
Mechanistic and Therapeutic Insights into Ataxic Disorders with Pentanucleotide Expansions


Abstract
:1. Introduction
2. SCA10
2.1. Patient Demographics, Clinical Presentation, and Neuropathology
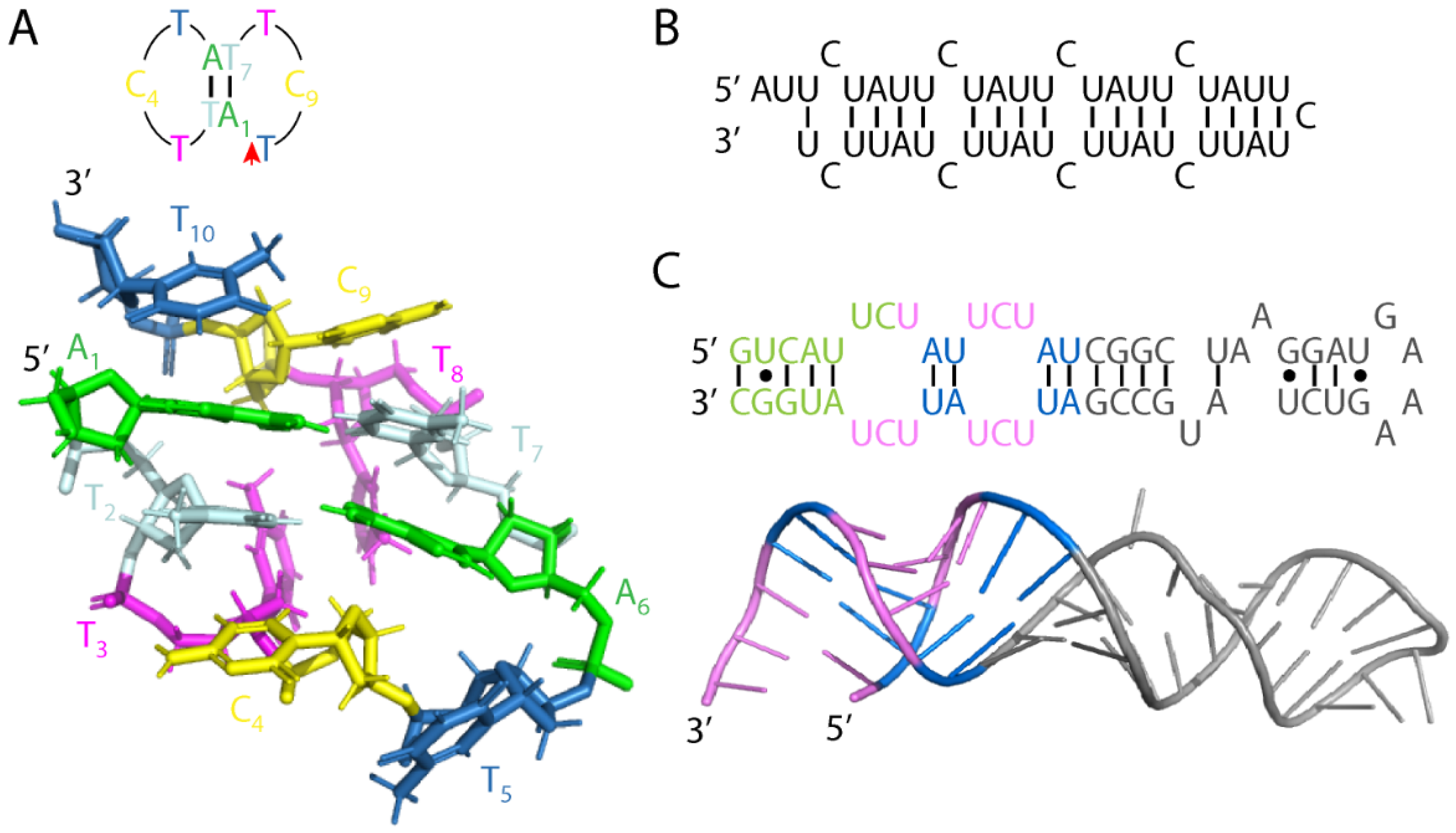
2.2. SCA10 Repeat Origin and Structures
2.3. RNA Gain-of-Function in SCA10 Cells and Animal Models
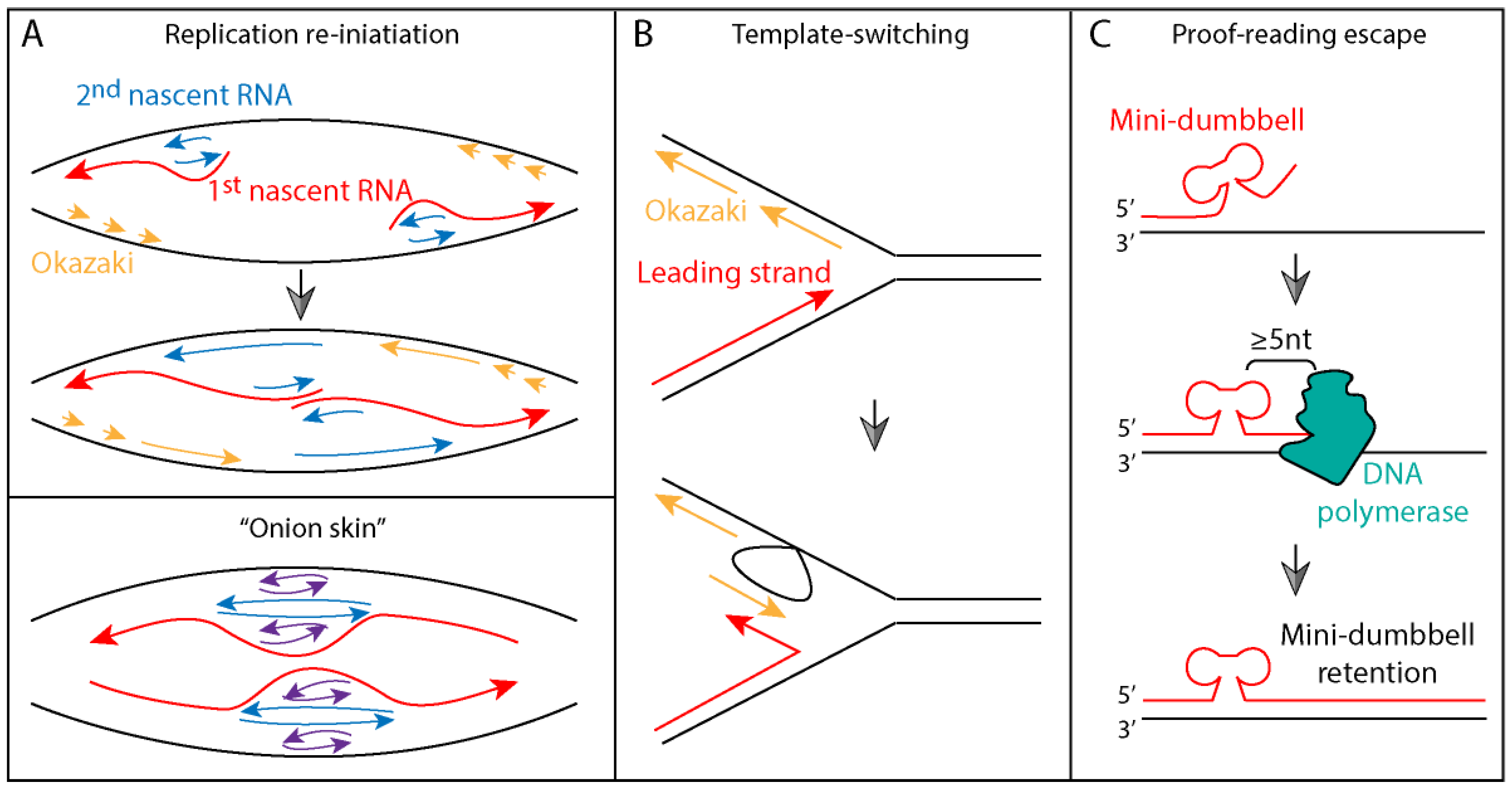
2.4. Proposed Models of SCA10 Repeat Expansion
2.5. Theranostic Development for SCA10
3. SCA31
3.1. Patient Demographics and Neuropathological Features
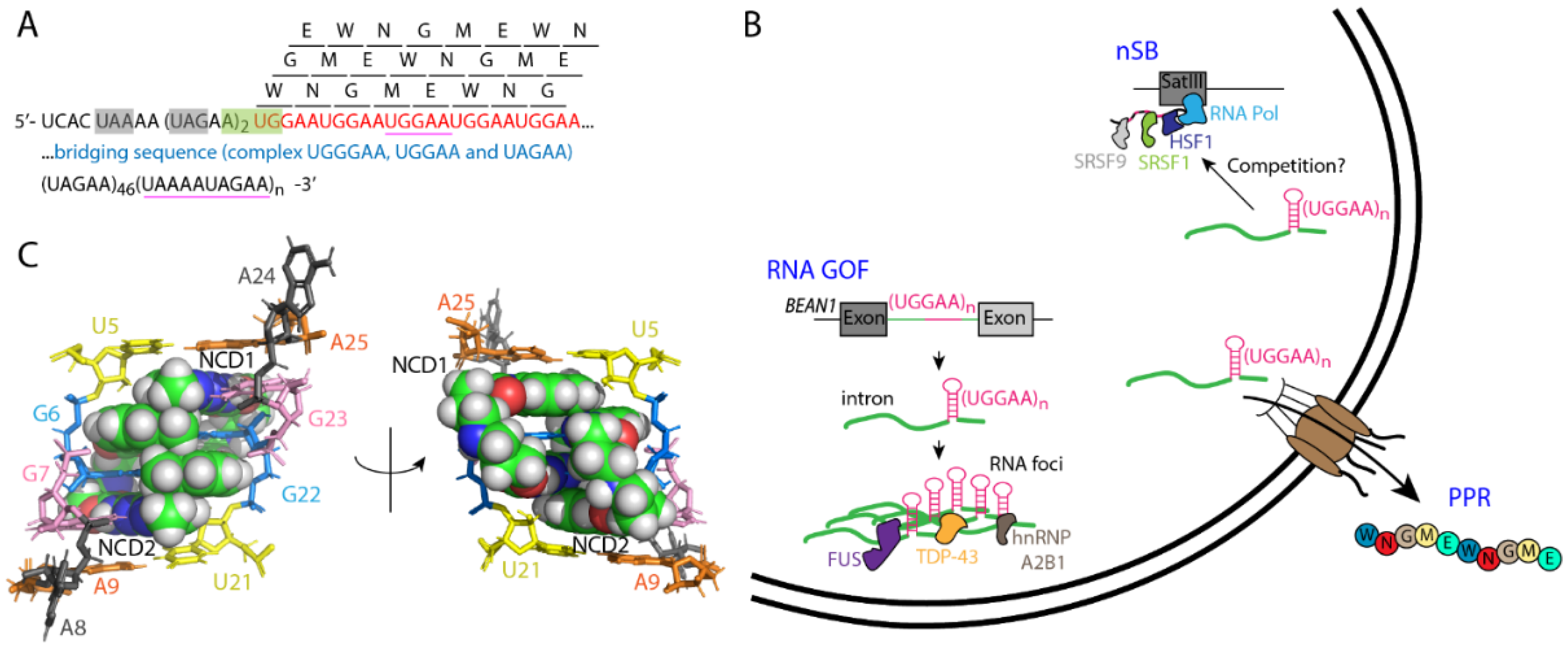
3.2. RNA Gain-of-Function and Pentapeptide Repeat Protein in SCA31
3.3. Interplay between SCA31 RNA Expansion and RNA Binding Proteins
3.4. Small Molecule Targeting UGGAA in SCA31
4. SCA37
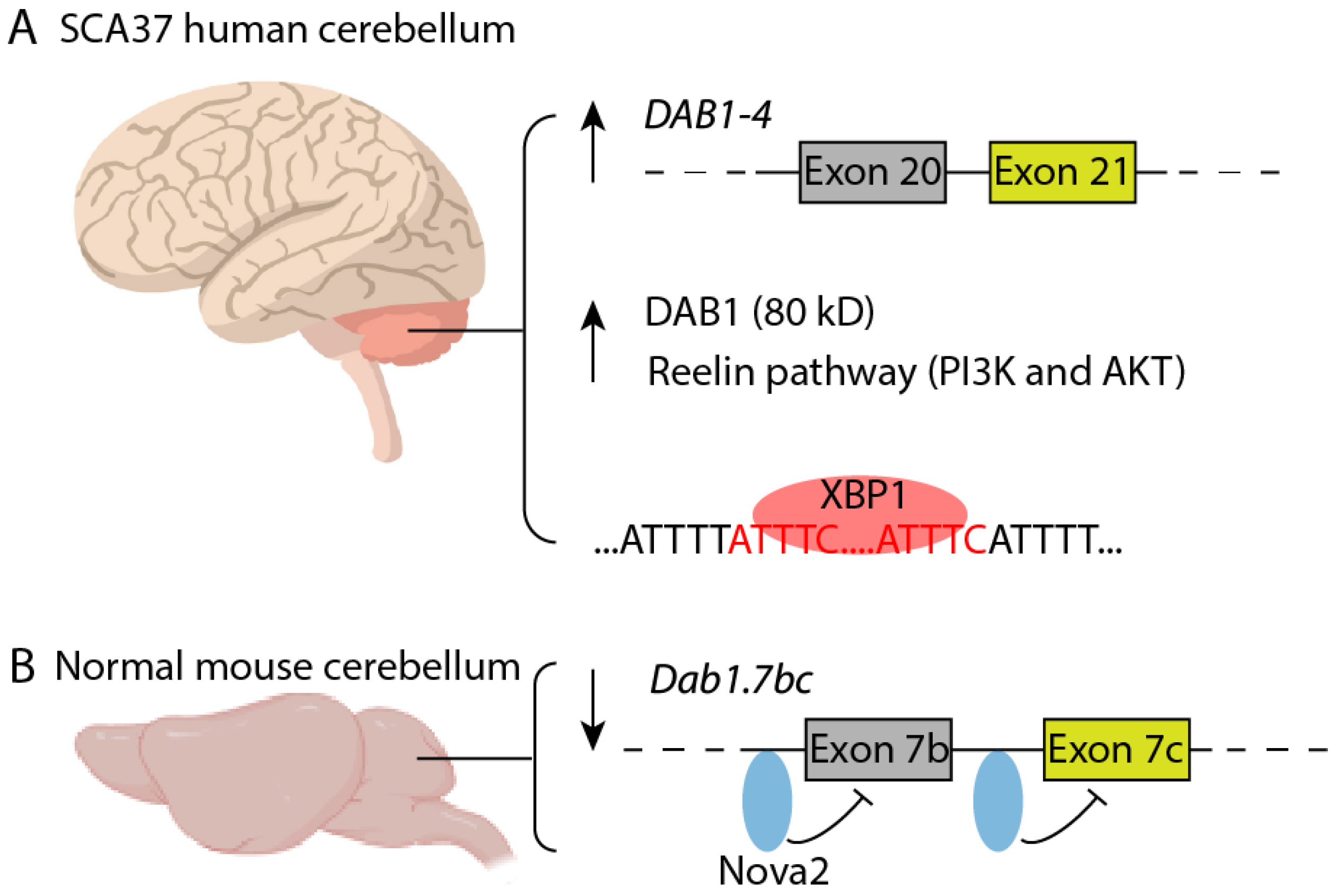
4.1. Clinical Presentation and Neuropathology
4.2. RNA Toxicity and Dysregulation of DAB1 Expression in SCA37
5. CANVAS
5.1. Clinical Presentation and Neuropathology
5.2. Repeat Polymorphism and Intron Retention for CANVAS
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fondon, J.W., 3rd; Hammock, E.A.; Hannan, A.J.; King, D.G. Simple sequence repeats: Genetic modulators of brain function and behavior. Trends Neurosci. 2008, 31, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Jansen, A.; Gemayel, R.; Verstrepen, K.J. Unstable microsatellite repeats facilitate rapid evolution of coding and regulatory sequences. Genome Dyn. 2012, 7, 108–125. [Google Scholar] [CrossRef] [PubMed]
- Chintalaphani, S.R.; Pineda, S.S.; Deveson, I.W.; Kumar, K.R. An update on the neurological short tandem repeat expansion disorders and the emergence of long-read sequencing diagnostics. Acta Neuropathol. Commun. 2021, 9, 98. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Ashizawa, T. RNA toxicity and foci formation in microsatellite expansion diseases. Curr. Opin. Genet. Dev. 2017, 44, 17–29. [Google Scholar] [CrossRef]
- Peskett, T.R.; Rau, F.; O’Driscoll, J.; Patani, R.; Lowe, A.R.; Saibil, H.R. A Liquid to Solid Phase Transition Underlying Pathological Huntingtin Exon1 Aggregation. Mol. Cell. 2018, 70, 588–601.e6. [Google Scholar] [CrossRef]
- Rodriguez, C.M.; Todd, P.K. New pathologic mechanisms in nucleotide repeat expansion disorders. Neurobiol. Dis. 2019, 130, 104515. [Google Scholar] [CrossRef]
- Chakraborty, A.; Tapryal, N.; Venkova, T.; Mitra, J.; Vasquez, V.; Sarker, A.H.; Duarte-Silva, S.; Huai, W.; Ashizawa, T.; Ghosh, G.; et al. Deficiency in classical nonhomologous end-joining-mediated repair of transcribed genes is linked to SCA3 pathogenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 8154–8165. [Google Scholar] [CrossRef]
- Gao, R.; Chakraborty, A.; Geater, C.; Pradhan, S.; Gordon, K.L.; Snowden, J.; Yuan, S.; Dickey, A.S.; Choudhary, S.; Ashizawa, T.; et al. Mutant huntingtin impairs PNKP and ATXN3, disrupting DNA repair and transcription. Elife 2019, 8, e42988. [Google Scholar] [CrossRef]
- Gerber, H.P.; Seipel, K.; Georgiev, O.; Hofferer, M.; Hug, M.; Rusconi, S.; Schaffner, W. Transcriptional activation modulated by homopolymeric glutamine and proline stretches. Science 1994, 263, 808–811. [Google Scholar] [CrossRef]
- Massey, T.H.; Jones, L. The central role of DNA damage and repair in CAG repeat diseases. Dis. Model. Mech. 2018, 11, dmm031930. [Google Scholar] [CrossRef] [Green Version]
- Cleary, J.D.; Pattamatta, A.; Ranum, L.P.W. Repeat-associated non-ATG (RAN) translation. J. Biol. Chem. 2018, 293, 16127–16141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, K.S.; Cooper, T.A. The roles of RNA processing in translating genotype to phenotype. Nat. Rev. Mol. Cell. Biol. 2017, 18, 102–114. [Google Scholar] [CrossRef]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.; et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouwer, J.R.; Willemsen, R.; Oostra, B.A. Microsatellite repeat instability and neurological disease. Bioessays 2009, 31, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Vale, R.D. RNA phase transitions in repeat expansion disorders. Nature 2017, 546, 243–247. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Ashizawa, T. SCA10 and ATTCT repeat expansion: Clinical features and molecular aspects. Cytogenet. Genome Res. 2003, 100, 184–188. [Google Scholar] [CrossRef]
- Matsuura, T.; Achari, M.; Khajavi, M.; Bachinski, L.L.; Zoghbi, H.Y.; Ashizawa, T. Mapping of the gene for a novel spinocerebellar ataxia with pure cerebellar signs and epilepsy. Ann. Neurol. 1999, 45, 407–411. [Google Scholar] [CrossRef]
- Matsuura, T.; Yamagata, T.; Burgess, D.L.; Rasmussen, A.; Grewal, R.P.; Watase, K.; Khajavi, M.; McCall, A.E.; Davis, C.F.; Zu, L.; et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat. Genet. 2000, 26, 191–194. [Google Scholar] [CrossRef]
- Zu, L.; Figueroa, K.P.; Grewal, R.; Pulst, S.M. Mapping of a new autosomal dominant spinocerebellar ataxia to chromosome 22. Am. J. Hum. Genet. 1999, 64, 594–599. [Google Scholar] [CrossRef] [Green Version]
- Bushara, K.; Bower, M.; Liu, J.; McFarland, K.N.; Landrian, I.; Hutter, D.; Teive, H.A.; Rasmussen, A.; Mulligan, C.J.; Ashizawa, T. Expansion of the Spinocerebellar ataxia type 10 (SCA10) repeat in a patient with Sioux Native American ancestry. PLoS ONE 2013, 8, e81342. [Google Scholar] [CrossRef] [Green Version]
- Alonso, I.; Jardim, L.B.; Artigalas, O.; Saraiva-Pereira, M.L.; Matsuura, T.; Ashizawa, T.; Sequeiros, J.; Silveira, I. Reduced penetrance of intermediate size alleles in spinocerebellar ataxia type 10. Neurology 2006, 66, 1602–1604. [Google Scholar] [CrossRef]
- Baizabal-Carvallo, J.F.; Xia, G.; Botros, P.; Laguna, J.; Ashizawa, T.; Jankovic, J. Bolivian kindred with combined spinocerebellar ataxia types 2 and 10. Acta Neurol. Scand. 2015, 132, 139–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonardi, L.; Marcotulli, C.; McFarland, K.N.; Tessa, A.; DiFabio, R.; Santorelli, F.M.; Pierelli, F.; Ashizawa, T.; Casali, C. Spinocerebellar ataxia type 10 in Peru: The missing link in the Amerindian origin of the disease. J. Neurol. 2014, 261, 1691–1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teive, H.A.; Roa, B.B.; Raskin, S.; Fang, P.; Arruda, W.O.; Neto, Y.C.; Gao, R.; Werneck, L.C.; Ashizawa, T. Clinical phenotype of Brazilian families with spinocerebellar ataxia 10. Neurology 2004, 63, 1509–1512. [Google Scholar] [CrossRef]
- Veliz-Otani, D.; Inca-Martinez, M.; Bampi, G.B.; Ortega, O.; Jardim, L.B.; Saraiva-Pereira, M.L.; Mazzetti, P.; Cornejo-Olivas, M. ATXN10 Microsatellite Distribution in a Peruvian Amerindian Population. Cerebellum 2019, 18, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Naito, H.; Takahashi, T.; Kamada, M.; Morino, H.; Yoshino, H.; Hattori, N.; Maruyama, H.; Kawakami, H.; Matsumoto, M. First report of a Japanese family with spinocerebellar ataxia type 10: The second report from Asia after a report from China. PLoS ONE 2017, 12, e0177955. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; McFarland, K.N.; Liu, J.; Zeng, D.; Landrian, I.; Xia, G.; Hao, Y.; Jin, M.; Mulligan, C.J.; Gu, W.; et al. Spinocerebellar ataxia type 10 in Chinese Han. Neurol. Genet. 2015, 1, e26. [Google Scholar] [CrossRef] [PubMed]
- Almeida, T.; Alonso, I.; Martins, S.; Ramos, E.M.; Azevedo, L.; Ohno, K.; Amorim, A.; Saraiva-Pereira, M.L.; Jardim, L.B.; Matsuura, T.; et al. Ancestral origin of the ATTCT repeat expansion in spinocerebellar ataxia type 10 (SCA10). PLoS ONE 2009, 4, e4553. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, T.; Fang, P.; Pearson, C.E.; Jayakar, P.; Ashizawa, T.; Roa, B.B.; Nelson, D.L. Interruptions in the expanded ATTCT repeat of spinocerebellar ataxia type 10: Repeat purity as a disease modifier? Am. J. Hum. Genet. 2006, 78, 125–129. [Google Scholar] [CrossRef] [Green Version]
- McFarland, K.N.; Liu, J.; Landrian, I.; Godiska, R.; Shanker, S.; Yu, F.; Farmerie, W.G.; Ashizawa, T. SMRT Sequencing of Long Tandem Nucleotide Repeats in SCA10 Reveals Unique Insight of Repeat Expansion Structure. PLoS ONE 2015, 10, e0135906. [Google Scholar] [CrossRef]
- McFarland, K.N.; Liu, J.; Landrian, I.; Zeng, D.; Raskin, S.; Moscovich, M.; Gatto, E.M.; Ochoa, A.; Teive, H.A.; Rasmussen, A.; et al. Repeat interruptions in spinocerebellar ataxia type 10 expansions are strongly associated with epileptic seizures. Neurogenetics 2014, 15, 59–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Findlay Black, H.; Wright, G.E.B.; Collins, J.A.; Caron, N.; Kay, C.; Xia, Q.; Arning, L.; Bijlsma, E.K.; Squitieri, F.; Nguyen, H.P.; et al. Frequency of the loss of CAA interruption in the HTT CAG tract and implications for Huntington disease in the reduced penetrance range. Genet. Med. 2020, 22, 2108–2113. [Google Scholar] [CrossRef] [PubMed]
- Pesovic, J.; Peric, S.; Brkusanin, M.; Brajuskovic, G.; Rakocevic-Stojanovic, V.; Savic-Pavicevic, D. Repeat Interruptions Modify Age at Onset in Myotonic Dystrophy Type 1 by Stabilizing DMPK Expansions in Somatic Cells. Front. Genet. 2018, 9, 601. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.E.B.; Black, H.F.; Collins, J.A.; Gall-Duncan, T.; Caron, N.S.; Pearson, C.E.; Hayden, M.R. Interrupting sequence variants and age of onset in Huntington’s disease: Clinical implications and emerging therapies. Lancet Neurol. 2020, 19, 930–939. [Google Scholar] [CrossRef]
- Rasmussen, A.; Matsuura, T.; Ruano, L.; Yescas, P.; Ochoa, A.; Ashizawa, T.; Alonso, E. Clinical and genetic analysis of four Mexican families with spinocerebellar ataxia type 10. Ann. Neurol. 2001, 50, 234–239. [Google Scholar] [CrossRef]
- Hernandez-Castillo, C.R.; Diaz, R.; Vaca-Palomares, I.; Torres, D.L.; Chirino, A.; Campos-Romo, A.; Ochoa, A.; Rasmussen, A.; Fernandez-Ruiz, J. Extensive cerebellar and thalamic degeneration in spinocerebellar ataxia type 10. Parkinsonism Relat. Disord. 2019, 66, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.; McFarland, K.N.; Wang, K.; Sarkar, P.S.; Yachnis, A.T.; Ashizawa, T. Purkinje cell loss is the major brain pathology of spinocerebellar ataxia type 10. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1409–1411. [Google Scholar] [CrossRef] [Green Version]
- Grewal, R.P.; Achari, M.; Matsuura, T.; Durazo, A.; Tayag, E.; Zu, L.; Pulst, S.M.; Ashizawa, T. Clinical features and ATTCT repeat expansion in spinocerebellar ataxia type 10. Arch. Neurol. 2002, 59, 1285–1290. [Google Scholar] [CrossRef] [Green Version]
- Ashizawa, T.; Matsuura, T. Spinocerebellar ataxia type 10 (SCA10): A disease caused by a novel pentanucleotide repeat expansion. Rinsho Shinkeigaku 2001, 41, 1120–1122. [Google Scholar]
- Kurosaki, T.; Matsuura, T.; Ohno, K.; Ueda, S. Alu-mediated acquisition of unstable ATTCT pentanucleotide repeats in the human ATXN10 gene. Mol. Biol. Evol. 2009, 26, 2573–2579. [Google Scholar] [CrossRef]
- Potaman, V.N.; Bissler, J.J.; Hashem, V.I.; Oussatcheva, E.A.; Lu, L.; Shlyakhtenko, L.S.; Lyubchenko, Y.L.; Matsuura, T.; Ashizawa, T.; Leffak, M.; et al. Unpaired structures in SCA10 (ATTCT)n.(AGAAT)n repeats. J. Mol. Biol. 2003, 326, 1095–1111. [Google Scholar] [CrossRef]
- Handa, V.; Yeh, H.J.; McPhie, P.; Usdin, K. The AUUCU repeats responsible for spinocerebellar ataxia type 10 form unusual RNA hairpins. J. Biol. Chem. 2005, 280, 29340–29345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, P.; Lam, S.L. Minidumbbell structures formed by ATTCT pentanucleotide repeats in spinocerebellar ataxia type 10. Nucleic Acids Res. 2020, 48, 7557–7568. [Google Scholar] [CrossRef] [PubMed]
- De Mezer, M.; Wojciechowska, M.; Napierala, M.; Sobczak, K.; Krzyzosiak, W.J. Mutant CAG repeats of Huntingtin transcript fold into hairpins, form nuclear foci and are targets for RNA interference. Nucleic Acids Res. 2011, 39, 3852–3863. [Google Scholar] [CrossRef] [Green Version]
- Napierala, M.; Krzyzosiak, W.J. CUG repeats present in myotonin kinase RNA form metastable “slippery” hairpins. J. Biol. Chem. 1997, 272, 31079–31085. [Google Scholar] [CrossRef] [Green Version]
- Reddy, K.; Zamiri, B.; Stanley, S.Y.R.; Macgregor, R.B., Jr.; Pearson, C.E. The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J. Biol. Chem. 2013, 288, 9860–9866. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Gonzalez, A.L.; Yildirim, I.; Tran, T.; Lohman, J.R.; Fang, P.; Guo, M.; Disney, M.D. Crystallographic and Computational Analyses of AUUCU Repeating RNA That Causes Spinocerebellar Ataxia Type 10 (SCA10). Biochemistry 2015, 54, 3851–3859. [Google Scholar] [CrossRef] [Green Version]
- Komar, A.A.; Hatzoglou, M. Internal ribosome entry sites in cellular mRNAs: Mystery of their existence. J. Biol. Chem. 2005, 280, 23425–23428. [Google Scholar] [CrossRef] [Green Version]
- Komar, A.A.; Hatzoglou, M. Cellular IRES-mediated translation: The war of ITAFs in pathophysiological states. Cell Cycle 2011, 10, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Green, K.M.; Glineburg, M.R.; Kearse, M.G.; Flores, B.N.; Linsalata, A.E.; Fedak, S.J.; Goldstrohm, A.C.; Barmada, S.J.; Todd, P.K. RAN translation at C9orf72-associated repeat expansions is selectively enhanced by the integrated stress response. Nat. Commun. 2017, 8, 2005. [Google Scholar] [CrossRef] [Green Version]
- Wakamiya, M.; Matsuura, T.; Liu, Y.; Schuster, G.C.; Gao, R.; Xu, W.; Sarkar, P.S.; Lin, X.; Ashizawa, T. The role of ataxin 10 in the pathogenesis of spinocerebellar ataxia type 10. Neurology 2006, 67, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Sznajder, L.J.; Thomas, J.D.; Carrell, E.M.; Reid, T.; McFarland, K.N.; Cleary, J.D.; Oliveira, R.; Nutter, C.A.; Bhatt, K.; Sobczak, K.; et al. Intron retention induced by microsatellite expansions as a disease biomarker. Proc. Natl. Acad. Sci. USA 2018, 115, 4234–4239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keren, B.; Jacquette, A.; Depienne, C.; Leite, P.; Durr, A.; Carpentier, W.; Benyahia, B.; Ponsot, G.; Soubrier, F.; Brice, A.; et al. Evidence against haploinsuffiency of human ataxin 10 as a cause of spinocerebellar ataxia type 10. Neurogenet. 2010, 11, 273–274. [Google Scholar] [CrossRef] [PubMed]
- White, M.C.; Gao, R.; Xu, W.; Mandal, S.M.; Lim, J.G.; Hazra, T.K.; Wakamiya, M.; Edwards, S.F.; Raskin, S.; Teive, H.A.; et al. Inactivation of hnRNP K by expanded intronic AUUCU repeat induces apoptosis via translocation of PKCdelta to mitochondria in spinocerebellar ataxia 10. PLoS Genet. 2010, 6, e1000984. [Google Scholar] [CrossRef] [PubMed]
- McFarland, K.N.; Ashizawa, T. Transgenic models of spinocerebellar ataxia type 10: Modeling a repeat expansion disorder. Genes 2012, 3, 481–491. [Google Scholar] [CrossRef]
- White, M.; Xia, G.; Gao, R.; Wakamiya, M.; Sarkar, P.S.; McFarland, K.; Ashizawa, T. Transgenic mice with SCA10 pentanucleotide repeats show motor phenotype and susceptibility to seizure: A toxic RNA gain-of-function model. J. Neurosci. Res. 2012, 90, 706–714. [Google Scholar] [CrossRef] [Green Version]
- Bomsztyk, K.; Denisenko, O.; Ostrowski, J. hnRNP K: One protein multiple processes. Bioessays 2004, 26, 629–638. [Google Scholar] [CrossRef]
- Lu, H.; Sun, J.; Liang, W.; Chang, Z.; Rom, O.; Zhao, Y.; Zhao, G.; Xiong, W.; Wang, H.; Zhu, T.; et al. Cyclodextrin Prevents Abdominal Aortic Aneurysm via Activation of Vascular Smooth Muscle Cell TFEB. Circulation 2020. [Google Scholar] [CrossRef]
- Auweter, S.D.; Oberstrass, F.C.; Allain, F.H. Sequence-specific binding of single-stranded RNA: Is there a code for recognition? Nucleic Acids Res. 2006, 34, 4943–4959. [Google Scholar] [CrossRef] [Green Version]
- Gallardo, M.; Hornbaker, M.J.; Zhang, X.; Hu, P.; Bueso-Ramos, C.; Post, S.M. Aberrant hnRNP K expression: All roads lead to cancer. Cell Cycle 2016, 15, 1552–1557. [Google Scholar] [CrossRef] [Green Version]
- Schultz, D.B.; Nascimento, F.A.; Camargo, C.H.F.; Ashizawa, T.; Teive, H.A.G. Cancer frequency in patients with spinocerebellar ataxia type 10. Parkinsonism Relat. Disord. 2020, 76, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Cherng, N.; Shishkin, A.A.; Schlager, L.I.; Tuck, R.H.; Sloan, L.; Matera, R.; Sarkar, P.S.; Ashizawa, T.; Freudenreich, C.H.; Mirkin, S.M. Expansions, contractions, and fragility of the spinocerebellar ataxia type 10 pentanucleotide repeat in yeast. Proc. Natl. Acad. Sci. USA 2011, 108, 2843–2848. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Bissler, J.J.; Sinden, R.R.; Leffak, M. Unstable spinocerebellar ataxia type 10 (ATTCT*(AGAAT) repeats are associated with aberrant replication at the ATX10 locus and replication origin-dependent expansion at an ectopic site in human cells. Mol. Cell. Biol. 2007, 27, 7828–7838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashem, V.; Tiwari, A.; Bewick, B.; Teive, H.A.G.; Moscovich, M.; Schuele, B.; Bushara, K.; Bower, M.; Rasmussen, A.; Tsai, Y.C.; et al. Pulse-Field capillary electrophoresis of repeat-primed PCR amplicons for analysis of large repeats in Spinocerebellar Ataxia Type 10. PLoS ONE 2020, 15, e0228789. [Google Scholar] [CrossRef]
- Yang, W.Y.; Gao, R.; Southern, M.; Sarkar, P.S.; Disney, M.D. Design of a bioactive small molecule that targets r(AUUCU) repeats in spinocerebellar ataxia 10. Nat. Commun. 2016, 7, 11647. [Google Scholar] [CrossRef] [PubMed]
- Rzuczek, S.G.; Colgan, L.A.; Nakai, Y.; Cameron, M.D.; Furling, D.; Yasuda, R.; Disney, M.D. Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat. Chem. Biol. 2017, 13, 188–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.H.; Park, H.Y.; Jeong, S.Y.; Hong, J.H.; Kim, H.J. 16q-linked autosomal dominant cerebellar ataxia in a Korean family. Eur. J. Neurol. 2007, 14, e16–e17. [Google Scholar] [CrossRef]
- Lee, Y.C.; Liu, C.S.; Lee, T.Y.; Lo, Y.C.; Lu, Y.C.; Soong, B.W. SCA31 is rare in the Chinese population on Taiwan. Neurobiol. Aging 2012, 33, 426.e23–426.e24. [Google Scholar] [CrossRef]
- Ouyang, Y.; He, Z.; Li, L.; Qin, X.; Zhao, Y.; Yuan, L. Spinocerebellar ataxia type 31 exists in northeast China. J. Neurol. Sci. 2012, 316, 164–167. [Google Scholar] [CrossRef]
- Yang, K.; Zeng, S.; Liu, Z.; Shi, S.; Sun, W.; Yuan, Y.; Weng, L.; Jiang, H.; Shen, L.; Tang, B.; et al. Analysis of spinocerebellar ataxia type 31 related mutations among patients from mainland China. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2018, 35, 309–313. [Google Scholar] [CrossRef]
- Ishikawa, K.; Durr, A.; Klopstock, T.; Muller, S.; De Toffol, B.; Vidailhet, M.; Vighetto, A.; Marelli, C.; Wichmann, H.E.; Illig, T.; et al. Pentanucleotide repeats at the spinocerebellar ataxia type 31 (SCA31) locus in Caucasians. Neurol. Aging 2011, 77, 1853–1855. [Google Scholar] [CrossRef] [PubMed]
- Pedroso, J.L.; Abrahao, A.; Ishikawa, K.; Raskin, S.; de Souza, P.V.; de Rezende Pinto, W.B.; Braga-Neto, P.; de Albuquerque, M.V.; Mizusawa, H.; Barsottini, O.G. When should we test patients with familial ataxias for SCA31? A misdiagnosed condition outside Japan? J. Neurol. Sci. 2015, 355, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Yoshida, K.; Matsushima, A.; Shimizu, Y.; Sato, S.; Yahikozawa, H.; Ohara, S.; Yazawa, M.; Ushiyama, M.; Sato, M.; et al. Natural History of Spinocerebellar Ataxia Type 31: A 4-Year Prospective Study. Cerebellum 2017, 16, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Owada, K.; Ishikawa, K.; Toru, S.; Ishida, G.; Gomyoda, M.; Tao, O.; Noguchi, Y.; Kitamura, K.; Kondo, I.; Noguchi, E.; et al. A clinical, genetic, and neuropathologic study in a family with 16q-linked ADCA type III. Neurology 2005, 65, 629–632. [Google Scholar] [CrossRef]
- Itaya, S.; Kobayashi, Z.; Ozaki, K.; Sato, N.; Numasawa, Y.; Ishikawa, K.; Yokota, T.; Matsuda, H.; Shintani, S. Spinocerebellar Ataxia Type 31 with Blepharospasm. Intern. Med. 2018, 57, 1651–1654. [Google Scholar] [CrossRef] [Green Version]
- Norioka, R.; Sugaya, K.; Murayama, A.; Kawazoe, T.; Tobisawa, S.; Kawata, A.; Takahashi, K. Midbrain atrophy related to parkinsonism in a non-coding repeat expansion disorder: Five cases of spinocerebellar ataxia type 31 with nigrostriatal dopaminergic dysfunction. Cerebellum Ataxias 2021, 8, 11. [Google Scholar] [CrossRef]
- Ishikawa, K.; Mizusawa, H. The chromosome 16q-linked autosomal dominant cerebellar ataxia (16q-ADCA): A newly identified degenerative ataxia in Japan showing peculiar morphological changes of the Purkinje cell: The 50th Anniversary of Japanese Society of Neuropathology. Neuropathology 2010, 30, 490–494. [Google Scholar] [CrossRef]
- Niimi, Y.; Takahashi, M.; Sugawara, E.; Umeda, S.; Obayashi, M.; Sato, N.; Ishiguro, T.; Higashi, M.; Eishi, Y.; Mizusawa, H.; et al. Abnormal RNA structures (RNA foci) containing a penta-nucleotide repeat (UGGAA)n in the Purkinje cell nucleus is associated with spinocerebellar ataxia type 31 pathogenesis. Neuropathology 2013, 33, 600–611. [Google Scholar] [CrossRef]
- Yoshida, K.; Asakawa, M.; Suzuki-Kouyama, E.; Tabata, K.; Shintaku, M.; Ikeda, S.; Oyanagi, K. Distinctive features of degenerating Purkinje cells in spinocerebellar ataxia type 31. Neuropathology 2014, 34, 261–267. [Google Scholar] [CrossRef]
- Sato, N.; Amino, T.; Kobayashi, K.; Asakawa, S.; Ishiguro, T.; Tsunemi, T.; Takahashi, M.; Matsuura, T.; Flanigan, K.M.; Iwasaki, S.; et al. Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA)n. Am. J. Hum. Genet. 2009, 85, 544–557. [Google Scholar] [CrossRef] [Green Version]
- Ishiguro, T.; Sato, N.; Ueyama, M.; Fujikake, N.; Sellier, C.; Kanegami, A.; Tokuda, E.; Zamiri, B.; Gall-Duncan, T.; Mirceta, M.; et al. Regulatory Role of RNA Chaperone TDP-43 for RNA Misfolding and Repeat-Associated Translation in SCA31. Neuron 2017, 94, 108–124.e107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valgardsdottir, R.; Chiodi, I.; Giordano, M.; Rossi, A.; Bazzini, S.; Ghigna, C.; Riva, S.; Biamonti, G. Transcription of Satellite III non-coding RNAs is a general stress response in human cells. Nucleic Acids Res. 2008, 36, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Valgardsdottir, R.; Chiodi, I.; Giordano, M.; Cobianchi, F.; Riva, S.; Biamonti, G. Structural and functional characterization of noncoding repetitive RNAs transcribed in stressed human cells. Mol. Biol. Cell. 2005, 16, 2597–2604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goenka, A.; Sengupta, S.; Pandey, R.; Parihar, R.; Mohanta, G.C.; Mukerji, M.; Ganesh, S. Human satellite-III non-coding RNAs modulate heat-shock-induced transcriptional repression. J. Cell. Sci. 2016, 129, 3541–3552. [Google Scholar] [CrossRef] [Green Version]
- Chiodi, I.; Corioni, M.; Giordano, M.; Valgardsdottir, R.; Ghigna, C.; Cobianchi, F.; Xu, R.M.; Riva, S.; Biamonti, G. RNA recognition motif 2 directs the recruitment of SF2/ASF to nuclear stress bodies. Nucleic Acids Res. 2004, 32, 4127–4136. [Google Scholar] [CrossRef] [Green Version]
- Metz, A.; Soret, J.; Vourc’h, C.; Tazi, J.; Jolly, C. A key role for stress-induced satellite III transcripts in the relocalization of splicing factors into nuclear stress granules. J. Cell. Sci. 2004, 117, 4551–4558. [Google Scholar] [CrossRef] [Green Version]
- Ishiguro, T.; Nagai, Y.; Ishikawa, K. Insight Into Spinocerebellar Ataxia Type 31 (SCA31) From Drosophila Model. Front. Neurosci. 2021, 15, 648133. [Google Scholar] [CrossRef]
- McLoughlin, H.S.; Moore, L.R.; Paulson, H.L. Pathogenesis of SCA3 and implications for other polyglutamine diseases. Neurobiol. Dis. 2020, 134, 104635. [Google Scholar] [CrossRef]
- Kumar, A.; Vaish, M.; Ratan, R.R. Transcriptional dysregulation in Huntington’s disease: A failure of adaptive transcriptional homeostasis. Drug Discov. Today 2014, 19, 956–962. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.C.; Lin, K.F.; He, R.Y.; Tu, P.H.; Koubek, J.; Hsu, Y.C.; Huang, J.J. Inhibition of TDP-43 aggregation by nucleic acid binding. PLoS ONE 2013, 8, e64002. [Google Scholar] [CrossRef] [Green Version]
- Mann, J.R.; Gleixner, A.M.; Mauna, J.C.; Gomes, E.; DeChellis-Marks, M.R.; Needham, P.G.; Copley, K.E.; Hurtle, B.; Portz, B.; Pyles, N.J.; et al. RNA Binding Antagonizes Neurotoxic Phase Transitions of TDP-43. Neuron 2019, 102, 321–338.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, T.; Nagano, K.; Ueyama, M.; Ninomiya, K.; Hirose, T.; Nagai, Y.; Ishikawa, K.; Kawai, G.; Nakatani, K. Small molecule targeting r(UGGAA)n disrupts RNA foci and alleviates disease phenotype in Drosophila model. Nat. Commun. 2021, 12, 236. [Google Scholar] [CrossRef]
- Seixas, A.I.; Loureiro, J.R.; Costa, C.; Ordonez-Ugalde, A.; Marcelino, H.; Oliveira, C.L.; Loureiro, J.L.; Dhingra, A.; Brandao, E.; Cruz, V.T.; et al. A Pentanucleotide ATTTC Repeat Insertion in the Non-coding Region of DAB1, Mapping to SCA37, Causes Spinocerebellar Ataxia. Am. J. Hum. Genet. 2017, 101, 87–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Munuera, C.; Corral-Juan, M.; Stevanin, G.; San Nicolas, H.; Roig, C.; Corral, J.; Campos, B.; de Jorge, L.; Morcillo-Suarez, C.; Navarro, A.; et al. New subtype of spinocerebellar ataxia with altered vertical eye movements mapping to chromosome 1p32. JAMA Neurol. 2013, 70, 764–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corral-Juan, M.; Serrano-Munuera, C.; Rabano, A.; Cota-Gonzalez, D.; Segarra-Roca, A.; Ispierto, L.; Cano-Orgaz, A.T.; Adarmes, A.D.; Mendez-Del-Barrio, C.; Jesus, S.; et al. Clinical, genetic and neuropathological characterization of spinocerebellar ataxia type 37. Brain 2018, 141, 1981–1997. [Google Scholar] [CrossRef]
- Ayala, R.; Shu, T.; Tsai, L.H. Trekking across the brain: The journey of neuronal migration. Cell 2007, 128, 29–43. [Google Scholar] [CrossRef] [Green Version]
- Goldowitz, D.; Cushing, R.C.; Laywell, E.; D’Arcangelo, G.; Sheldon, M.; Sweet, H.O.; Davisson, M.; Steindler, D.; Curran, T. Cerebellar disorganization characteristic of reeler in scrambler mutant mice despite presence of reelin. J. Neurosci. 1997, 17, 8767–8777. [Google Scholar] [CrossRef]
- Sheldon, M.; Rice, D.S.; D’Arcangelo, G.; Yoneshima, H.; Nakajima, K.; Mikoshiba, K.; Howell, B.W.; Cooper, J.A.; Goldowitz, D.; Curran, T. Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature 1997, 389, 730–733. [Google Scholar] [CrossRef]
- Sweet, H.O.; Bronson, R.T.; Johnson, K.R.; Cook, S.A.; Davisson, M.T. Scrambler, a new neurological mutation of the mouse with abnormalities of neuronal migration. Mamm. Genome 1996, 7, 798–802. [Google Scholar] [CrossRef]
- Yano, M.; Hayakawa-Yano, Y.; Mele, A.; Darnell, R.B. Nova2 regulates neuronal migration through an RNA switch in disabled-1 signaling. Neuron 2010, 66, 848–858. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [Green Version]
- Cortese, A.; Tozza, S.; Yau, W.Y.; Rossi, S.; Beecroft, S.J.; Jaunmuktane, Z.; Dyer, Z.; Ravenscroft, G.; Lamont, P.J.; Mossman, S.; et al. Cerebellar ataxia, neuropathy, vestibular areflexia syndrome due to RFC1 repeat expansion. Brain 2020, 143, 480–490. [Google Scholar] [CrossRef]
- Szmulewicz, D.J.; Waterston, J.A.; MacDougall, H.G.; Mossman, S.; Chancellor, A.M.; McLean, C.A.; Merchant, S.; Patrikios, P.; Halmagyi, G.M.; Storey, E. Cerebellar ataxia, neuropathy, vestibular areflexia syndrome (CANVAS): A review of the clinical features and video-oculographic diagnosis. Ann. N. Y. Acad. Sci. 2011, 1233, 139–147. [Google Scholar] [CrossRef]
- Cortese, A.; Simone, R.; Sullivan, R.; Vandrovcova, J.; Tariq, H.; Yau, W.Y.; Humphrey, J.; Jaunmuktane, Z.; Sivakumar, P.; Polke, J.; et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat. Genet. 2019, 51, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Rafehi, H.; Szmulewicz, D.J.; Bennett, M.F.; Sobreira, N.L.M.; Pope, K.; Smith, K.R.; Gillies, G.; Diakumis, P.; Dolzhenko, E.; Eberle, M.A.; et al. Bioinformatics-Based Identification of Expanded Repeats: A Non-reference Intronic Pentamer Expansion in RFC1 Causes CANVAS. Am. J. Hum. Genet. 2019, 105, 151–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szmulewicz, D.J.; McLean, C.A.; Rodriguez, M.L.; Chancellor, A.M.; Mossman, S.; Lamont, D.; Roberts, L.; Storey, E.; Halmagyi, G.M. Dorsal root ganglionopathy is responsible for the sensory impairment in CANVAS. Neurology 2014, 82, 1410–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, R.; Kaiyrzhanov, R.; Houlden, H. Cerebellar ataxia, neuropathy, vestibular areflexia syndrome: Genetic and clinical insights. Curr. Opin. Neurol. 2021, 34, 556–564. [Google Scholar] [CrossRef]
- Pelosi, L.; Mulroy, E.; Leadbetter, R.; Kilfoyle, D.; Chancellor, A.M.; Mossman, S.; Wing, L.; Wu, T.Y.; Roxburgh, R.H. Peripheral nerves are pathologically small in cerebellar ataxia neuropathy vestibular areflexia syndrome: A controlled ultrasound study. Eur. J. Neurol. 2018, 25, 659–665. [Google Scholar] [CrossRef]
- Reyes-Leiva, D.; Aldecoa, I.; Gelpi, E.; Rojas-Garcia, R. Motor neuron involvement expands the neuropathological phenotype of late-onset ataxia in RFC1 mutation (CANVAS). Brain Pathol. 2022, e13051. [Google Scholar] [CrossRef]
- Huin, V.; Coarelli, G.; Guemy, C.; Boluda, S.; Debs, R.; Mochel, F.; Stojkovic, T.; Grabli, D.; Maisonobe, T.; Gaymard, B.; et al. Motor neuron pathology in CANVAS due to RFC1 expansions. Brain 2021, awab449. [Google Scholar] [CrossRef]
- Akcimen, F.; Ross, J.P.; Bourassa, C.V.; Liao, C.; Rochefort, D.; Gama, M.T.D.; Dicaire, M.J.; Barsottini, O.G.; Brais, B.; Pedroso, J.L.; et al. Investigation of the RFC1 Repeat Expansion in a Canadian and a Brazilian Ataxia Cohort: Identification of Novel Conformations. Front. Genet. 2019, 10, 1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scriba, C.K.; Beecroft, S.J.; Clayton, J.S.; Cortese, A.; Sullivan, R.; Yau, W.Y.; Dominik, N.; Rodrigues, M.; Walker, E.; Dyer, Z.; et al. A novel RFC1 repeat motif (ACAGG) in two Asia-Pacific CANVAS families. Brain 2020, 143, 2904–2910. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, M.; Nan, H.; Koh, K.; Ichinose, Y.; Gao, L.; Shimozono, K.; Hata, T.; Kim, Y.J.; Ohtsuka, T.; Cortese, A.; et al. RFC1 repeat expansion in Japanese patients with late-onset cerebellar ataxia. J. Hum. Genet. 2020, 65, 1143–1147. [Google Scholar] [CrossRef] [PubMed]
- Beecroft, S.J.; Cortese, A.; Sullivan, R.; Yau, W.Y.; Dyer, Z.; Wu, T.Y.; Mulroy, E.; Pelosi, L.; Rodrigues, M.; Taylor, R.; et al. A Maori specific RFC1 pathogenic repeat configuration in CANVAS, likely due to a founder allele. Brain 2020, 143, 2673–2680. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Loureiro, J.R.; Castro, A.F.; Figueiredo, A.S.; Silveira, I. Molecular Mechanisms in Pentanucleotide Repeat Diseases. Cells 2022, 11, 205. [Google Scholar] [CrossRef]
- Hesselberth, J.R. Lives that introns lead after splicing. Wires RNA 2013, 4, 677–691. [Google Scholar] [CrossRef]
- Hautbergue, G.M.; Castelli, L.M.; Ferraiuolo, L.; Sanchez-Martinez, A.; Cooper-Knock, J.; Higginbottom, A.; Lin, Y.H.; Bauer, C.S.; Dodd, J.E.; Myszczynska, M.A.; et al. SRSF1-dependent nuclear export inhibition of C9ORF72 repeat transcripts prevents neurodegeneration and associated motor deficits. Nat. Commun. 2017, 8, 16063. [Google Scholar] [CrossRef]
- Campuzano, V.; Montermini, L.; Lutz, Y.; Cova, L.; Hindelang, C.; Jiralerspong, S.; Trottier, Y.; Kish, S.J.; Faucheux, B.; Trouillas, P.; et al. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum. Mol. Genet. 1997, 6, 1771–1780. [Google Scholar] [CrossRef] [Green Version]
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- Garcia, J.C.; Bustos, R.H. The Genetic Diagnosis of Neurodegenerative Diseases and Therapeutic Perspectives. Brain Sci. 2018, 8, 222. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hao, L.; Wang, H.; Santostefano, K.; Thapa, A.; Cleary, J.; Li, H.; Guo, X.; Terada, N.; Ashizawa, T.; et al. Therapeutic Genome Editing for Myotonic Dystrophy Type 1 Using CRISPR/Cas9. Mol. Ther. 2018, 26, 2617–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteys, A.M.; Ebanks, S.A.; Keiser, M.S.; Davidson, B.L. CRISPR/Cas9 Editing of the Mutant Huntingtin Allele In Vitro and In Vivo. Mol. Ther. 2017, 25, 12–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dastidar, S.; Ardui, S.; Singh, K.; Majumdar, D.; Nair, N.; Fu, Y.; Reyon, D.; Samara, E.; Gerli, M.F.M.; Klein, A.F.; et al. Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucleic Acids Res. 2018, 46, 8275–8298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, B.S.; Saxena, T.; Oliveira, R.; Mendez-Gomez, H.R.; Cleary, J.D.; Denes, L.T.; McConnell, O.; Arboleda, J.; Xia, G.; Swanson, M.S.; et al. Impeding Transcription of Expanded Microsatellite Repeats by Deactivated Cas9. Mol. Cell. 2017, 68, 479–490.e5. [Google Scholar] [CrossRef] [Green Version]
- Batra, R.; Nelles, D.A.; Pirie, E.; Blue, S.M.; Marina, R.J.; Wang, H.; Chaim, I.A.; Thomas, J.D.; Zhang, N.; Nguyen, V.; et al. Elimination of Toxic Microsatellite Repeat Expansion RNA by RNA-Targeting Cas9. Cell 2017, 170, 899–912.e10. [Google Scholar] [CrossRef]
- Zhang, N.; Bewick, B.; Xia, G.; Furling, D.; Ashizawa, T. A CRISPR-Cas13a Based Strategy That Tracks and Degrades Toxic RNA in Myotonic Dystrophy Type 1. Front. Genet. 2020, 11, 594576. [Google Scholar] [CrossRef]
- Van Agtmaal, E.L.; Andre, L.M.; Willemse, M.; Cumming, S.A.; van Kessel, I.D.G.; van den Broek, W.; Gourdon, G.; Furling, D.; Mouly, V.; Monckton, D.G.; et al. CRISPR/Cas9-Induced (CTGCAG)n Repeat Instability in the Myotonic Dystrophy Type 1 Locus: Implications for Therapeutic Genome Editing. Mol. Ther. 2017, 25, 24–43. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, T.M.; Sobczak, K.; Lueck, J.D.; Osborne, R.J.; Lin, X.; Dirksen, R.T.; Thornton, C.A. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science 2009, 325, 336–339. [Google Scholar] [CrossRef] [Green Version]
- Manning, K.S.; Rao, A.N.; Castro, M.; Cooper, T.A. BNA(NC) Gapmers Revert Splicing and Reduce RNA Foci with Low Toxicity in Myotonic Dystrophy Cells. ACS Chem. Biol. 2017, 12, 2503–2509. [Google Scholar] [CrossRef] [Green Version]
- Scoles, D.R.; Meera, P.; Schneider, M.D.; Paul, S.; Dansithong, W.; Figueroa, K.P.; Hung, G.; Rigo, F.; Bennett, C.F.; Otis, T.S.; et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature 2017, 544, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, T.M.; Leger, A.J.; Pandey, S.K.; MacLeod, A.R.; Nakamori, M.; Cheng, S.H.; Wentworth, B.M.; Bennett, C.F.; Thornton, C.A. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 2012, 488, 111–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jauvin, D.; Chretien, J.; Pandey, S.K.; Martineau, L.; Revillod, L.; Bassez, G.; Lachon, A.; MacLeod, A.R.; Gourdon, G.; Wheeler, T.M.; et al. Targeting DMPK with Antisense Oligonucleotide Improves Muscle Strength in Myotonic Dystrophy Type 1 Mice. Mol. Ther. Nucleic Acids 2017, 7, 465–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLoughlin, H.S.; Moore, L.R.; Chopra, R.; Komlo, R.; McKenzie, M.; Blumenstein, K.G.; Zhao, H.; Kordasiewicz, H.B.; Shakkottai, V.G.; Paulson, H.L. Oligonucleotide therapy mitigates disease in spinocerebellar ataxia type 3 mice. Ann. Neurol. 2018, 84, 64–77. [Google Scholar] [CrossRef]
- Moore, L.R.; Keller, L.; Bushart, D.D.; Delatorre, R.G.; Li, D.; McLoughlin, H.S.; do Carmo Costa, M.; Shakkottai, V.G.; Smith, G.D.; Paulson, H.L. Antisense oligonucleotide therapy rescues aggresome formation in a novel spinocerebellar ataxia type 3 human embryonic stem cell line. Stem Cell Res. 2019, 39, 101504. [Google Scholar] [CrossRef]
- Zhang, N.; Bewick, B.; Schultz, J.; Tiwari, A.; Krencik, R.; Zhang, A.; Adachi, K.; Xia, G.; Yun, K.; Sarkar, P.; et al. DNAzyme Cleavage of CAG Repeat RNA in Polyglutamine Diseases. Neurotherapeutics 2021, 18, 1710–1728. [Google Scholar] [CrossRef]
- Cinesi, C.; Aeschbach, L.; Yang, B.; Dion, V. Contracting CAG/CTG repeats using the CRISPR-Cas9 nickase. Nat. Commun. 2016, 7, 13272. [Google Scholar] [CrossRef] [Green Version]
- Ouellet, D.L.; Cherif, K.; Rousseau, J.; Tremblay, J.P. Deletion of the GAA repeats from the human frataxin gene using the CRISPR-Cas9 system in YG8R-derived cells and mouse models of Friedreich ataxia. Gene. Ther. 2017, 24, 265–274. [Google Scholar] [CrossRef]
- Ikeda, M.; Taniguchi-Ikeda, M.; Kato, T.; Shinkai, Y.; Tanaka, S.; Hagiwara, H.; Sasaki, N.; Masaki, T.; Matsumura, K.; Sonoo, M.; et al. Unexpected Mutations by CRISPR-Cas9 CTG Repeat Excision in Myotonic Dystrophy and Use of CRISPR Interference as an Alternative Approach. Mol. Ther. Methods Clin. Dev. 2020, 18, 131–144. [Google Scholar] [CrossRef]
- Mosbach, V.; Viterbo, D.; Descorps-Declere, S.; Poggi, L.; Vaysse-Zinkhofer, W.; Richard, G.F. Resection and repair of a Cas9 double-strand break at CTG trinucleotide repeats induces local and extensive chromosomal deletions. PLoS. Genet. 2020, 16, e1008924. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Maitland, M.L.; Lebwohl, D. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. Reply. N. Engl. J. Med. 2021, 385, 1722–1723. [Google Scholar] [CrossRef] [PubMed]
- Rong, Z.; Gong, X.; Hulleman, J.D.; Corey, D.R.; Mootha, V.V. Trinucleotide Repeat-Targeting dCas9 as a Therapeutic Strategy for Fuchs’ Endothelial Corneal Dystrophy. Transl. Vis. Sci. Technol. 2020, 9, 47. [Google Scholar] [CrossRef]
- Burstein, D.; Harrington, L.B.; Strutt, S.C.; Probst, A.J.; Anantharaman, K.; Thomas, B.C.; Doudna, J.A.; Banfield, J.F. New CRISPR-Cas systems from uncultivated microbes. Nature 2017, 542, 237–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, L.B.; Burstein, D.; Chen, J.S.; Paez-Espino, D.; Ma, E.; Witte, I.P.; Cofsky, J.C.; Kyrpides, N.C.; Banfield, J.F.; Doudna, J.A. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science 2018, 362, 839–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pausch, P.; Al-Shayeb, B.; Bisom-Rapp, E.; Tsuchida, C.A.; Li, Z.; Cress, B.F.; Knott, G.J.; Jacobsen, S.E.; Banfield, J.F.; Doudna, J.A. CRISPR-CasPhi from huge phages is a hypercompact genome editor. Science 2020, 369, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, X.; Zhou, J.; Yang, C.; Wang, G.; Tan, Y.; Wu, Y.; Zhang, S.; Yi, K.; Kang, C. The CRISPR-Cas13a Gene-Editing System Induces Collateral Cleavage of RNA in Glioma Cells. Adv. Sci. 2019, 6, 1901299. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Liu, L.; Lang, J.; Cheng, K.; Wang, Y.; Li, X.; Shi, J.; Wang, Y.; Nie, G. A CRISPR-Cas13a system for efficient and specific therapeutic targeting of mutant KRAS for pancreatic cancer treatment. Cancer Lett. 2018, 431, 171–181. [Google Scholar] [CrossRef]
- Abbott, T.R.; Dhamdhere, G.; Liu, Y.; Lin, X.; Goudy, L.; Zeng, L.; Chemparathy, A.; Chmura, S.; Heaton, N.S.; Debs, R.; et al. Development of CRISPR as an Antiviral Strategy to Combat SARS-CoV-2 and Influenza. Cell 2020, 181, 865–876.e812. [Google Scholar] [CrossRef]
- Zhou, H.; Su, J.; Hu, X.; Zhou, C.; Li, H.; Chen, Z.; Xiao, Q.; Wang, B.; Wu, W.; Sun, Y.; et al. Glia-to-Neuron Conversion by CRISPR-CasRx Alleviates Symptoms of Neurological Disease in Mice. Cell 2020. [Google Scholar] [CrossRef]
- Zhang, N. DNAzyme as a rising gene-silencing agent in theranostic settings. Neural Regen. Res. 2022, 17, 1989–1990. [Google Scholar] [CrossRef]
- Jafar-Nejad, P.; Powers, B.; Soriano, A.; Zhao, H.; Norris, D.A.; Matson, J.; DeBrosse-Serra, B.; Watson, J.; Narayanan, P.; Chun, S.J.; et al. The atlas of RNase H antisense oligonucleotide distribution and activity in the CNS of rodents and non-human primates following central administration. Nucleic Acids Res. 2021, 49, 657–673. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Gene (Locus) | Pathogenic Repeat Range |
|---|---|---|
| SCA10 | Intron 9 of ATXN10 (22q13.31) | ATTCT800–4500 |
| SCA31 | ntron shared by BEAN & TK2 (16q22.1) | TGGAAn |
| SCA37 | 5′-UTR intron 1 or 3 of DAB1 (1p32.2) | ATTTC31–75 |
| CANVAS | Intron 2 of RFC1 (4p14) | AAGGG400–2000 |
| ACAGG~1000 | ||
| AAAGG10–25AAGGGnAAAGGn |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, N.; Ashizawa, T. Mechanistic and Therapeutic Insights into Ataxic Disorders with Pentanucleotide Expansions. Cells 2022, 11, 1567. https://doi.org/10.3390/cells11091567
Zhang N, Ashizawa T. Mechanistic and Therapeutic Insights into Ataxic Disorders with Pentanucleotide Expansions. Cells. 2022; 11(9):1567. https://doi.org/10.3390/cells11091567
Chicago/Turabian StyleZhang, Nan, and Tetsuo Ashizawa. 2022. "Mechanistic and Therapeutic Insights into Ataxic Disorders with Pentanucleotide Expansions" Cells 11, no. 9: 1567. https://doi.org/10.3390/cells11091567
APA StyleZhang, N., & Ashizawa, T. (2022). Mechanistic and Therapeutic Insights into Ataxic Disorders with Pentanucleotide Expansions. Cells, 11(9), 1567. https://doi.org/10.3390/cells11091567