabassedas, N.; Rodriguez, B.L.; Gibbons, D.L.; Russell, W.K.; et al. A protumorigenic secretory pathway activated by p53 deficiency in lung adenocarcinoma. J. Clin. Investig. 2021, 131, e137186. [Google Scholar] [CrossRef] [PubMed]
- Rutaganira, F.U.; Fowler, M.L.; McPhail, J.A.; Gelman, M.A.; Nguyen, K.; Xiong, A.; Dornan, G.L.; Tavshanjian, B.; Glenn, J.S.; Shokat, K.M.; et al. Design and Structural Characterization of Potent and Selective Inhibitors of Phosphatidylinositol 4 Kinase IIIβ. J. Med. Chem. 2016, 59, 1830–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, J.H.; Shi, D.S.; Grossmann, A.H.; Odelberg, S.J.; Ostanin, K.; Li, D.Y. ARF6 is an actionable node that orchestrates oncogenic GNAQ signaling in uveal melanoma. Cancer Cell 2016, 29, 889–904. [Google Scholar] [CrossRef] [Green Version]
- Fu, D.; Roufogalis, B.D. Actin disruption inhibits endosomal traffic of P-glycoprotein-EGFP and resistance to daunorubicin accumulation. Am. J. Physiol. Cell Physiol. 2007, 292, C1543–C1552. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.H.; Beckett, P.; Brown, P.D.; Bone, E.A.; Davidson, A.H.; Galloway, W.A.; Gearing, A.J.H.; Huxley, P.; Laber, D.; McCourt, M.; et al. Preclinical and clinical studies of MMP inhibitors in cancer. Ann. N. Y. Acad. Sci. 2006, 878, 228–235. [Google Scholar] [CrossRef]
- Shah, M.A.; Starodub, A.; Sharma, S.; Silverman, J.A.; Lenz, H.; Bendell, J.C. Andecaliximab/GS-5745 alone and combined with mFOLFOX6 in advanced gastric and gastroesophageal junction adenocarcinoma: Results from a Phase I study. Clin. Cancer Res. 2018, 24, 3829–3837. [Google Scholar] [CrossRef] [Green Version]
- Huet, G.; Hennebicq-Reig, S.; Bolos, C.; Ulloa, F.; Lesuffleur, T.; Barbat, A.; Carriere, V.; Kim, I.; Real, F.X.; Delannoy, P.; et al. GalNAc-alpha-O-benzyl inhibits NeuAcalpha2-3 glycosylation and blocks the intracellular transport of apical glycoproteins and mucus in differentiated HT-29 cells. J. Cell Biol. 1998, 141, 1311–1322. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Linstedt, A.D. Inhibitor of ppGalNAc-T3-mediated O-glycosylation blocks cancer cell invasiveness and lowers FGF23 levels. Elife 2017, 6, e24051. [Google Scholar] [CrossRef] [Green Version]
- Madden, E.C.; Gorman, A.M.; Logue, S.E.; Samali, A. Tumour Cell Secretome in Chemoresistance and Tumour Recurrence. Trends Cancer 2020, 6, 489–505. [Google Scholar] [CrossRef] [Green Version]
- Grieve, A.G.; Rabouille, C. Golgi bypass: Skirting around the heart of classical secretion. Cold Spring Harb. Perspect. Biol. 2011, 3, a005298. [Google Scholar] [CrossRef] [Green Version]
- Almiron Bonnin, D.A.; Havrda, M.C.; Israel, M.A. Glioma Cell Secretion: A Driver of Tumor Progression and a Potential Therapeutic Target. Cancer Res. 2018, 78, 6031–6039. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bajaj, R.; Warner, A.N.; Fradette, J.F.; Gibbons, D.L. Dance of The Golgi: Understanding Golgi Dynamics in Cancer Metastasis. Cells 2022, 11, 1484. https://doi.org/10.3390/cells11091484
Bajaj R, Warner AN, Fradette JF, Gibbons DL. Dance of The Golgi: Understanding Golgi Dynamics in Cancer Metastasis. Cells. 2022; 11(9):1484. https://doi.org/10.3390/cells11091484
Chicago/Turabian StyleBajaj, Rakhee, Amanda N. Warner, Jared F. Fradette, and Don L. Gibbons. 2022. "Dance of The Golgi: Understanding Golgi Dynamics in Cancer Metastasis" Cells 11, no. 9: 1484. https://doi.org/10.3390/cells11091484
APA StyleBajaj, R., Warner, A. N., Fradette, J. F., & Gibbons, D. L. (2022). Dance of The Golgi: Understanding Golgi Dynamics in Cancer Metastasis. Cells, 11(9), 1484. https://doi.org/10.3390/cells11091484