
Circadian Governance of Cardiac Growth

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
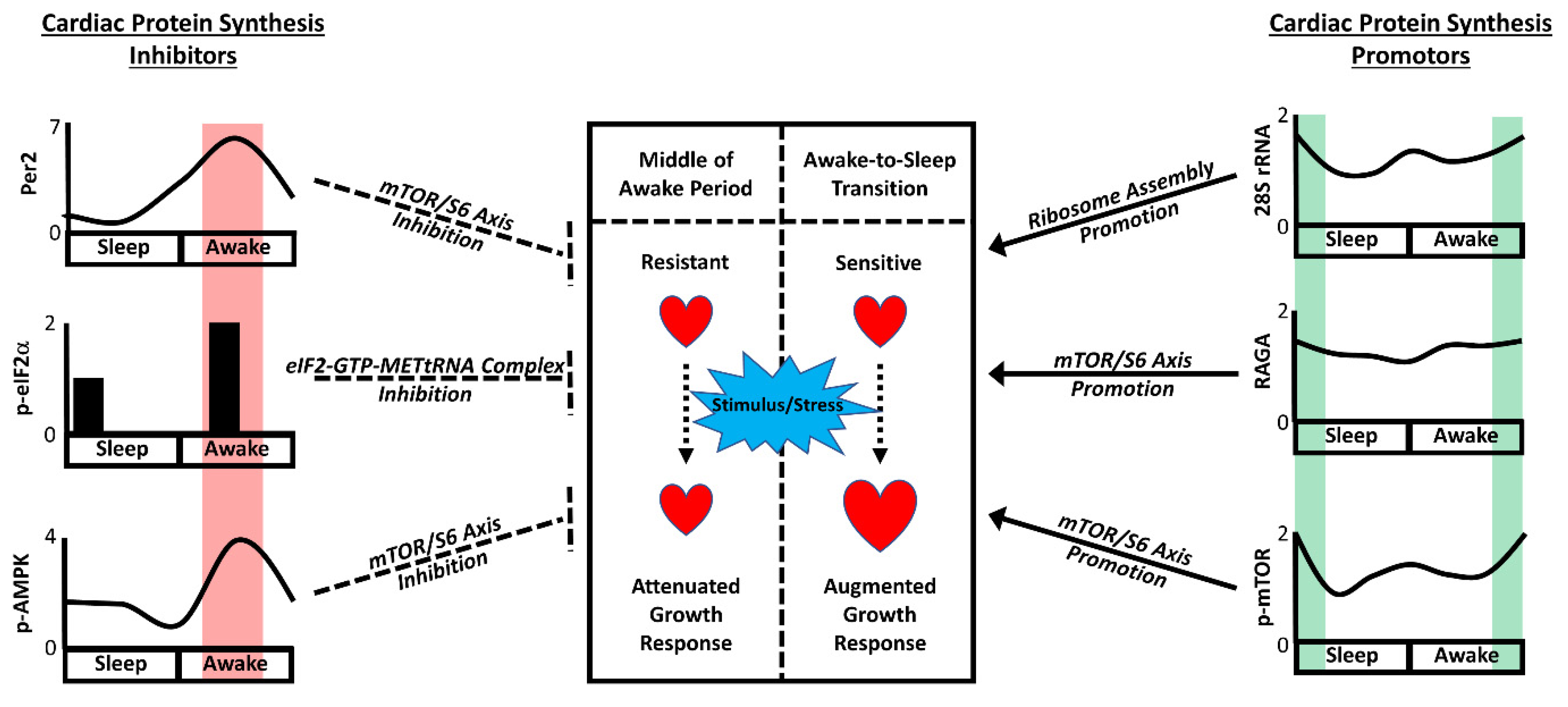
2. The Awake-to-Sleep Phase Transition Is a Period of Pro-Hypertrophic Growth
3. Contribution of Extrinsic versus Intrinsic Factors
4. Lessons Learned from Extra-Cardiac Tissues
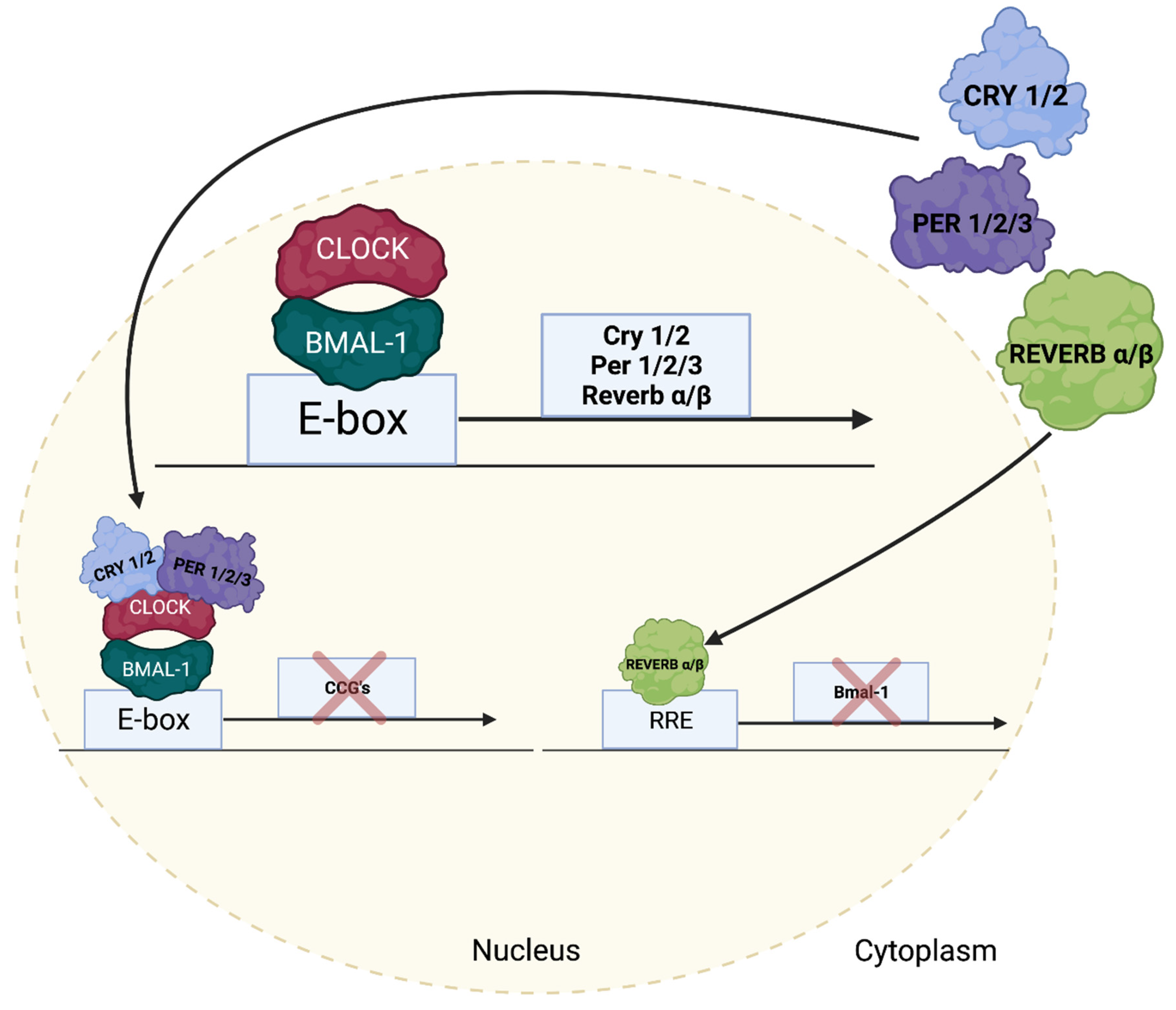
5. Regulation of Cardiac Growth Pathways by the Cardiomyocyte Circadian Clock
6. Translational Implications
7. Summary and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Takahashi, J.S.; Hong, H.-K.; Ko, C.H.; McDearmon, E.L. The genetics of mammalian circadian order and disorder: Implications for physiology and disease. Nat. Rev. Genet. 2008, 9, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Edery, I. Circadian rhythms in a nutshell. Physiol. Genom. 2000, 3, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Degaute, J.P.; van de Borne, P.; Linkowski, P.; Van Cauter, E. Quantitative analysis of the 24-hour blood pressure and heart rate patterns in young men. Hypertension 1991, 18, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voutilainen, S.; Kupari, M.; Hippelainen, M.; Karppinen, K.; Ventila, M. Circadian variation of left ventricular diastolic function in healthy people. Heart 1996, 75, 35–39. [Google Scholar] [CrossRef] [Green Version]
- Curtis, A.M.; Cheng, Y.; Kapoor, S.; Reilly, D.; Price, T.S.; FitzGerald, G.A. Circadian variation of blood pressure and the vascular response to asynchronous stress. Proc. Natl. Acad. Sci. USA 2007, 104, 3450–3455. [Google Scholar] [CrossRef] [Green Version]
- Muller, J.E.; Tofler, G.H.; Stone, P.H. Circadian variation and triggers of onset of acute cardiovascular disease. Circulation 1989, 79, 733–743. [Google Scholar] [CrossRef] [Green Version]
- Richards, A.M.; Nicholls, M.G.; Espiner, E.A.; Ikram, H.; Cullens, M.; Hinton, D. Diurnal Patterns of Blood Pressure, Heart Rate and Vasoactive Hormones in Normal Man. Clin. Exp. Hypertens. Part A Theory Pr. 1986, 8, 153–166. [Google Scholar] [CrossRef]
- Martino, T.A.; Young, M.E. Influence of the Cardiomyocyte Circadian Clock on Cardiac Physiology and Pathophysiology. J. Biol. Rhythm. 2015, 30, 183–205. [Google Scholar] [CrossRef]
- Rana, S.; Prabhu, S.D.; Young, M.E. Chronobiological Influence Over Cardiovascular Function: The Good, the Bad, and the Ugly. Circ. Res. 2020, 126, 258–279. [Google Scholar] [CrossRef]
- Young, M.E. The circadian clock within the heart: Potential influence on myocardial gene expression, metabolism, and function. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1–H16. [Google Scholar] [CrossRef] [Green Version]
- Young, M.E. Temporal partitioning of cardiac metabolism by the cardiomyocyte circadian clock. Exp. Physiol. 2016, 101, 1035–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.E. Circadian Control of Cardiac Metabolism: Physiologic Roles and Pathologic Implications. Methodist DeBakey Cardiovasc. J. 2017, 13, 15–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.; Razeghi, P.; Cedars, A.; Guthrie, P.; Taegtmeyer, H. Intrinsic diurnal variations in cardiac metabolism and con-tractile function. Circ. Res. 2001, 89, 1199–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durgan, D.J.; Pat, B.M.; Laczy, B.; Bradley, J.A.; Tsai, J.-Y.; Grenett, M.H.; Ratcliffe, W.F.; Brewer, R.A.; Nagendran, J.; Villegas-Montoya, C.; et al. O-GlcNAcylation, Novel Post-Translational Modification Linking Myocardial Metabolism and Cardiomyocyte Circadian Clock. J. Biol. Chem. 2011, 286, 44606–44619. [Google Scholar] [CrossRef] [Green Version]
- Bray, M.S.; Shaw, C.A.; Moore, M.W.S.; Garcia, R.A.P.; Zanquetta, M.M.; Durgan, D.J.; Jeong, W.J.; Tsai, J.-Y.; Bugger, H.; Zhang, D.; et al. Disruption of the circadian clock within the cardiomyocyte influences myocardial contractile function, metabolism, and gene expression. Am. J. Physiol. Circ. Physiol. Heart 2008, 294, H1036–H1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, J.-Y.; Kienesberger, P.C.; Pulinilkunnil, T.; Sailors, M.H.; Durgan, D.J.; Villegas-Montoya, C.; Jahoor, A.; Gonzalez, R.; Garvey, M.E.; Boland, B.; et al. Direct Regulation of Myocardial Triglyceride Metabolism by the Cardiomyocyte Circadian Clock. J. Biol. Chem. 2010, 285, 2918–2929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGinnis, G.; Tang, Y.; Brewer, R.A.; Brahma, M.; Stanley, H.L.; Shanmugam, G.; Rajasekaran, N.S.; Rowe, G.; Frank, S.J.; Wende, A.; et al. Genetic disruption of the cardiomyocyte circadian clock differentially influences insulin-mediated processes in the heart. J. Mol. Cell. Cardiol. 2017, 110, 80–95. [Google Scholar] [CrossRef]
- Brewer, R.A.; Collins, H.E.; Berry, R.D.; Brahma, M.; Tirado, B.; Peliciari-Garcia, R.A.; Stanley, H.L.; Wende, A.R.; Taegtmeyer, H.; Rajasekaran, N.S.; et al. Temporal partitioning of adaptive responses of the murine heart to fasting. Life Sci. 2018, 197, 30–39. [Google Scholar] [CrossRef]
- Peliciari-Garcia, R.A.; Darley-Usmar, V.; Young, M.E. An overview of the emerging interface between cardiac metabolism, redox biology and the circadian clock. Free Radic. Biol. Med. 2018, 119, 75–84. [Google Scholar] [CrossRef]
- Taegtmeyer, H.; Harinstein, M.E.; Gheorghiade, M. More Than Bricks and Mortar: Comments on Protein and Amino Acid Metabolism in the Heart. Am. J. Cardiol. 2008, 101, S3–S7. [Google Scholar] [CrossRef]
- Durgan, D.J.; Tsai, J.Y.; Grenett, M.H.; Pat, B.M.; Ratcliffe, W.F.; Villegas-Montoya, C.; Garvey, M.E.; Nagendran, J.; Dyck, J.R.; Bray, M.S. Young, Evidence suggesting that the cardiomyocyte circadian clock modulates re-sponsiveness of the heart to hypertrophic stimuli in mice. Chronobiol. Int. 2011, 28, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Latimer, M.N.; Sonkar, R.; Mia, S.; Frayne, I.R.; Carter, K.J.; Johnson, C.A.; Rana, S.; Xie, M.; Rowe, G.C.; Wende, A.R.; et al. Branched chain amino acids selectively promote cardiac growth at the end of the awake period. J. Mol. Cell. Cardiol. 2021, 157, P31–P44. [Google Scholar] [CrossRef]
- Gibb, A.A.; McNally, L.A.; Riggs, D.W.; Conklin, D.J.; Bhatnagar, A.; Hill, B.G. FVB/NJ Mice Are a Useful Model for Exam-ining Cardiac Adaptations to Treadmill Exercise. Front. Physiol. 2016, 7, 636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardo, B.C.; Weeks, K.; Pretorius, L.; McMullen, J.R. Molecular distinction between physiological and pathological cardiac hypertrophy: Experimental findings and therapeutic strategies. Pharmacol. Ther. 2010, 128, 191–227. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.B.; Rourke, B.C.; Caiozzo, V.J.; Bennett, A.F.; Hicks, J.W. Physiology: Postprandial cardiac hypertrophy in pythons. Nature 2005, 434, 37–38. [Google Scholar]
- Rockman, H.; Mao, L.; Thomas, S.; Esposito, G.; Caron, K. Important role of endogenous norepinephrine and epinephrine in the development of in vivo pressure-overload cardiac hypertrophy. J. Am. Coll. Cardiol. 2001, 38, 876–882. [Google Scholar] [CrossRef]
- Ren, R.; Oakley, R.H.; Cruz-Topete, D.; Cidlowski, J.A. Dual Role for Glucocorticoids in Cardiomyocyte Hypertrophy and Apoptosis. Endocrinology 2012, 153, 5346–5360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abel, E.D. Insulin signaling in the heart. Am. J. Physiol. Endocrinol. Metab. 2021, 321, E130–E145. [Google Scholar] [CrossRef]
- Dillmann, W. Cardiac hypertrophy and thyroid hormone signaling. Heart Fail. Rev. 2009, 15, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, G.; Colao, A.; Ferone, D.; Marzullo, P.; Orio, F.; Longobardi, S.; Merola, B. Effect of Growth Hormone on Cardiac Function. Horm. Res. 1997, 48 (Suppl. S4), 38–42. [Google Scholar] [CrossRef]
- Gamble, K.L.; Berry, R.; Frank, S.J.; Young, M.E. Circadian clock control of endocrine factors. Nat. Rev. Endocrinol. 2014, 10, 466–475. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Sur, S.H.; Mistlberger, R.E.; Morris, M. Circadian blood pressure and heart rate rhythms in mice. Am. J. Physiol. -Regul. Integr. Comp. Physiol. 1999, 276, R500–R504. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.W.; Yoshihara, T.; Machida, S.; Naito, H. Circadian rhythm of intracellular protein synthesis signaling in rat cardiac and skeletal muscles. Biochem. Biophys. Rep. 2017, 9, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Avram, A.M.; Jaffe, C.A.; Symons, K.V.; Barkan, A.L. Endogenous Circulating Ghrelin Does Not Mediate Growth Hormone Rhythmicity or Response to Fasting. J. Clin. Endocrinol. Metab. 2005, 90, 2982–2987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaffe, C.A.; Ocampo-Lim, B.; Guo, W.; Krueger, K.; Sugahara, I.; DeMott-Friberg, R.; Bermann, M.; Barkan, A.L. Regulatory mechanisms of growth hormone secretion are sexually dimorphic. J. Clin. Investig. 1998, 102, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Naito, Y.; Tsujino, T.; Fujioka, Y.; Ohyanagi, M.; Iwasaki, T. Augmented diurnal variations of the cardiac renin-angiotensin system in hypertensive rats. Hypertension 2002, 40, 827–833. [Google Scholar] [CrossRef] [Green Version]
- Muoio, D.M.; Newgard, C.B. Mechanisms of disease:Molecular and metabolic mechanisms of insulin resistance and β-cell failure in type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 193–205. [Google Scholar] [CrossRef]
- Durgan, D.J.; Hotze, M.A.; Tomlin, T.M.; Egbejimi, O.; Graveleau, C.; Abel, E.D.; Shaw, C.A.; Bray, M.S.; Hardin, P.E.; Young, M.E. The intrinsic circadian clock within the cardiomyocyte. Am. J. Physiol. Circ. Physiol. 2005, 289, H1530–H1541. [Google Scholar] [CrossRef] [Green Version]
- Gekakis, N.; Staknis, D.; Nguyen, H.B.; Davis, F.C.; Wilsbacher, L.D.; King, D.P.; Takahashi, J.S.; Weitz, C.J. Role of the CLOCK protein in the mammalian circadian mechanism. Science 1998, 280, 1564–1569. [Google Scholar] [CrossRef]
- Hogenesch, J.B.; Gu, Y.-Z.; Jain, S.; Bradfield, C.A. The basic-helix–loop–helix-PAS orphan MOP3 forms transcriptionally active complexes with circadian and hypoxia factors. Proc. Natl. Acad. Sci. USA 1998, 95, 5474–5479. [Google Scholar] [CrossRef] [Green Version]
- Zylka, M.J.; Shearman, L.P.; Weaver, D.; Reppert, S.M. Three period Homologs in Mammals: Differential Light Responses in the Suprachiasmatic Circadian Clock and Oscillating Transcripts Outside of Brain. Neuron 1998, 20, 1103–1110. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, Y.; Sancar, A. Vitamin B 2 -based blue-light photoreceptors in the retinohypothalamic tract as the photoactive pigments for setting the circadian clock in mammals. Proc. Natl. Acad. Sci. USA 1998, 95, 6097–6102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preitner, N.; Damiola, F.; Lopez-Molina, L.; Zakany, J.; Duboule, D.; Albrecht, U.; Schibler, U. The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 2002, 110, 251–260. [Google Scholar] [CrossRef]
- Kume, K.; Zylka, M.J.; Sriram, S.; Shearman, L.P.; Weaver, D.; Jin, X.; Maywood, E.S.; Hastings, M.H.; Reppert, S.M. mCRY1 and mCRY2 Are Essential Components of the Negative Limb of the Circadian Clock Feedback Loop. Cell 1999, 98, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Shearman, L.P.; Sriram, S.; Weaver, D.R.; Maywood, E.S.; Chaves, I.; Zheng, B.; Kume, K.; Lee, C.C.; van der Horst, G.T.; Hastings, M.H.; et al. Interacting Molecular Loops in the Mammalian Circadian Clock. Science 2000, 288, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Lahens, N.F.; Ballance, H.I.; Hughes, M.E.; Hogenesch, J.B. A circadian gene expression atlas in mammals: Im-plications for biology and medicine. Proc. Natl. Acad. Sci. USA 2014, 111, 16219–16224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.E.; Brewer, R.A.; Peliciari-Garcia, R.A.; Collins, H.E.; He, L.; Birky, T.L.; Peden, B.W.; Thompson, E.; Ammons, B.-J.; Bray, M.S.; et al. Cardiomyocyte-Specific BMAL1 Plays Critical Roles in Metabolism, Signaling, and Maintenance of Contractile Function of the Heart. J. Biol. Rhythm. 2014, 29, 257–276. [Google Scholar] [CrossRef]
- Ingle, K.A.; Kain, V.; Goel, M.; Prabhu, S.D.; Young, M.E.; Halade, G.V. Cardiomyocyte-specific Bmal1 deletion in mice triggers diastolic dysfunction, extracellular matrix response, and impaired resolution of inflammation. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1827–H1836. [Google Scholar] [CrossRef]
- Wang, Z. Regulation of Cell Cycle Progression by Growth Factor-Induced Cell Signaling. Cells 2021, 10, 3327. [Google Scholar] [CrossRef]
- Mitra, S. Does evening sun increase the risk of skin cancer? Proc. Natl. Acad. Sci. USA 2011, 108, 18857–18858. [Google Scholar] [CrossRef] [Green Version]
- Gaddameedhi, S.; Selby, C.P.; Kaufmann, W.K.; Smart, R.C.; Sancar, A. Control of skin cancer by the circadian rhythm. Proc. Natl. Acad. Sci. USA 2011, 108, 18790–18795. [Google Scholar] [CrossRef] [Green Version]
- Papagiannakopoulos, T.; Bauer, M.R.; Davidson, S.M.; Heimann, M.; Subbaraj, L.; Bhutkar, A.; Bartlebaugh, J.; Heiden, M.G.V.; Jacks, T. Circadian Rhythm Disruption Promotes Lung Tumorigenesis. Cell Metab. 2016, 24, 324–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manouchehri, E.; Taghipour, A.; Ghavami, V.; Ebadi, A.; Homaei, F.; Roudsari, R.L. Night-shift work duration and breast cancer risk: An updated systematic review and meta-analysis. BMC Womens Health 2021, 21, 89. [Google Scholar] [CrossRef] [PubMed]
- Yousef, E.; Mitwally, N.; Noufal, N.; Tahir, M.R. Shift work and risk of skin cancer: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 89. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.B.; Karp, N.A.; Maywood, E.S.; Sage, E.A.; Deery, M.; O’Neill, J.S.; Wong, G.K.; Chesham, J.; Odell, M.; Lilley, K.S.; et al. Circadian Orchestration of the Hepatic Proteome. Curr. Biol. 2006, 16, 1107–1115. [Google Scholar] [CrossRef] [Green Version]
- Sinturel, F.; Gerber, A.; Mauvoisin, D.; Wang, J.; Gatfield, D.; Stubblefield, J.J.; Green, C.B.; Gachon, F.; Schibler, U. Diurnal Oscillations in Liver Mass and Cell Size Accompany Ribosome Assembly Cycles. Cell 2017, 169, 651–663.e14. [Google Scholar] [CrossRef] [Green Version]
- Lipton, J.O.; Yuan, E.D.; Boyle, L.M.; Ebrahimi-Fakhari, D.; Kwiatkowski, E.; Nathan, A.; Güttler, T.; Davis, F.; Asara, J.M.; Sahin, M. The Circadian Protein BMAL1 Regulates Translation in Response to S6K1-Mediated Phosphorylation. Cell 2015, 161, 1138–1151. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.; Dang, F.; Li, P.; Wang, P.; Xu, Q.; Liu, Z.; Li, Y.; Wu, Y.; Chen, Y.; Liu, Y. The Circadian Protein Period2 Suppresses mTORC1 Activity via Recruiting Tsc1 to mTORC1 Complex. Cell Metab. 2018, 29, 653–667.e6. [Google Scholar] [CrossRef] [Green Version]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [Green Version]
- Vazquez de Aldana, C.R.; Wek, R.C.; Segundo, P.S.; Truesdell, A.G.; Hinnebusch, A.G. Multicopy tRNA genes functionally suppress mutations in yeast eIF-2 alpha kinase GCN2: Evidence for separate pathways coupling GCN4 expression to un-changed tRNA. Mol. Cell. Biol. 1994, 14, 7920–7932. [Google Scholar]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of Translation Initiation in Eukaryotes: Mechanisms and Biological Targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef] [Green Version]
- Karki, S.; Castillo, K.; Ding, Z.; Kerr, O.; Lamb, T.M.; Wu, C.; Sachs, M.S.; Bell-Pedersen, D. Circadian clock control of eIF2α phosphorylation is necessary for rhythmic translation initiation. Proc. Natl. Acad. Sci. USA 2020, 117, 10935–10945. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Jiang, X.; Bao, P.; Qin, M.; Xu, J. Circadian control of stress granules by oscillating EIF2α. Cell Death Dis. 2019, 10, 215. [Google Scholar] [CrossRef] [PubMed]
- Robles, M.; Humphrey, S.; Mann, M. Phosphorylation Is a Central Mechanism for Circadian Control of Metabolism and Physiology. Cell Metab. 2017, 25, 118–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamia, K.A.; Sachdeva, U.M.; DiTacchio, L.; Williams, E.C.; Alvarez, J.G.; Egan, D.F.; Vasquez, D.S.; Juguilon, H.; Panda, S.; Shaw, R.J.; et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science 2009, 326, 437–440. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Young, M.E.; Razeghi, P.; Taegtmeyer, H. Clock genes in the heart: Characterization and attenuation with hypertrophy. J. Mol. Cell. Cardiol. 2001, 33, A135. [Google Scholar] [CrossRef] [Green Version]
- Mia, S.; Kane, M.S.; Latimer, M.N.; Reitz, C.J.; Sonkar, R.; Benavides, G.A.; Smith, S.R.; Frank, S.J.; Martino, T.A.; Zhang, J. Young, Differential effects of REV-ERBalpha/beta agonism on cardiac gene expression, metabolism, and contractile function in a mouse model of circadian disruption. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H1487–H1508. [Google Scholar] [CrossRef]
- Sonkar, R.; Berry, R.; Latimer, M.N.; Prabhu, S.D.; Young, M.E.; Frank, S.J. Augmented Cardiac Growth Hormone Signaling Contributes to Cardiomyopathy Following Genetic Disruption of the Cardiomyocyte Circadian Clock. Front. Pharmacol. 2022, 13, 836725. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell Sci. 2013, 126 Pt 8, 1713–1719. [Google Scholar] [CrossRef] [Green Version]
- González-Terán, B.; López, J.A.; Rodríguez, E.; Leiva, L.; Martínez-Martínez, S.; Bernal, J.A.; Jiménez-Borreguero, L.J.; Redondo, J.M.; Vazquez, J.; Sabio, G. p38γ and δ promote heart hypertrophy by targeting the mTOR-inhibitory protein DEPTOR for degradation. Nat. Commun. 2016, 7, 10477. [Google Scholar] [CrossRef] [Green Version]
- De La Sierra, A.; Gorostidi, M.; Banegas, J.R.; Segura, J.; De La Cruz, J.J.; Ruilope, L.M. Nocturnal Hypertension or Nondipping: Which Is Better Associated With the Cardiovascular Risk Profile? Am. J. Hypertens. 2013, 27, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Verdecchia, P.; Schillaci, G.; Guerrieri, M.; Gatteschi, C.; Benemio, G.; Boldrini, F.; Porcellati, C. Circadian blood pressure changes and left ventricular hypertrophy in essential hypertension. Circulation 1990, 81, 528–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, T.; Floras, J. Sleep apnea and heart failure: Part I: Obstructive sleep apnea. Circulation 2003, 107, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Vyas, M.V.; Garg, A.X.; Iansavichus, A.V.; Costella, J.; Donner, A.; Laugsand, L.E.; Janszky, I.; Mrkobrada, M.; Parraga, G.; Hackam, D.G. Shift work and vascular events: Systematic review and meta-analysis. BMJ 2012, 345, e4800. [Google Scholar] [CrossRef] [Green Version]
- Knutsson, A.; Jonsson, B.; Akerstedt, T.; Orth-Gomer, K. Increased Risk of Ischaemic Heart Disease in Shift Workers. Lancet 1986, 328, 89–92. [Google Scholar] [CrossRef]
- Ekstrand, K.; Bostrom, P.A.; Arborelius, M.; Nilsson, J.A.; Lindell, S.E. Cardiovascular risk factors in commercial flight air-crew officers compared with those in the general population. Angiology 1996, 47, 1089–1094. [Google Scholar] [CrossRef]
- Hermida, R.C.; Ayala, D.E.; Mojon, A.; Fernandez, J.R. Influence of circadian time of hypertension treatment on cardio-vascular risk: Results of the MAPEC study. Chronobiol. Int. 2010, 27, 1629–1651. [Google Scholar] [CrossRef]
- Martino, T.A.; Tata, N.; Simpson, J.A.; Vanderlaan, R.; Dawood, F.; Kabir, M.G.; Khaper, N.; Cifelli, C.; Podobed, P.; Liu, P.P. The primary benefits of angiotensin-converting enzyme inhibition on cardiac re-modeling occur during sleep time in murine pressure overload hypertrophy. J. Am. Coll. Cardiol. 2011, 57, 2020–2028. [Google Scholar] [CrossRef]
- Betts, T.A.; Alford, C. Beta-blockers and sleep: A controlled trial. Eur. J. Clin. Pharmacol. 1985, 28, 65–68. [Google Scholar] [CrossRef]
- Sachan, N.; Dey, A.; Rotter, D.; Grinsfelder, D.B.; Battiprolu, P.K.; Sikder, D.; Copeland, V.; Oh, M.; Bush, E.; Shelton, J.M.; et al. Sustained Hemodynamic Stress Disrupts Normal Circadian Rhythms in Calcineurin-Dependent Signaling and Protein Phosphorylation in the Heart. Circ. Res. 2011, 108, 437–445. [Google Scholar] [CrossRef]
- St-Onge, M.P.; Ard, J.; Baskin, M.L.; Chiuve, S.E.; Johnson, H.M.; Kris-Etherton, P.; Varady, K.; American Heart Association Obesity Committee of the Council on Lifestyle and Cardiometabolic Health; Council on Cardiovascular Disease in the Young; Council on Clinical Cardiology; et al. Meal Timing and Frequency: Implications for Cardiovascular Disease Prevention: A Scientific Statement From the American Heart Association. Circulation 2017, 135, e96–e121. [Google Scholar] [CrossRef] [PubMed]
- Mia, S.; Sonkar, R.; Williams, L.; Latimer, M.N.; Robillard, I.F.; Diwan, A.; Frank, S.J.; Rosiers, C.D.; Young, M.E. Impact of obesity on day-night differences in cardiac metabolism. FASEB J. 2021, 35, e21298. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Dyar, K.A.; Treebak, J.T.; Jepsen, S.L.; Ehrlich, A.M.; Ashcroft, S.P.; Trost, K.; Kunzke, T.; Prade, V.M.; Small, L.; et al. Sassone-Corsi, Atlas of exercise metabolism reveals time-dependent signatures of metabolic homeostasis. Cell Metab. 2022, 34, 329–345.e8. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Latimer, M.N.; Young, M.E. Circadian Governance of Cardiac Growth. Cells 2022, 11, 1494. https://doi.org/10.3390/cells11091494
Latimer MN, Young ME. Circadian Governance of Cardiac Growth. Cells. 2022; 11(9):1494. https://doi.org/10.3390/cells11091494
Chicago/Turabian StyleLatimer, Mary N., and Martin E. Young. 2022. "Circadian Governance of Cardiac Growth" Cells 11, no. 9: 1494. https://doi.org/10.3390/cells11091494
APA StyleLatimer, M. N., & Young, M. E. (2022). Circadian Governance of Cardiac Growth. Cells, 11(9), 1494. https://doi.org/10.3390/cells11091494