
Lipocalin 2 Deficiency Alters Prostaglandin Biosynthesis and mTOR Signaling Regulation of Thermogenesis and Lipid Metabolism in Adipocytes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cell Culture and Differentiation of Primary Stromal-Vascular Cells
2.3. Measurement of Prostaglandins
2.4. Quantitative Real-Time PCR
2.5. Western Blot Analysis
2.6. Glycerol Assay
2.7. Statistical Analysis
3. Results
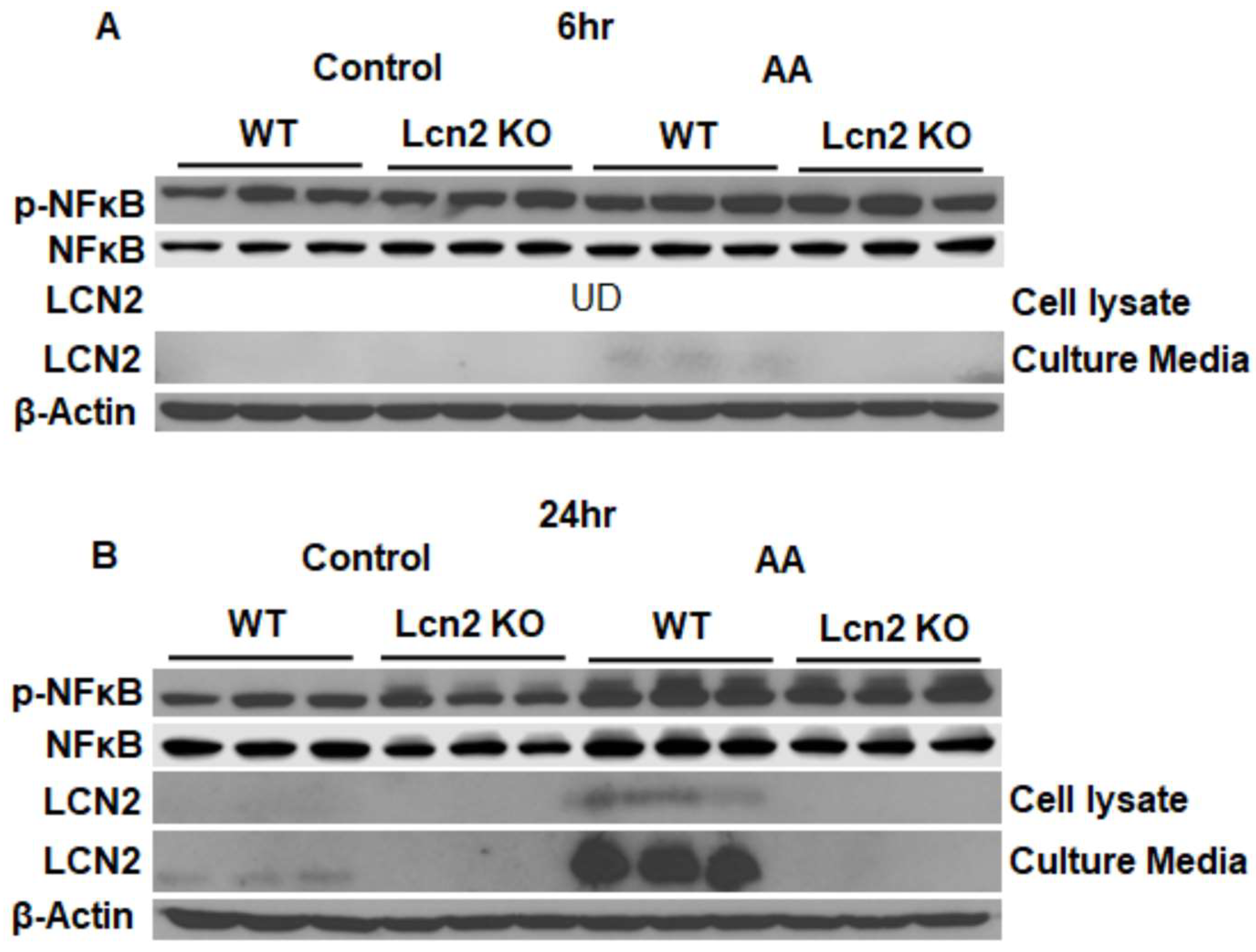
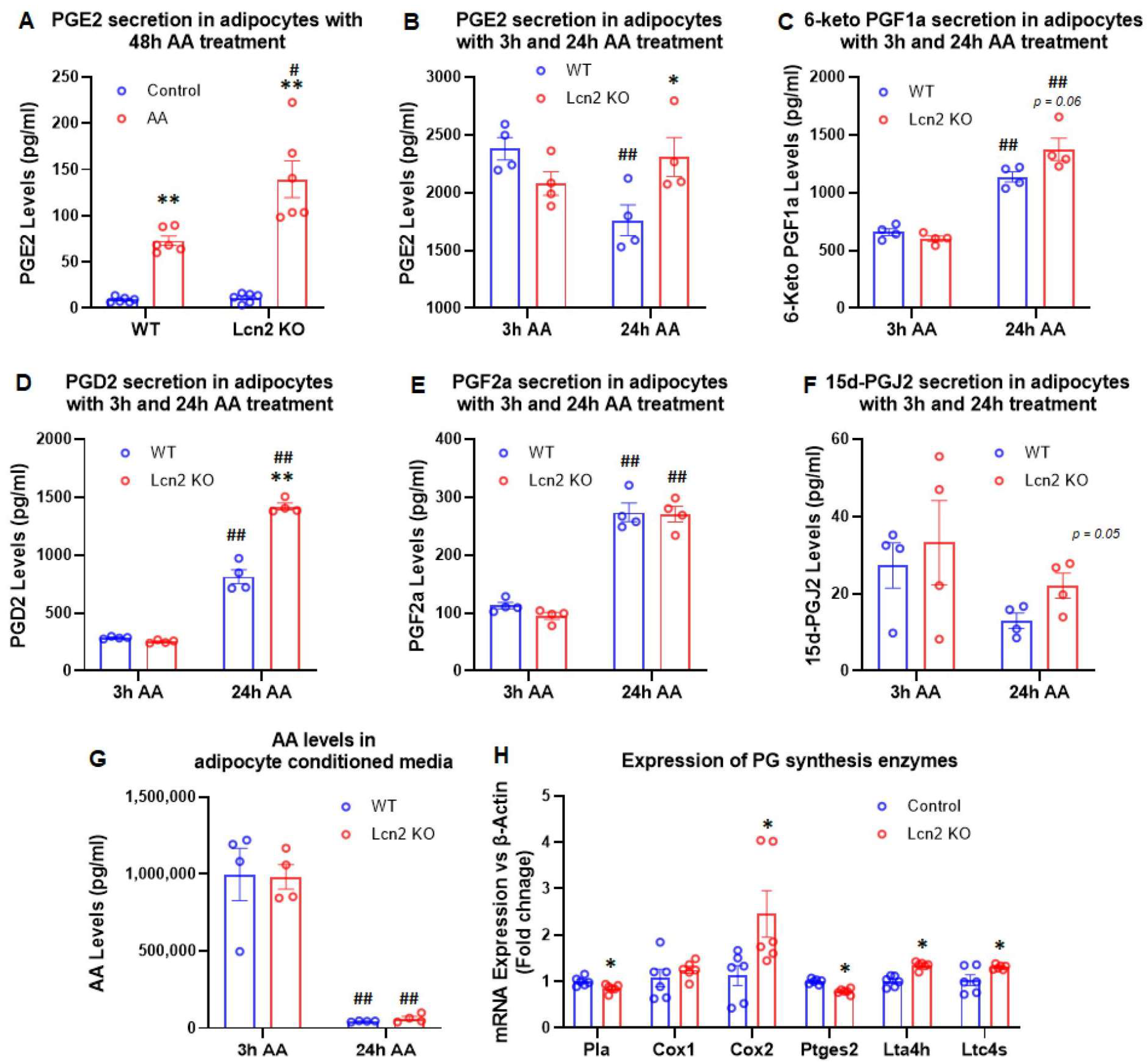
3.1. Lcn2 Regulates AA-Induced Prostaglandin Biosynthesis and Secretion in Beige Adipocytes
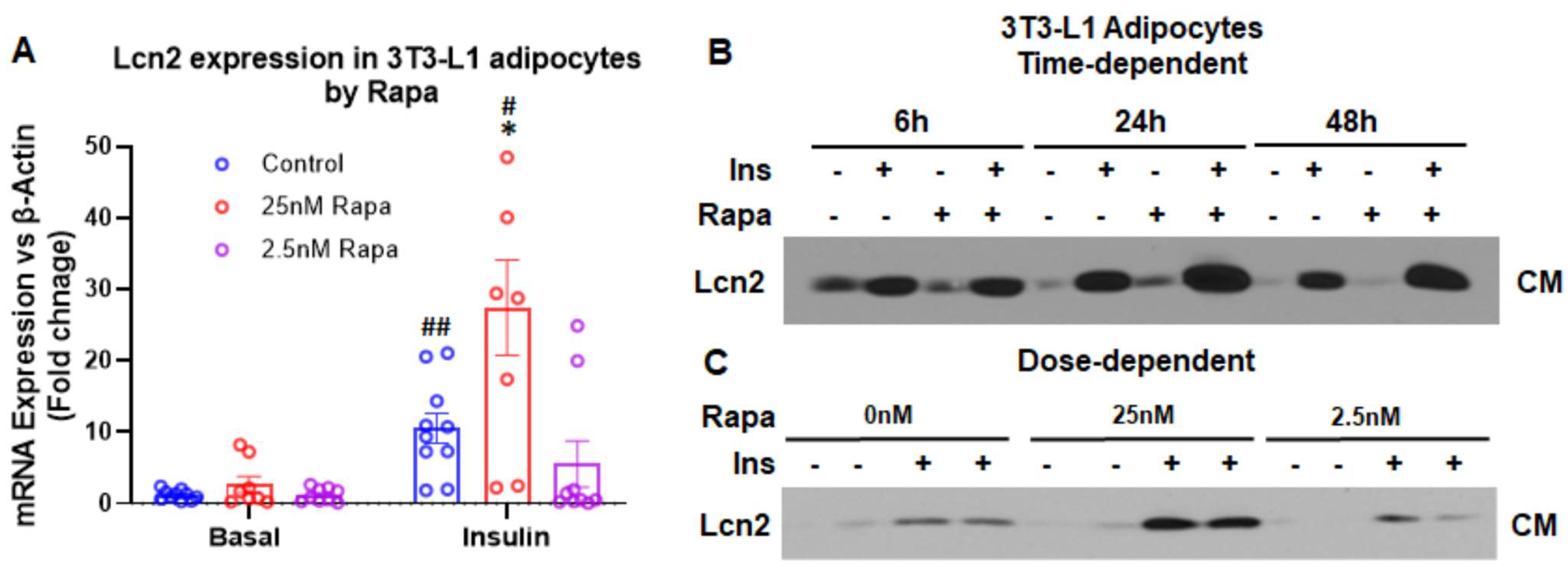
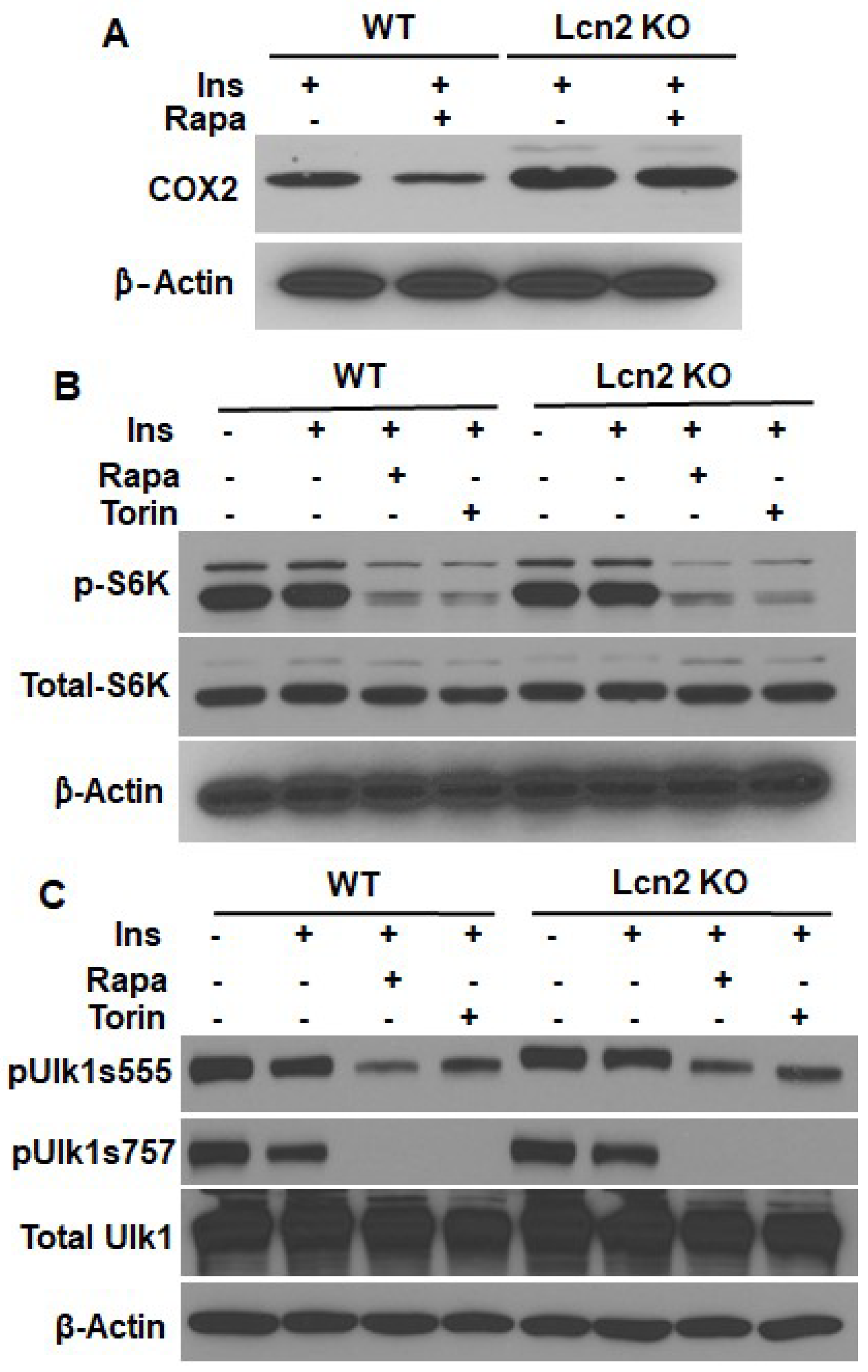
3.2. Lcn2 Mediates the Regulation of mTOR Signaling in Prostaglandin Biosynthesis
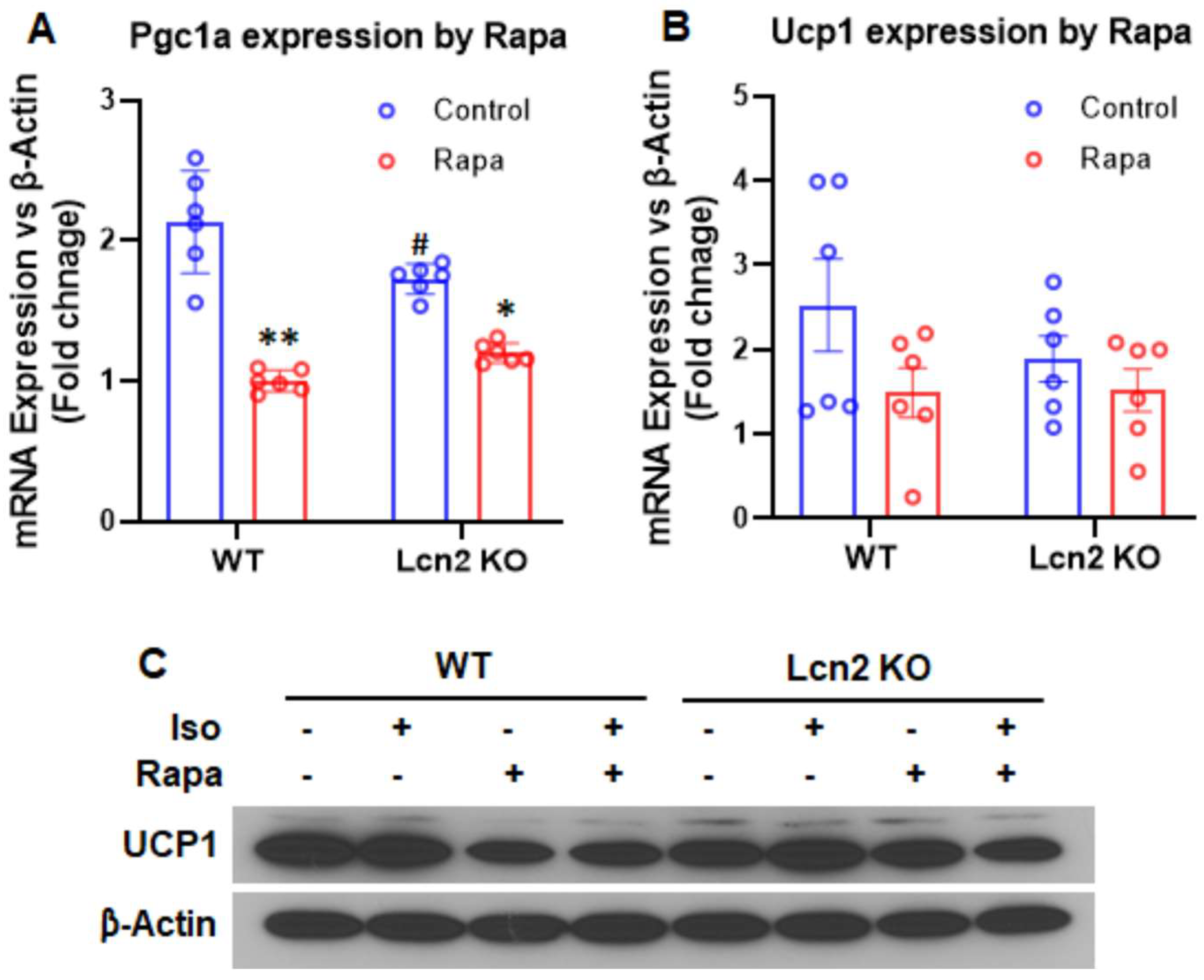
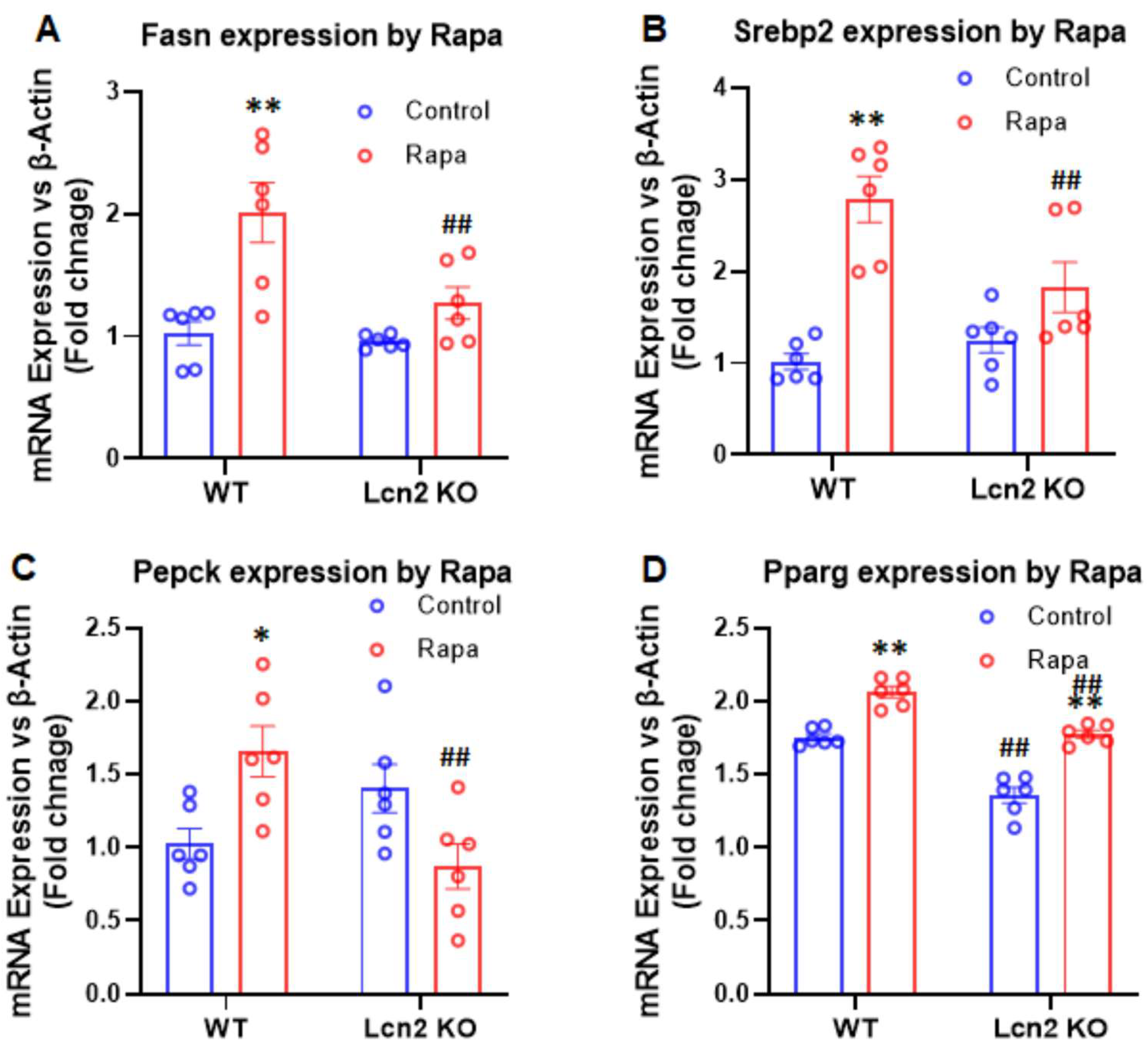
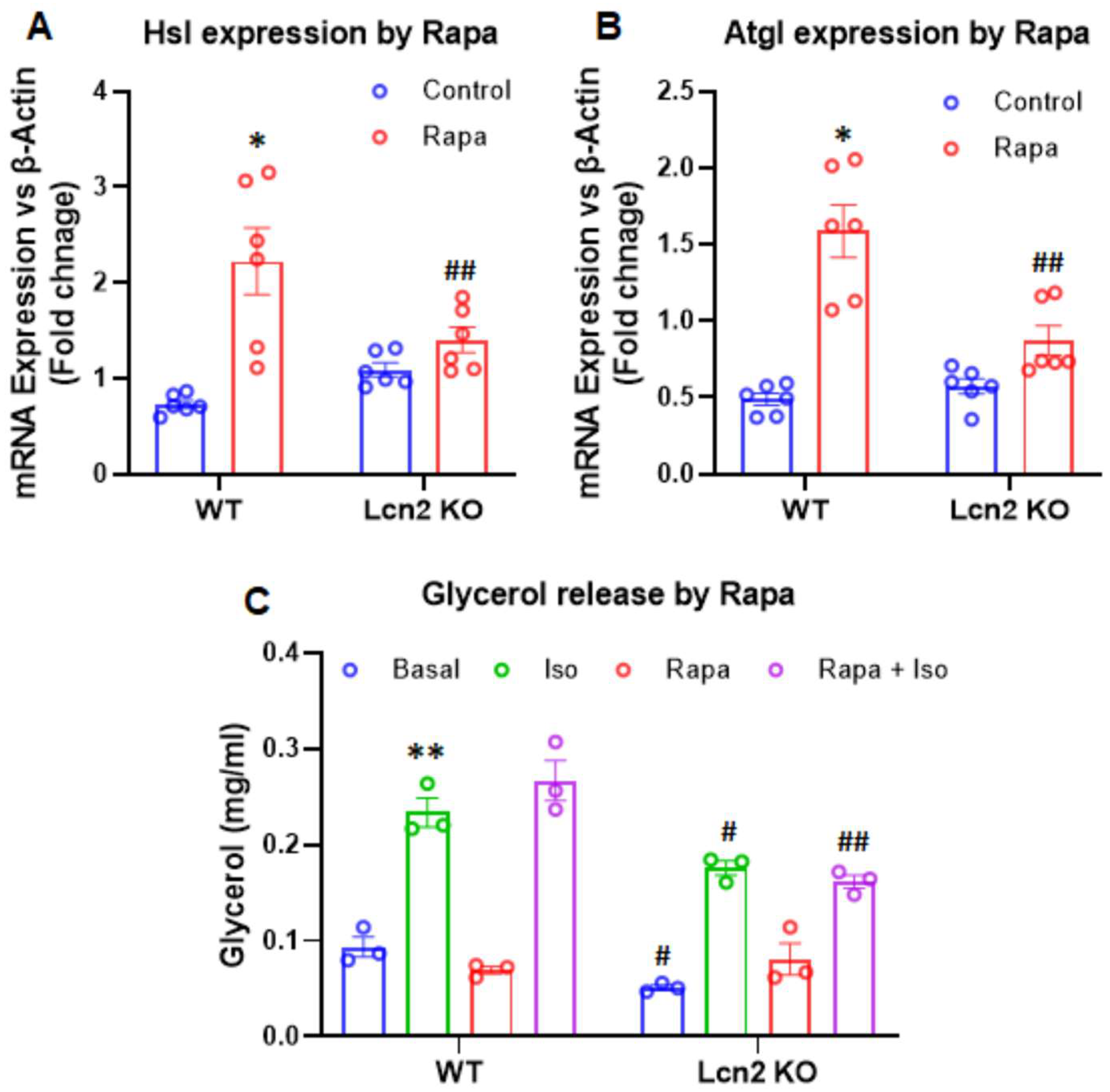
3.3. Lcn2 Deficiency Attenuates the Effect of mTORC1 Inhibition on Thermogenesis, Lipogenesis, and Lipolysis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kjeldsen, L.; Johnsen, A.H.; Sengelov, H.; Borregaard, N. Isolation and Primary Structure of NGAL, a Novel Protein Associated with Human Neutrophil Gelatinase. J. Biol. Chem. 1993, 268, 10425–10432. [Google Scholar] [CrossRef]
- Flower, D.R. Beyond the Superfamily: The Lipocalin Receptors. Biochim. Biophys. Acta-Protein Struct. Mol. Enzymol. 2000, 1482, 327–336. [Google Scholar] [CrossRef]
- Flower, D.R. The Lipocalin Protein Family: Structure and Function. Biochem. J. 1996, 318, 1–14. [Google Scholar] [CrossRef]
- Berger, T.; Togawa, A.; Duncan, G.S.; Elia, A.J.; You-Ten, A.; Wakeham, A.; Fong, H.E.H.; Cheung, C.C.; Mak, T.W. Lipocalin 2-Deficient Mice Exhibit Increased Sensitivity to Escherichia Coli Infection but Not to Ischemia-Reperfusion Injury. Proc. Natl. Acad. Sci. USA 2006, 103, 1834–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flo, T.H.; Smith, K.D.; Sato, S.; Rodriguez, D.J.; Holmes, M.A.; Strong, R.K.; Akira, S.; Aderem, A. Lipocalin 2 Mediates an Innate Immune Response to Bacterial Infection by Sequestrating Iron. Nature 2004, 432, 917–921. [Google Scholar] [CrossRef]
- Cowland, J.B.; Borregaard, N. Molecular Characterization and Pattern of Tissue Expression of the Gene for Neutrophil Gelatinase-Associated Lipocalin from Humans. Genomics 1997, 45, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Hu, Z.; Goswami, J.; Gaffen, S.L. Identification of Common Transcriptional Regulatory Elements in Interleukin-17 Target Genes. J. Biol. Chem. 2006, 281, 24138–24148. [Google Scholar] [CrossRef] [Green Version]
- Garay-Rojas, E.; Harper, M.; Hraba-Renevey, S.; Kress, M. An Apparent Autocrine Mechanism Amplifies the Dexamethasone- and Retinoic Acid-Induced Expression of Mouse Lipocalin-Encoding Gene 24p3. Gene 1996, 170, 173–180. [Google Scholar] [CrossRef]
- Seth, P.; Porter, D.; Lahti-Domenici, J.; Geng, Y.; Richardson, A.; Polyak, K. Cellular and Molecular Targets of Estrogen in Normal Human Breast Tissue. Cancer Res. 2002, 62, 4540–4544. [Google Scholar]
- Zhang, Y.; Foncea, R.; Deis, J.A.; Guo, H.; Bernlohr, D.A.; Chen, X. Lipocalin 2 Expression and Secretion Is Highly Regulated by Metabolic Stress, Cytokines, and Nutrients in Adipocytes. PLoS ONE 2014, 9, e96997. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Borregaard, N.; Kjeldsen, L.; Moses, M.A. The High Molecular Weight Urinary Matrix Metalloproteinase (MMP) Activity Is a Complex of Gelatinase B/MMP-9 and Neutrophil Gelatinase-Associated Lipocalin (NGAL): Modulation of MMP-9 Activity by NGAL. J. Biol. Chem. 2001, 276, 37258–37265. [Google Scholar] [CrossRef] [Green Version]
- Meheus, L.A.; Fransen, L.M.; Raymackers, J.G.; Blockx, H.A.; Van Beeumen, J.J.; Van Bun, S.M.; Van de Voorde, A. Identification by Microsequencing of Lipopolysaccharide-Induced Proteins Secreted by Mouse Macrophages. J. Immunol. 1993, 151, 1535–1547. [Google Scholar] [PubMed]
- Goetz, D.H.; Holmes, M.A.; Borregaard, N.; Bluhm, M.E.; Raymond, K.N.; Strong, R.K. The Neutrophil Lipocalin NGAL Is a Bacteriostatic Agent That Interferes with Siderophore-Mediated Iron Acquisition. Mol. Cell 2002, 10, 1033–1043. [Google Scholar] [CrossRef]
- Qiu, X.; Macchietto, M.G.; Liu, X.; Lu, Y.; Ma, Y.; Guo, H.; Saqui-Salces, M.; Bernlohr, D.A.; Chen, C.; Shen, S.; et al. Identification of Gut Microbiota and Microbial Metabolites Regulated by an Antimicrobial Peptide Lipocalin 2 in High Fat Diet-Induced Obesity. Int. J. Obes. 2021, 45, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Jin, D.; Zhang, Y.; Wright, W.; Bazuine, M.; Brockman, D.A.; Bernlohr, D.A.; Chen, X. Lipocalin-2 Deficiency Impairs Thermogenesis and Potentiates Diet-Induced Insulin Resistance in Mice. Diabetes 2010, 59, 1376–1385. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Guo, H.; Deis, J.A.; Mashek, M.G.; Zhao, M.; Ariyakumar, D.; Armien, A.G.; Bernlohr, D.A.; Mashek, D.G.; Chen, X. Lipocalin 2 Regulates Brown Fat Activation via a Nonadrenergic Activation Mechanism. J. Biol. Chem. 2014, 289, 22063–22077. [Google Scholar] [CrossRef] [Green Version]
- Deis, J.A.; Guo, H.; Wu, Y.; Liu, C.; Bernlohr, D.A.; Chen, X. Adipose Lipocalin 2 Overexpression Protects against Age-Related Decline in Thermogenic Function of Adipose Tissue and Metabolic Deterioration. Mol. Metab. 2019, 24, 18–29. [Google Scholar] [CrossRef]
- Deis, J.A.; Guo, H.; Wu, Y.; Liu, C.; Bernlohr, D.A.; Chen, X. Lipocalin 2 Regulates Retinoic Acid-Induced Activation of Beige Adipocytes. J. Mol. Endocrinol. 2018, 61, 115–126. [Google Scholar] [CrossRef]
- Alhouayek, M.; Muccioli, G.G. COX-2-Derived Endocannabinoid Metabolites as Novel Inflammatory Mediators. Trends Pharmacol. Sci. 2014, 35, 284–292. [Google Scholar] [CrossRef]
- Kawahara, K.; Hohjoh, H.; Inazumi, T.; Tsuchiya, S.; Sugimoto, Y. Prostaglandin E2-Induced Inflammation: Relevance of Prostaglandin e Receptors. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2015, 1851, 414–421. [Google Scholar] [CrossRef]
- García-Alonso, V.; Titos, E.; Alcaraz-Quiles, J.; Rius, B.; Lopategi, A.; López-Vicario, C.; Jakobsson, P.J.; Delgado, S.; Lozano, J.; Clària, J. Prostaglandin E2 Exerts Multiple Regulatory Actions on Human Obese Adipose Tissue Remodeling, Inflammation, Adaptive Thermogenesis and Lipolysis. PLoS ONE 2016, 11, e0153751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, E.K.; Hellberg, K.; Foncea, R.; Hertzel, A.V.; Suttles, J.; Bernlohr, D.A. Fatty Acids Induce Leukotriene C4 Synthesis in Macrophages in a Fatty Acid Binding Protein-Dependent Manner. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2013, 1831, 1199–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuehn, H.S.; Jung, M.Y.; Beaven, M.A.; Metcalfe, D.D.; Gilfillan, A.M. Prostaglandin E2 Activates and Utilizes MTORC2 as a Central Signaling Locus for the Regulation of Mast Cell Chemotaxis and Mediator Release. J. Biol. Chem. 2011, 286, 391–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, A.H.; Young, S.H.; Sinnett-Smith, J.; Ne Chou, C.E.; Moro, A.; Hertzer, K.M.; Hines, O.J.; Rozengurt, E.; Eibl, G. Prostaglandin E2 Activates the MTORC1 Pathway through an EP4/CAMP/PKA-and EP1/CA2+-Mediated Mechanism in the Human Pancreatic Carcinoma Cell Line PANC-1. Am. J. Physiol.-Cell Physiol. 2015, 309, C639–C649. [Google Scholar] [CrossRef] [Green Version]
- Labbé, S.M.; Mouchiroud, M.; Caron, A.; Secco, B.; Freinkman, E.; Lamoureux, G.; Gélinas, Y.; Lecomte, R.; Bossé, Y.; Chimin, P.; et al. MTORC1 Is Required for Brown Adipose Tissue Recruitment and Metabolic Adaptation to Cold. Sci. Rep. 2016, 6, 37223. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Bordicchia, M.; Zhang, C.; Fang, H.; Wei, W.; Li, J.L.; Guilherme, A.; Guntur, K.; Czech, M.P.; Collins, S. Activation of MTORC1 Is Essential for β-Adrenergic Stimulation of Adipose Browning. J. Clin. Investig. 2016, 126, 1704–1716. [Google Scholar] [CrossRef] [Green Version]
- Tran, C.M.; Mukherjee, S.; Ye, L.; Frederick, D.W.; Kissig, M.; Davis, J.G.; Lamming, D.W.; Seale, P.; Baur, J.A. Rapamycin Blocks Induction of the Thermogenic Program in White Adipose Tissue. Diabetes 2016, 65, 927–941. [Google Scholar] [CrossRef] [Green Version]
- Wada, S.; Neinast, M.; Jang, C.; Ibrahim, Y.H.; Lee, G.; Babu, A.; Li, J.; Hoshino, A.; Rowe, G.C.; Rhee, J.; et al. The Tumor Suppressor FLCN Mediates an Alternate MTOR Pathway to Regulate Browning of Adipose Tissue. Genes Dev. 2016, 30, 2551–2564. [Google Scholar] [CrossRef] [Green Version]
- Bayindir, I.; Babaeikelishomi, R.; Kocanova, S.; Sousa, I.S.; Lerch, S.; Hardt, O.; Wild, S.; Bosio, A.; Bystricky, K.; Herzig, S.; et al. Transcriptional Pathways in CPGI2-Induced Adipocyte Progenitor Activation for Browning. Front. Endocrinol. 2015, 6, 129. [Google Scholar] [CrossRef] [Green Version]
- Vegiopoulos, A.; Müller-Decker, K.; Strzoda, D.; Schmitt, I.; Chichelnitskiy, E.; Ostertag, A.; Diaz, M.B.; Rozman, J.; De Angelis, M.H.; Nüsing, R.M.; et al. Cyclooxygenase-2 Controls Energy Homeostasis in Mice by de Novo Recruitment of Brown Adipocytes. Science 2010, 328, 1158–1161. [Google Scholar] [CrossRef]
- Zhang, X.; Luo, Y.; Wang, C.; Ding, X.; Yang, X.; Wu, D.; Silva, F.; Yang, Z.; Zhou, Q.; Wang, L.; et al. Adipose MTORC1 Suppresses Prostaglandin Signaling and Beige Adipogenesis via the CRTC2-COX-2 Pathway. Cell Rep. 2018, 24, 3180–3193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuelsson, B. Arachidonic Acid Metabolism: Role in Inflammation. Z. Rheumatol. 1991, 50, 3–6. [Google Scholar] [PubMed]
- Chen, S.; Mei, X.; Yin, A.; Yin, H.; Cui, X.B.; Chen, S.Y. Response Gene to Complement 32 Suppresses Adipose Tissue Thermogenic Genes through Inhibiting B3-Adrenergic Receptor/MTORC1 Signaling. FASEB J. 2018, 32, 4836–4847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.H.; Kim, S.N.; Kwon, H.J.; Granneman, J.G. Metabolic Heterogeneity of Activated Beige/Brite Adipocytes in Inguinal Adipose Tissue. Sci. Rep. 2017, 7, 39794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mottillo, E.P.; Balasubramanian, P.; Lee, Y.H.; Weng, C.; Kershaw, E.E.; Granneman, J.G. Coupling of Lipolysis and de Novo Lipogenesis in Brown, Beige, and White Adipose Tissues during Chronic Β3-Adrenergic Receptor Activation. J. Lipid Res. 2014, 55, 2276–2286. [Google Scholar] [CrossRef] [Green Version]
- Guilherme, A.; Yenilmez, B.; Bedard, A.H.; Henriques, F.; Liu, D.; Lee, A.; Goldstein, L.; Kelly, M.; Nicoloro, S.M.; Chen, M.; et al. Control of Adipocyte Thermogenesis and Lipogenesis through Β3-Adrenergic and Thyroid Hormone Signal Integration. Cell Rep. 2020, 31, 107598. [Google Scholar] [CrossRef]
- Paolella, L.M.; Mukherjee, S.; Tran, C.M.; Bellaver, B.; Hugo, M.; Luongo, T.S.; Shewale, S.V.; Lu, W.; Chellappa, K.; Baur, J.A. MTORC1 Restrains Adipocyte Lipolysis to Prevent Systemic Hyperlipidemia. Mol. Metab. 2020, 32, 136–147. [Google Scholar] [CrossRef]
- Soliman, G.A.; Acosta-Jaquez, H.A.; Fingar, D.C. MTORC1 Inhibition via Rapamycin Promotes Triacylglycerol Lipolysis and Release of Free Fatty Acids in 3T3-L1 Adipocytes. Lipids 2010, 45, 1089–1100. [Google Scholar] [CrossRef]
- Ricoult, S.J.H.; Manning, B.D. The Multifaceted Role of MTORC1 in the Control of Lipid Metabolism. EMBO Rep. 2013, 14, 242–251. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deis, J.; Lin, T.-Y.; Bushman, T.; Chen, X. Lipocalin 2 Deficiency Alters Prostaglandin Biosynthesis and mTOR Signaling Regulation of Thermogenesis and Lipid Metabolism in Adipocytes. Cells 2022, 11, 1535. https://doi.org/10.3390/cells11091535
Deis J, Lin T-Y, Bushman T, Chen X. Lipocalin 2 Deficiency Alters Prostaglandin Biosynthesis and mTOR Signaling Regulation of Thermogenesis and Lipid Metabolism in Adipocytes. Cells. 2022; 11(9):1535. https://doi.org/10.3390/cells11091535
Chicago/Turabian StyleDeis, Jessica, Te-Yueh Lin, Theresa Bushman, and Xiaoli Chen. 2022. "Lipocalin 2 Deficiency Alters Prostaglandin Biosynthesis and mTOR Signaling Regulation of Thermogenesis and Lipid Metabolism in Adipocytes" Cells 11, no. 9: 1535. https://doi.org/10.3390/cells11091535
APA StyleDeis, J., Lin, T. -Y., Bushman, T., & Chen, X. (2022). Lipocalin 2 Deficiency Alters Prostaglandin Biosynthesis and mTOR Signaling Regulation of Thermogenesis and Lipid Metabolism in Adipocytes. Cells, 11(9), 1535. https://doi.org/10.3390/cells11091535