1. Introduction
Trimeric G proteins are composed of α, β and γ subunits, which constitute the top hierarchy of determinants controlling contraction and relaxation of vascular smooth muscle cells (VSMCs) [
1]. Upon binding of stimulatory agonists to G-protein coupled receptors (GPCRs), VSMC contraction or relaxation is regulated via one or several types of Gα subunits, including Gα
q/11, Gα
12/13, Gα
i/o and Gα
s. Upon stimulation Gα subunits replace guanosine diphosphate (GDP) by guanosine triphosphate (GTP) and activate specific target effector proteins [
2]. This process is accompanied by the dissociation of βγ-subunits, which are capable of stimulating further signaling cascades comprising multiple kinases, such as mitogen-activated protein kinases (MAPKs) [
3]. As can be deduced from the complexity of this signal transduction mechanism and considering that several types of α-subunits may be activated by one agonist, contractile responses of VSMCs are regulated via multiple input pathways. As such, endothelin-1 (ET-1) may stimulate several signaling cascades in aortic VSMCs at once upon binding to type-A ET-1 receptors (ET
A). This promotes the intracellular release of calcium via the Gα
q/11-PLC-IP-3 pathway, activation of Rho-associated kinase via Gα
12/13-RhoGEF [
4,
5], stimulation of protein kinase A (PKA) via Gα
i-coupled βγ-subunits [
6], and activation of several MAPKs [
5]. On the functional level, these signaling events converge to promote a robust and long-lasting constriction of arterial VSMCs.
The outcome of G-protein-mediated signaling events critically depends on the expression of GPCRs, which is altered by environmental conditions promoting the dedifferentiation of VSMCs [
7]. As such, cell culture conditions optimized to feature the growth of VSMCs while suppressing a contractile phenotype challenge analyses of the G-protein-dependent signaling events and the assessment of the therapeutic capacity of pharmacological agents. Moreover, the high rigidity of culture surfaces limit or even prohibit the observation of regular contractile responses to G-protein-activating agonists. Especially VSMC relaxation is difficult to assess as tensile stress originating from the environment is required to reshape the VSMC morphology upon a decrease in the tension generated by the contractile apparatus. Several experimental setups have been developed to overcome these analytical limitations, including the determination of the percentage of VSMCs changing their morphological shape in response to a stimulus [
4]. By growing VSMCs on artificial silicone-based substrates, the formation of distortions of the extracellular matrix has been utilized as indirect readout to determine the increase or decrease in VSMC tension [
8]. In a more recent study, elegant magnetic three-dimensional (3D) bioprinting-based techniques were applied to generate vasoactive ring-like VSMC structures for drug testing purposes [
9]. Assessment of contractile responses is meanwhile supported by modern computational techniques coping with the phenotype heterogeneity of cultured VSMCs promoted by the cell culture environment [
10].
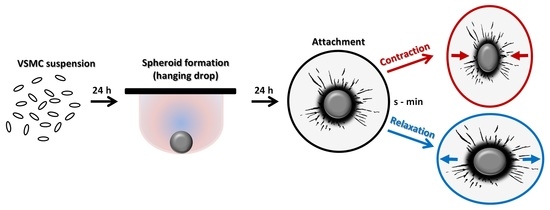
While all these models allow for the analyses of contractile responses, most of them either rely on proliferating VSMCs cultured as monolayer, require long-term culture of VSMCs or sophisticated technical equipment. In this study, we cultured VSMCs in three-dimensional spheroids, which were shown to promote cellular quiescence without applying starvation conditions and maintain their contractile phenotype [
11]. They responded to G-protein-activating agonists, such as angiotensin II and norepinephrine, by increasing intracellular calcium levels while allowing for analyses of downstream signaling events [
12]. VSMC spheroids are generated from suspended VSMCs within 24 h in hanging drops or in non-adhesive U-shaped 96-well plates if specific treatment options for subsets of spheroids are required. Here, we tested the general suitability of VSMC spheroids as a tool for the analysis and modification of G-protein-mediated responses on the level of signaling and function. In this context, we intended to develop a simple assay allowing the quantification of both VSMC contraction and relaxation without sophisticated equipment or specialized analytical tools.
2. Materials and Methods
2.1. Antibodies and Reagents
The rabbit anti-human/mouse Endothelin Receptor A polyclonal and rabbit anti-human/mouse Valosin-Containing Protein (VCP) antibody was purchased from Invitrogen (Carlsbad, CA, USA, PA3-065) and Novus Biologicals (Centennial, CO, USA. NB100-1558), respectively. Gq/11 protein inhibitor YM-25490 was purchased from Fujifilm Wako Chemicals (Neuss, Germany, 257-00631). Endothelin-1 (1160) and Iloprost (2038) from Tocris (Bristol, UK) and Caffeine from Sigma-Aldrich (Waltham, MA, USA, C0750). All other used chemicals were of analytical grade.
2.2. Cell Culture
Murine aortic SMCs (aoSMC) were isolated from the aorta of 6–8 weeks old
Rgs5fl/fl mice, as previously reported [
12,
13]. In brief, after carefully removing the adventitia, the aorta was washed twice in Dulbecco’s PBS (without calcium and magnesium). It was cut in 1-mm–sized rings and digested overnight with 1% collagenase (C5138, Merck GmbH, Darmstadt, Germany). aoSMC were resuspended, seeded on culture plates, and routinely checked for expression of SMC marker proteins (e.g., αSMA, SMMHC) and utilized until passage 5 for experiments. Human umbilical arterial smooth muscle cells (HUASMCs) were purchased from ProVitro ( Berlin, Germany, 1110611) and cultured up to passage 8 for all experiments. aoSMC and HUASMC were cultured in low-glucose DMEM (Thermo Fisher Scientific, Waltham, MA, USA) containing 15% fetal calf serum (Pelobiotech, Planegg, Germany).
2.3. Adenoviral Transfection
Murine SMCs (aoSMCs) isolated from Rgs5fl/fl mice were transduced with adenoviruses (MOI 1000) carrying empty vector (Ad-CMV-Null, Vector Biolabs, 1300, Malvern, PA, USA) or Rgs5 (Ad-CMV-Rgs5, Vector Biolabs, 270494, Malvern, PA, USA) to overexpress Rgs5. mRNA analysis was performed 72 h after transduction by qPCR to verify the overexpression of Rgs5.
2.4. Generation of 3D Spheroids
Murine aoSMC or HUASMCs were detached with trypsin, centrifuged at 1000 rpm for 5 min and counted in a Neubauer chamber or an automatic cell counter (CASY, OLS, Germany). Droplets of 25 µL culture medium with 0.24% w/v methyl cellulose (M0650, Sigma-Aldrich/Merck, Darmstadt, Germany) and 15% FCS containing 3000 (HUASMC) or 500 (aoSMC) cells were pipetted onto squared petri dishes, which were then turned bottom up for generating hanging drops. VSMCs aggregate to form 3D spheroids within 24 h. They were harvested by flushing the plate with 10 mL of DPBS per dish (14040091, Thermo-Fisher), centrifuged at 1000 rpm for 5 min, and processed for further application.
2.5. Collagen Hydrogel Preparation
Collagen stock solution (4.5 mL, ~2 mg/mL type I collagen prepared from rat tails in 0.1% acetic acid [
14]) was carefully mixed with 500 μL of 10X M199 (Sigma-Aldrich, M0605) and quickly neutralized by adding approx. 375 μL of sterile 0.2 M NaOH. Subsequently, the same volume DMEM supplemented with 30% FCS was added. Hydrogels were prepared by rapidly distributing 0.5 mL of the final collagen hydrogel mixture in each well of a pre-warmed 24-well plate, which was incubated for 30 min at 37 °C to support polymerization of the hydrogels. Homogeneous polymerization and collagen concentration are critical determinants of hydrogel rigidity and spheroid attachment.
2.6. Contraction/Relaxation Assay
Approximately 10 spheroids in 0.5 mL growth medium were homogeneously distributed on the surface of each hydrogel. Hydrogels were incubated at 37 °C/5% CO
2 in a humidified chamber for 18–24 h to allow attachment of the VSMC spheroids. Five to eight spheroids with no other spheroids located in their perimeter (>500 µm) were selected for observation per treatment group. Structural changes of attached spheroids were imaged by combining time-lapse (interval: 10 s for up to 7 min) and phase–contrast microscopy (Olympus IX3, Hamamatsu C10600 camera, Tokyo, Japan). Agonists (or solvent as control) were applied in 0.5 mL pre-warmed DMEM supplemented with 15% FCS (DMEM/FCS). Automated frame-based structural segmentation analyses of VSMC spheroids were performed by utilizing the cellSens
® software (Olympus, Tokyo, Japan, version 1.18). Area changes were assessed and quantified, as shown in
Figure S3.
2.7. IP-1 Assay
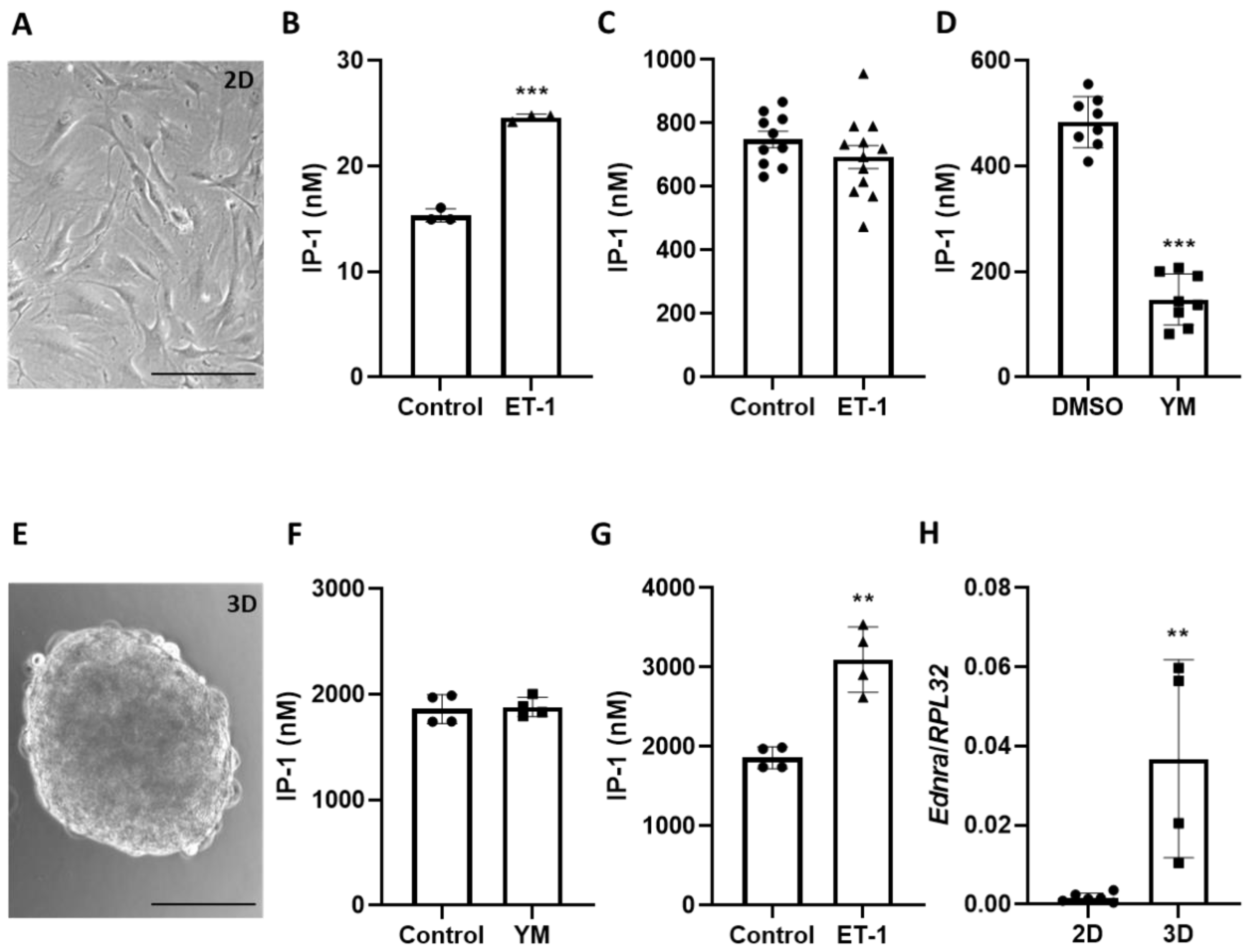
Intracellular concentrations of IP-1 were quantified in aoSMC, 3D spheroids or murine aortas using the HTRF-IP1 kit (62IPAPEB, Cisbio, Codolet, France) following the manufacturer’s instructions. Briefly, aoSMC were trypsinized, cell pellet was washed with PBS, and then resuspended in stimulation buffer containing 50 mM LiCl. aoSMC and transferred to a 384-well microtiter plate at a density of 10,000 cells per well in 7 µL of stimulation buffer. After 10 min of incubation, 7 µL of stimulation buffer containing indicated substances (100 nM of ET-1 or 10 μM of YM-254890) was added to the wells for 1 h. 3 μL of IP-1-d2 conjugate followed by 3 µL of europium cryptate-labelled anti-IP-1 antibody both dissolved in lysis buffer were then added to the wells. After a further dark incubation of 1 h at room temperature, time-resolved fluorescence was measured at 620 and 665 nm with the PheraStar (BMG Labtech, Ortenberg, Germany) multimode plate-reader. Using a standard curve generated from the IP-1 standard solution provided by the manufacturer, HTRF ratios were converted to IP-1 concentrations in nM for unknown samples.
For measurement in aortas and 3D spheroids, aortas/spheroids were stimulated in stimulation buffer with YM-254890 or ET-1 for 1 h. After centrifugation, they were lysed in lysis buffer provide by the manufacturer and 7 µL of these lysates were used for the measurement. Total protein content in the lysates was determined to normalize IP-1 levels.
2.8. cAMP Assay
Intracellular concentrations of the second messenger cAMP were quantified in aoSMC or 3D spheroids using the HTRF-cAMP-Gi kit (62AM9PEB, Cisbio, Codolet, France) following the manufacturer’s instructions. Briefly, aoSMC were trypsinized, cell pellet was washed with PBS, and then resuspended in stimulation buffer provided by the manufacturer. aoSMC were transferred to a 384-well microtiter plate at a density of 5000 cells per well in 5 µL of stimulation buffer. After 10 min of incubation, 5 µL of stimulation buffer containing 100 nM ET-1 was added for 1 h. 5 μL cAMP d2-labeled antibody followed by 5 µL of europium cryptate-labelled cAMP both dissolved in lysis buffer were added to the wells. After a further dark incubation of 1 h at room temperature, time-resolved fluorescence was measured at 620 and 665 nm with the PheraStar (BMG LabTech) multimode plate reader. A standard curve was generated with the cAMP standard solution provided by the manufacturer, and all HTRF ratios were converted to cAMP concentrations in nM for unknown samples. For measurement in 3D spheroids, spheroids were stimulated in stimulation buffer containing 100 nM ET-1 for 1 h. After centrifugation, they were lysed in lysis buffer provide by the manufacturer, and 5 µL of these lysates were used for the measurement. Total protein content in the lysates was determined to normalize cAMP levels.
2.9. Quantitative Real Time RT-PCR (qPCR) Analysis
RLT Buffer (79216, Qiagen) containing 1% β-Mercaptoethanol was used to lyse aoSMC. Total RNA was isolated using the RNeasy Mini Kit (74601, Qiagen, Hilden, Germany) according to the manufacturer’s instructions. cDNA was synthesized using the Omniscript Reverse Transcription Kit (205113, Qiagen) and quantitative real-time PCR for the target sequences was performed at the Rotor-Gene Q (Qiagen) device using the EvaGreen Master Mix (BS76.590.5000, Bio & SELL, Feucht, Germany). Amplification of the 60S ribosomal protein L32 (RPL32) cDNA served as an internal reference. Fluorescence was monitored (excitation at 470 nm and emission at 530 nm) at the end of the annealing phase (60 °C), and the threshold cycle (Ct) was set within the exponential phase of the PCR. Using the ΔΔCt method, quantification of the PCR product was performed. The following primer pairs were used: mRgs5 5′ GCGGAGAAGGCAAAGCAA 3′, 5′ GTGGTCAATGTTCACCTCTTTAGG 3′; mRpl32 5′ GGGAGCAACAAGAAAACCAA 3′, 5′ ATTGTGGACCAGGAACTTGC 3′; mEdnra 5′-GCTGGTTCCCTCTTCACTTAAGC-3′, 5′-TCATGGTTGCCAGGTTAATGC -3
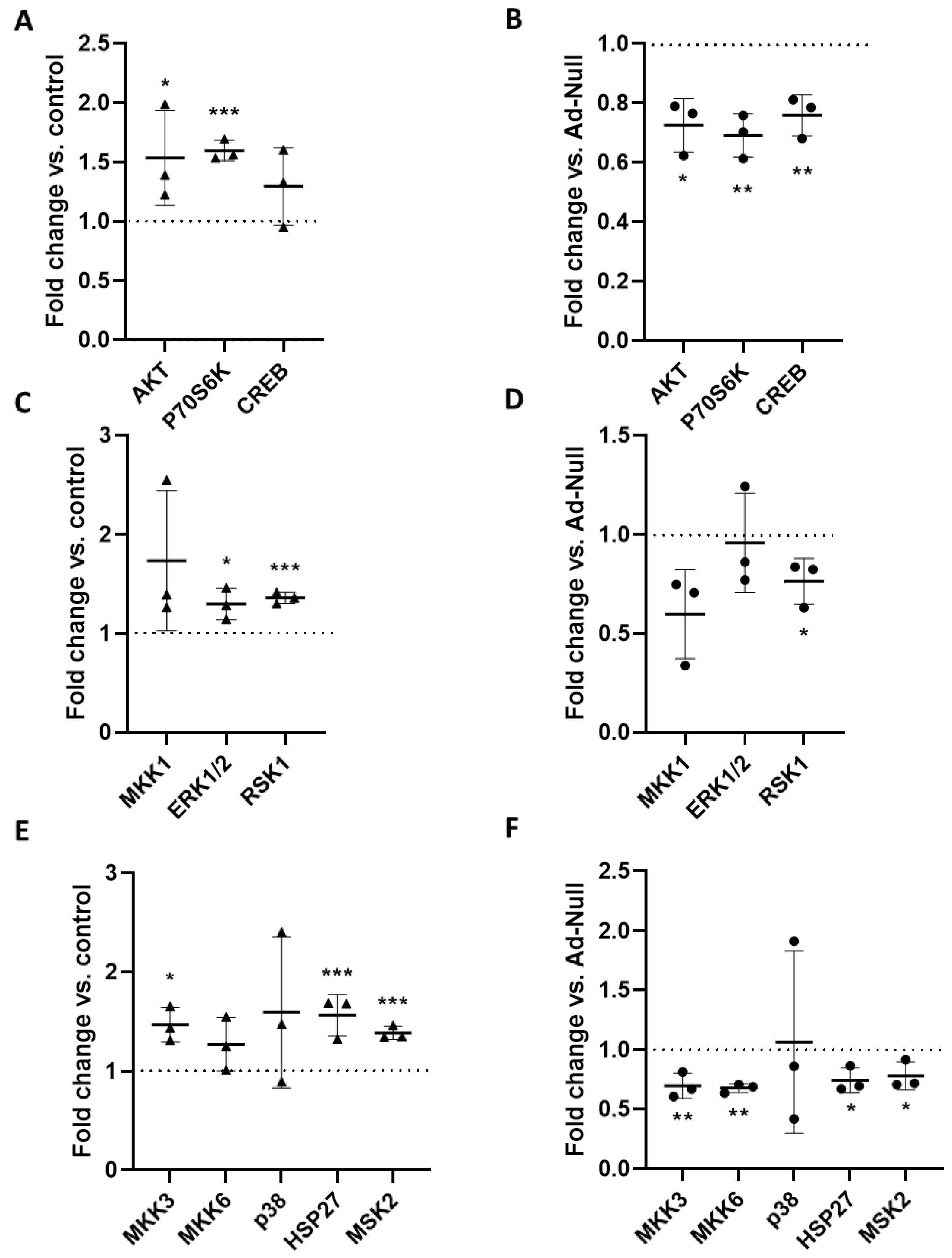
2.10. Analysis of Kinase Phosphorylation
To simultaneously determine the relative phosphorylation level of multiple kinases, a MAPK Pathway phosphorylation Array Kit (RayBiotech, Cologne, Germany, AAH-MAPK) was applied according to manufacturer’s instructions. Briefly, 3D aoSMC lysates were incubated with the membranes, where (phospho)kinase-specific primary antibodies have been spotted in a predefined manner. After washing, membranes were incubated with biotinylated detection antibodies. Streptavidin-HRP was added for chemiluminescent detection, which was performed with Image Quant™ TM LAS 4000 MINI (GE Healthcare). Grey intensities were quantified using the Image Quant TL Software (GE Healthcare, Waukesha, WI, USA, Version 8.1).
2.11. Statistical Analysis
All results are expressed as means ± SD as indicated and statistically analyzed by utilizing GraphPad Prism (Version 9.1). Outliers were identified by application of the Grubbs’ test with α set to 0.05. Differences among normally distributed values of two individual experimental groups were analyzed by unpaired Student’s t-test. Differences of one parameter between normally distributed values (Shapiro-Wilk test) of three experimental columns were analyzed by one-way ANOVA followed by Šídák’s multiple comparisons test for selected pairs of columns. Grouped data were analyzed using two-way ANOVA followed by Šídák’s multiple comparisons test to compare selected pair of columns. p < 0.05 was considered statistically significant (* p < 0.05, ** p < 0.01, *** p < 0.001).
4. Discussion
In vitro culture of VSMCs is usually achieved by growing primary arterial smooth muscle cells on plastic surfaces as two-dimensional monolayers while being exposed to medium formulations supplemented with growth factors and fetal calf serum. These conditions are fundamentally different from their natural environment and support proliferation while decreasing the degree of differentiation, as evidenced by the successive loss of markers defining their contractile phenotype, such as alpha smooth muscle actin (αSMA) and smooth muscle myosin heavy chain (SMMHC) [
16]. These proteins constitute structural elements of the contractile apparatus that in vivo generates the baseline tone of VSMCs and thus regulates the diameter of blood vessels. Consequently, the contractile capacity of VSMCs may be limited under standard cell culture conditions. Moreover, cultured VSMCs may also change the expression pattern of GPCRs, regulating the baseline tone, as was shown for rat aortic smooth muscle cells, which invert the ET
A/ET
B receptor ratio while being propagated in vitro [
17].
In light of these findings, we investigated whether VSMCs cultured in three-dimensional spheroids may serve as a suitable tool for tracing functional responses to G-protein-mediated stimuli. Earlier studies indicated that the differentiation of VSMCs is altered by organizing them in three-dimensional spheroids. In these aggregates, proliferation of VSMCs ceases, which is partially mediated by the autocrine release of transforming growth factor beta (TGFβ) [
11]—an important regulator of VSMC differentiation [
18,
19]. Moreover, gene expression associated with protein biosynthesis is downregulated while the level of proteins of the contractile apparatus is elevated or maintained. Collectively, these observations indicated that culturing of VSMCs in spheroids support their quiescent differentiated phenotype, which is why these were initially described as ‘in vitro analogue of the arterial wall’ [
20].
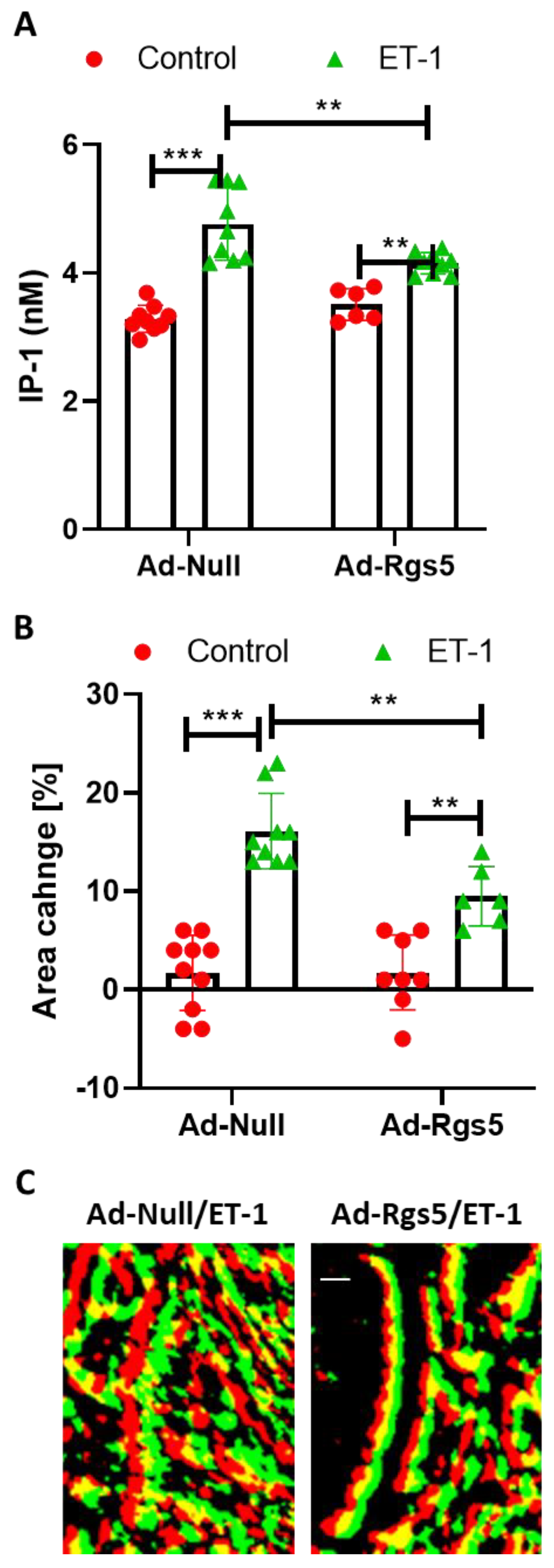
Here, we show that VSMCs in three-dimensional spheroids regain their sensitivity to ET-1 at least with respect to the Gα
q/11-PLC-IP-3 signaling axis. Increased expression of the ET
A receptor serves as the most likely explanation for this finding, which may partially be controlled by TGFβ considering the significant drop in ET
A receptor expression after inhibition of the corresponding signaling pathway in human VSMCs (
Figure S2, [
11]). Interestingly, a significant Gα
q/11-PLC-IP-3 baseline activity was detected in VSMCs under standard culture conditions, as was also shown for several types of Gα subunits in cultured bovine aortic smooth muscle cells [
4]. This agonist-independent G-protein activity was fully silenced in three-dimensional spheroids possibly caused by the elevated level of RGS5 detected in VSMC spheroids [
12] that is able to terminate Gα
q/11 activity by its GTPase activating properties. On the contrary, loss of RGS5 in VSMC spheroids was associated with an elevated intracellular calcium level as the result of unrestricted Gα
q/11-PLCβ-IP-3 activity [
12]. As such, RGS5 appears to be relevant for the control of baseline G-protein activity in general.
Despite being tempered by endogenous RGS5, ET-1 stimulated Gα
q/11-PLCβ-IP-3 activity in VSMC spheroids. Given that this stimulus did not affect the cAMP levels, triggering of Gα
i/o-dependent signaling appears unlikely in this context. Consequently, the observed ET-1-induced modification of downstream kinase activity is primarily mediated by the activation of Gα
q/11- and corresponding βγ-subunits, both of which may directly or indirectly promote phosphorylation and activation of AKT and MAPKs [
21,
22,
23]. Moreover, binding of agonists to GPCRs also supports the formation of scaffolds based on β-arrestins to support the G-protein independent interaction with and activation of other determinants of intracellular signaling cascades, including MAP kinases, PI3 kinase (PI3K), and AKT [
24]. However, as the phosphorylation of most of these kinases was attenuated upon overexpression of RGS5, Gα
q/11-dependent signaling appears to be the most likely source of MAPK and PI3K/AKT stimulation. Additionally, ET-1 triggers the activation of Gα
12/13-Rho signaling, which was not specifically addressed in this study. The relevance of this pathway for the contraction of VSMCs may nevertheless be indirectly deduced from the fact that RGS5 only partially blocked the contractile response to ET-1. The Gα
12/13-Rho signaling axis may, in fact, be amplified by RGS5, which was shown to promote RhoA activity in vitro and in vivo [
12,
13].
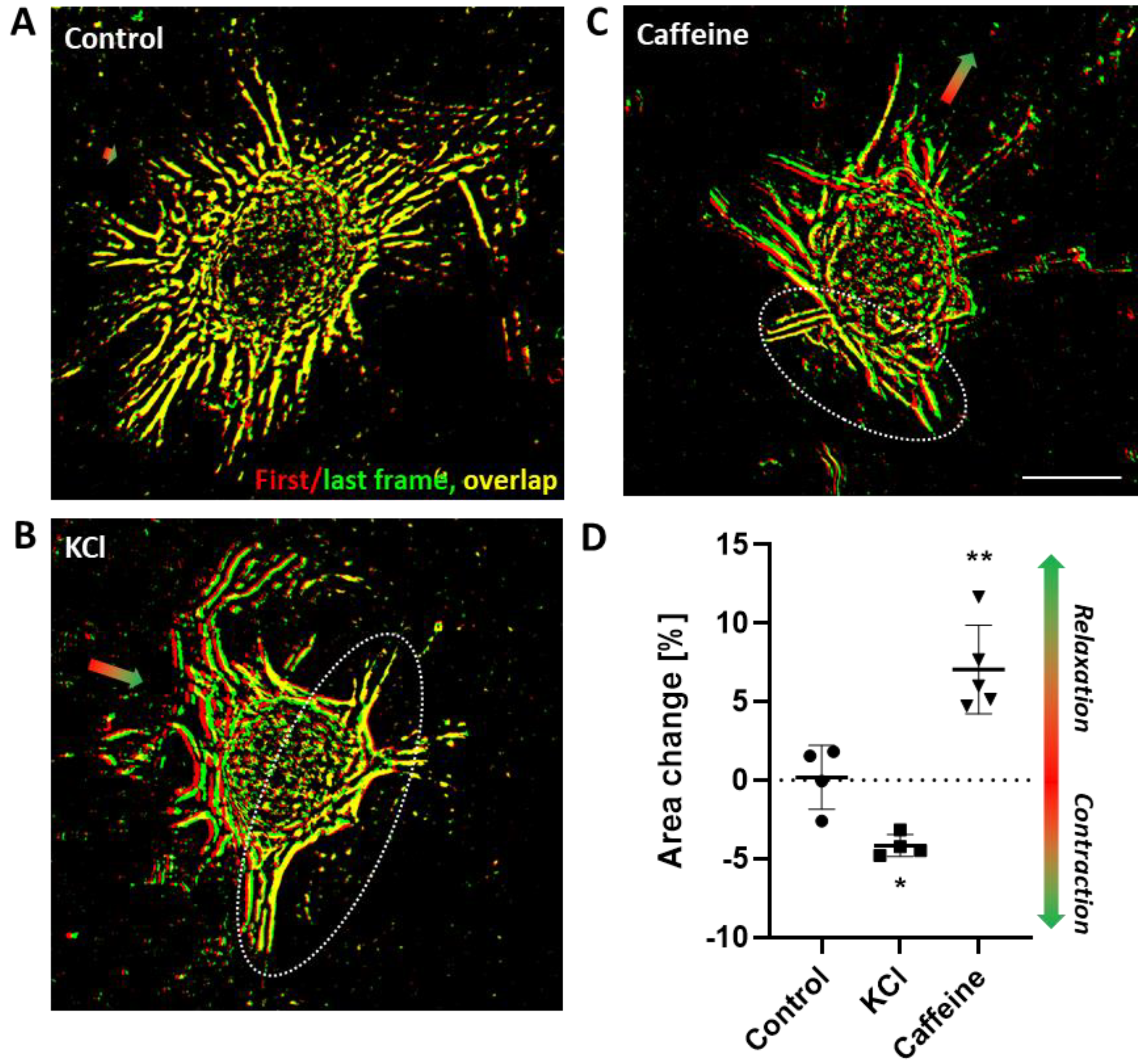
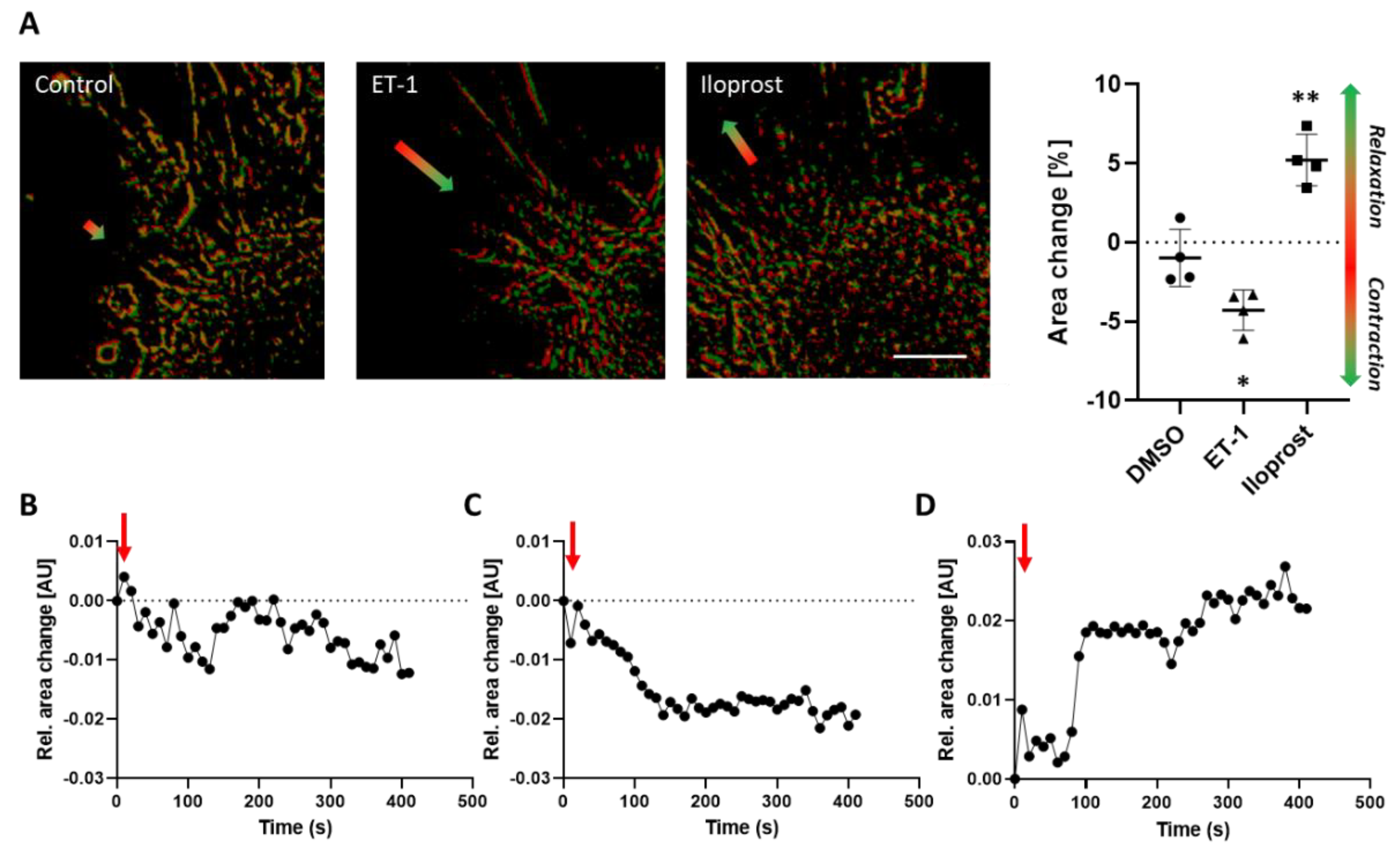
The diversity of signaling cascades triggered upon ET-1 stimulation underlines the relevance of assessing their functional impact on contractile responses of VSMCs. Especially in view of functional testing of pharmacological compounds, the development of corresponding assays is challenging as culture conditions usually promote cellular proliferation and a less contractile phenotype and may significantly diminish functional responses to distinct agonists as shown for ET-1. To overcome such limitations, we intended to develop a simple assay that supports both a stable quiescent contractile VSMC phenotype and assessment of both contraction and relaxation without the need of specialized equipment. While contraction-mediated shape changes of spheroids may in principle be directly traceable, their relaxation requires an intrinsic physical force sufficient to expand cells without interfering with their contraction. For that reason, we decided to attach the VSMC spheroids to collagen type I hydrogels, which were optimized to support cell attachment and ‘store‘ tensional forces generated by the overall tone of the focally accumulated VSMCs. In fact, forces generated by cellular traction are known to modify the mechanical properties of extracellular matrices [
25]. Those tensional forces originating from VSMCs were impressively visualized by the formation of wrinkles and distortions on polymerized silicon surfaces [
8]. Similarly, attachment of spheroid-derived endothelial cells was shown to generate tensional forces, which modify the structure of collagen hydrogels [
26]. The mechanical interaction of cells and the extracellular matrix is elegantly described by the tensegrity model [
27]. Cell-derived traction is generated by intracellular contractile elements and transferred to the matrix via focal adhesions. Consequently, any change in the force generated by the contractile apparatus of VSMCs is immediately transmitted to the matrix. Our model exploits this mechanism to visualize VSMC contraction or relaxation. Potassium chloride depolarizes VSMCs and triggers a calcium-calmodulin-dependent activation of the myosin light chain (MLC) kinase that phosphorylates the MLC to promote contraction [
28]. ET-1-induced activation of the ET
A receptor comprise a more complex interaction of the Gα
12/13- and Gα
q/11-dependent pathways promoting the phosphorylation of MLC by Rho-, calcium- as well as PKC-dependent signaling mechanisms. Relaxation of VSMC spheroids were also traceable, as shown for caffeine. Although this plant-derived alkaloid may in principle evoke a fast release of calcium from intracellular stores supporting a transient contraction in certain vascular beds [
29], relaxation appears to be the preferential response of VSMCs [
15]. Mechanistically, this may partially be based on the caffeine-mediated competitive inhibition of phosphodiesterases preserving the cAMP level as a prerequisite for protein kinase A (PKA)-regulated MLC dephosphorylation. Similarly, the prostacyclin analogue Iloprost binds to the prostacyclin receptor (IP1)—a GPCR that activates the Gα
s-cAMP-PKA signaling pathway to promote MLC dephosphorylation and thus VSMC relaxation [
30].
All aforementioned stimuli rapidly contracted or relaxed VSMCs as predicted by the literature and were traced intentionally by applying simple analytical techniques. We combined time-lapse recording and computer-assisted segmentation analyses to identify and quantify the corresponding morphological changes of VSMC spheroids. However, the analysis of their overall response may also be simplified by processing (e.g., by using open-source software ImageJ) individual phase contrast images taken before and some minutes after applying the stimulus when the response maximum has been reached. On the other hand, application of more sophisticated morphometric software tools may increase sensitivity and resolution of time-related analyses. With respect to increasing the throughput, hydrogels with a dished meniscus may also be generated in 96-well plates, which allow placing of single spheroids in the center of each gel for standardized recording protocols. Finally, endothelial cells (ECs) may be added to VSMC spheroids during their formation to generate vascular organoids composed of a superficial endothelial monolayer covering a core of VSMCs [
31,
32]. Such a model would broaden the options for analyses and include the endothelium as another important element controlling the tone of VSMCs.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




