
Impact of Combined Baricitinib and FTI Treatment on Adipogenesis in Hutchinson–Gilford Progeria Syndrome and Other Lipodystrophic Laminopathies

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Senescence Associated βeta-Galactosidase Assay
2.3. Western Blot
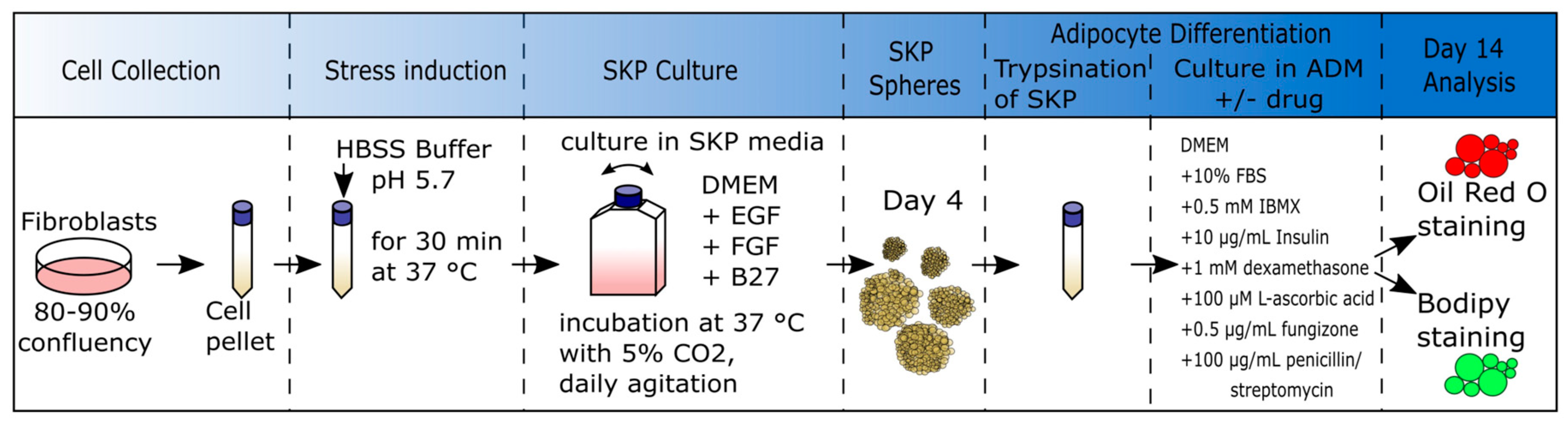
2.4. Low-pH SKP Isolation Method and Culture of SKPs
2.5. SKP Cell Differentiation into Adipocytes
2.6. Oil Red O (ORO) Staining
2.7. Bodipy Staining
2.8. Immunocytochemistry
2.9. Image Analysis
2.10. Statistical Evaluation and Graphics
3. Results
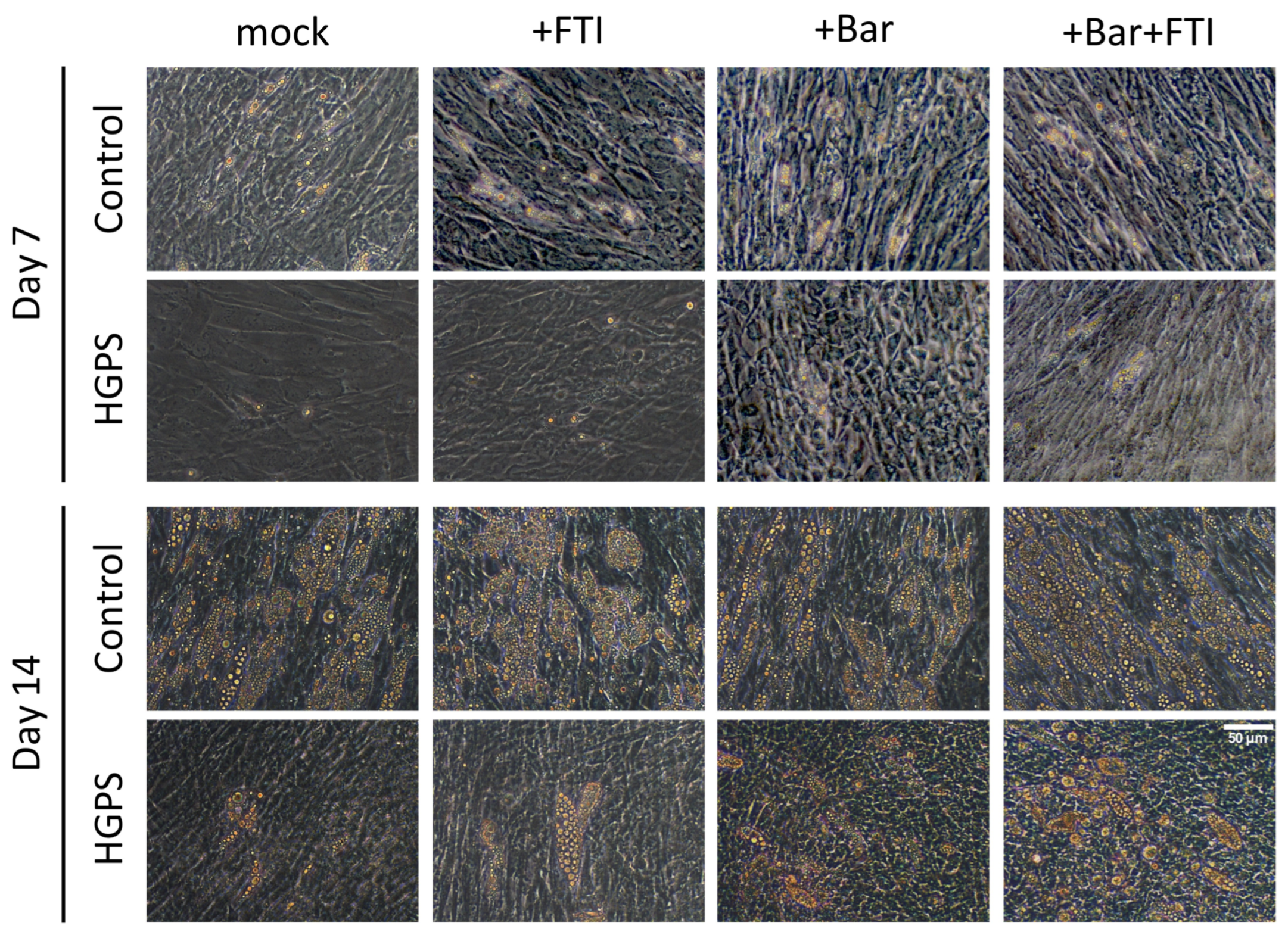
3.1. Adipocyte Differentiation of HGPS SKPs in the Presence of FTI and Baricitinib
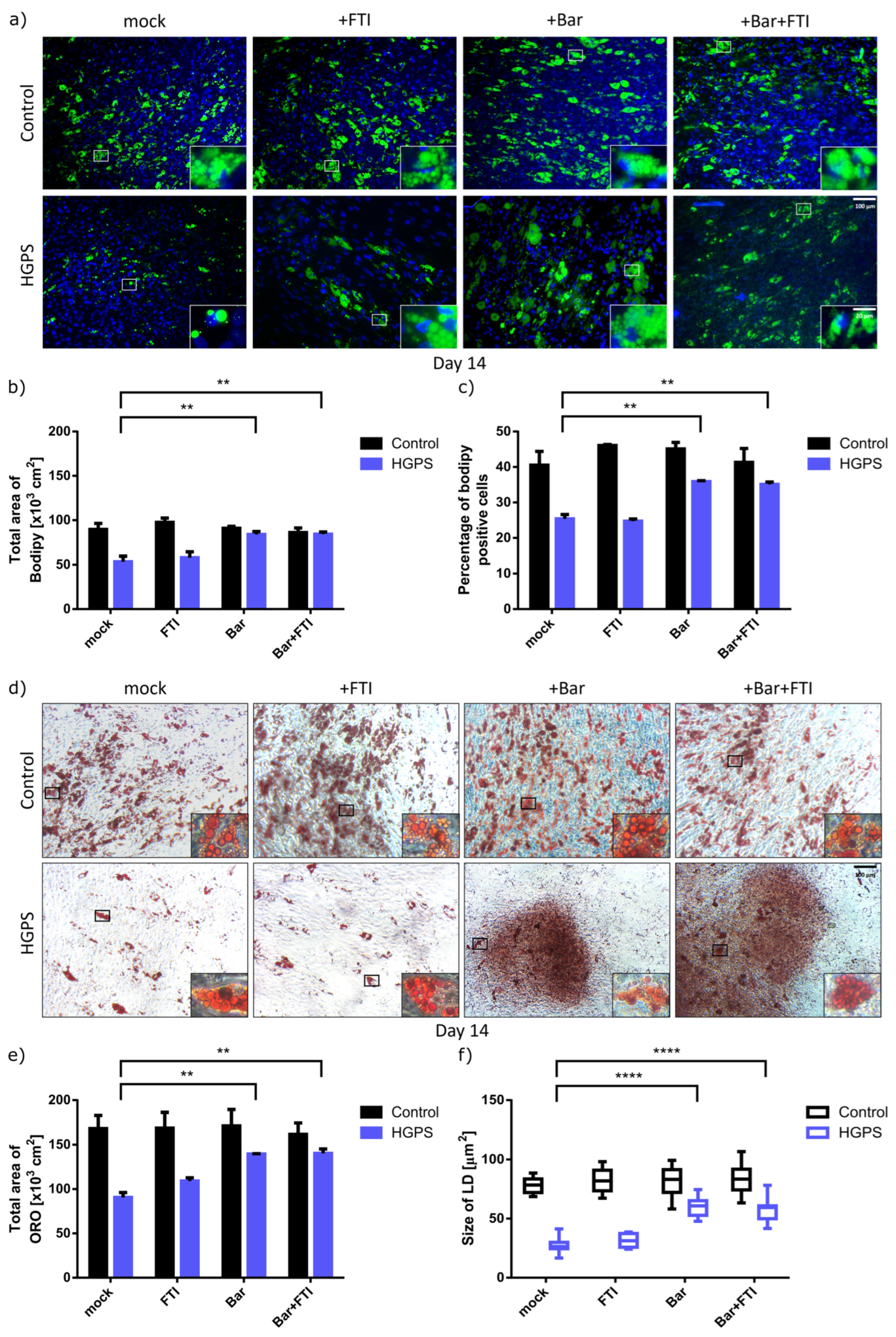
3.2. Baricitinib Alone or in Combination with FTI Improve Adipogenesis of HGPS SKPs
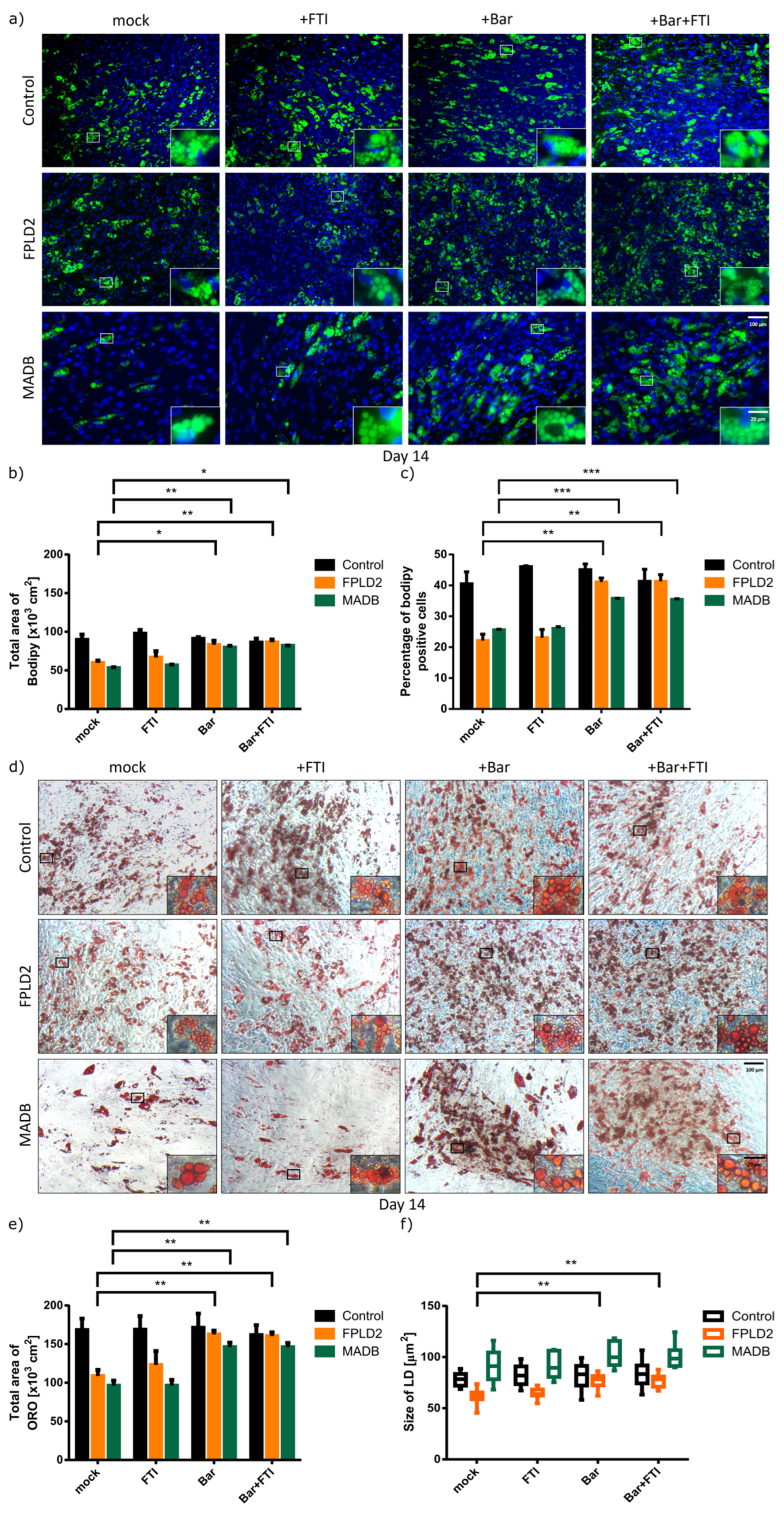
3.3. Effect of Baricitinib and FTI on FPLD2 and MADB Adipogenesis
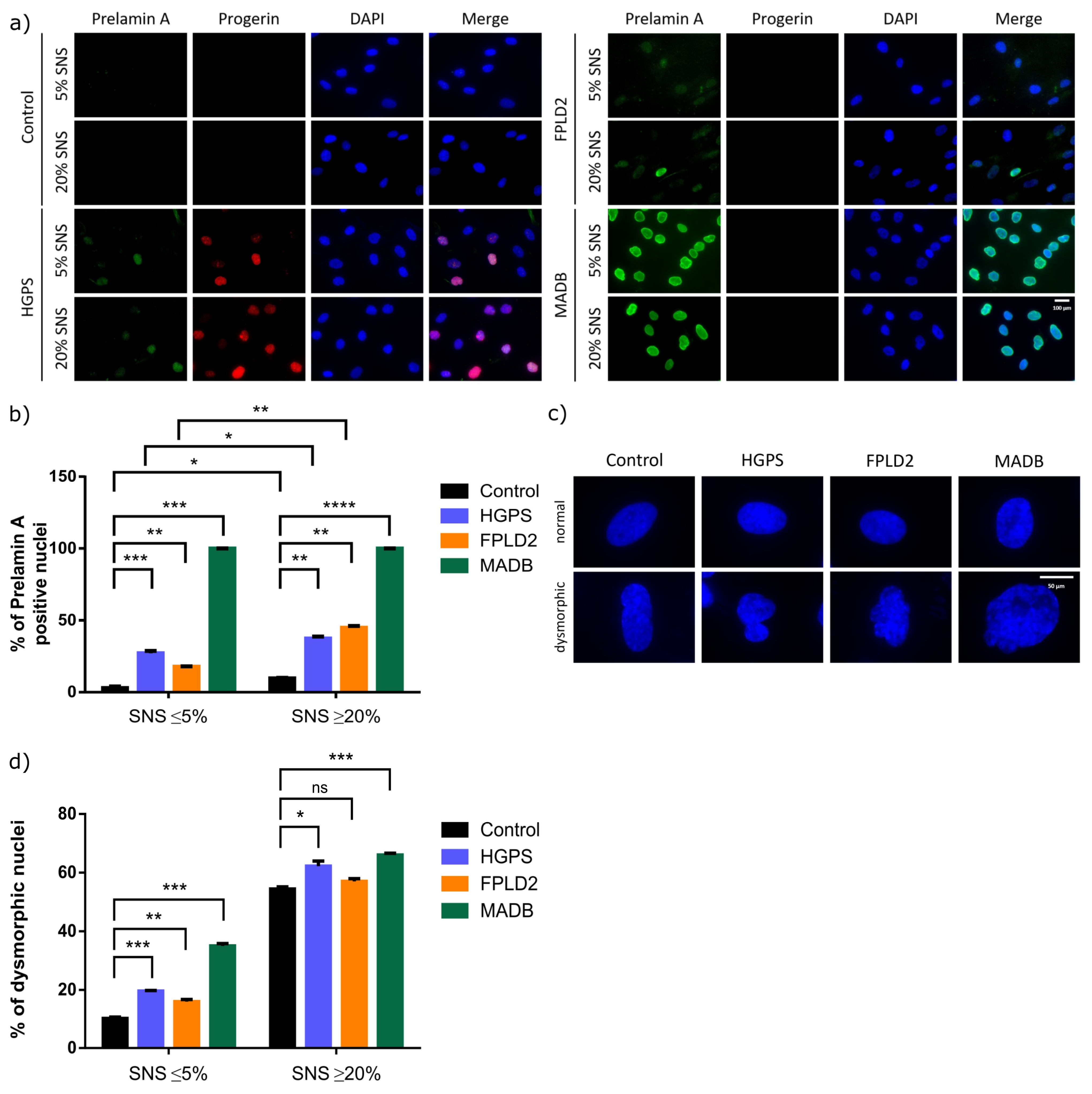
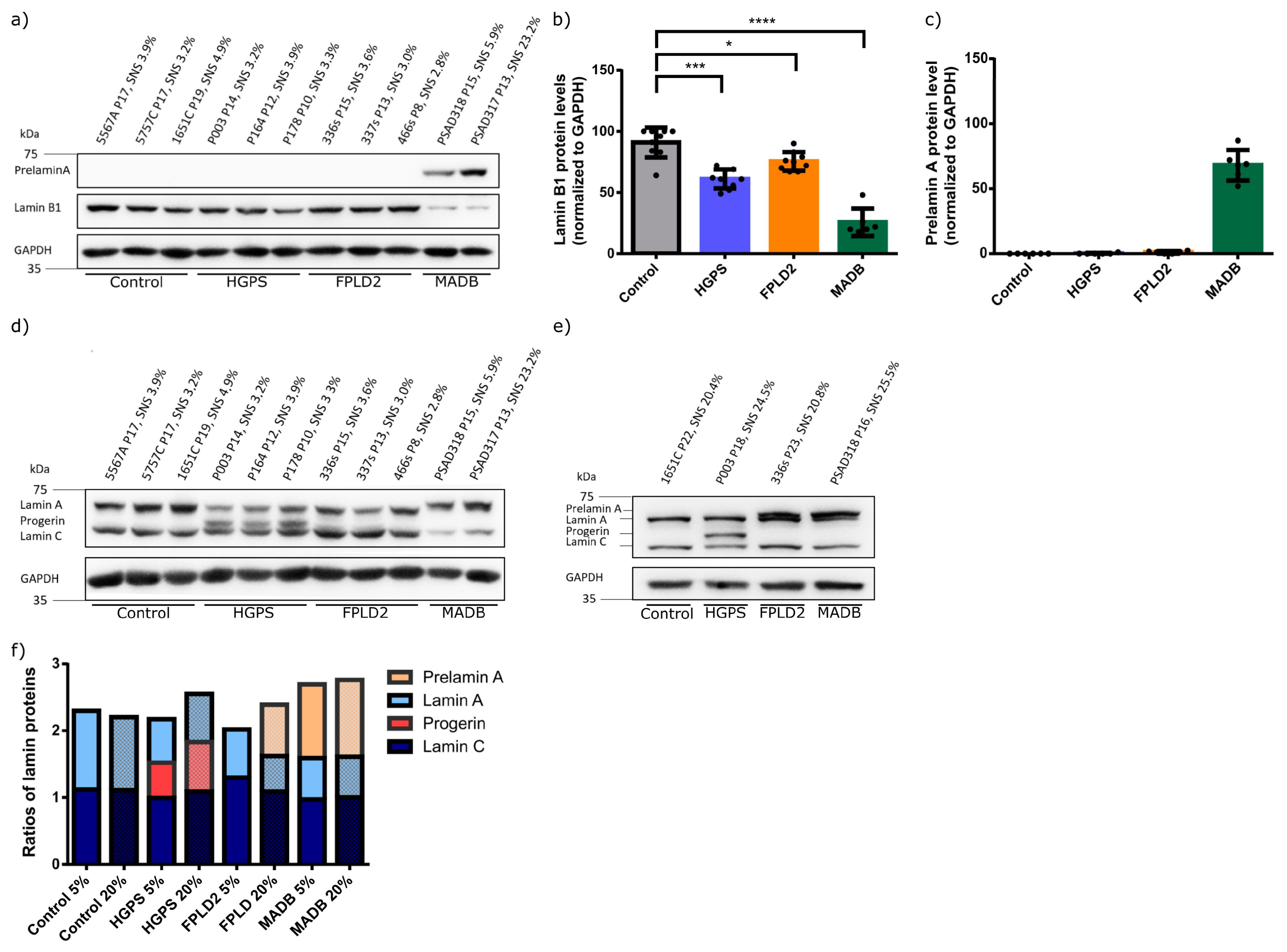
3.4. Lamin Status in HGPS, FFLD2, and MADB Primary Fibroblast Cultures
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gordon, L.B.; Shappell, H.; Massaro, J.; D’Agostino, R.B.; Brazier, J.; Campbell, S.E.; Kleinman, M.E.; Kieran, M.W. Association of Lonafarnib Treatment vs. No Treatment with Mortality Rate in Patients with Hutchinson-Gilford Progeria Syndrome. JAMA 2018, 319, 1687–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N. Engl. J. Med. 2008, 358, 592–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullrich, N.J.; Gordon, L.B. Hutchinson–Gilford progeria syndrome. In Handbook of Clinical Neurology: Neurocutaneous Syndromes; Elsevier: Amsterdam, The Netherlands, 2015; Volume 132, pp. 249–264. [Google Scholar]
- Gordon, L.B.; Brown, T.W.; Collins, F.S. Hutchinson Gilford Progeria Syndrome. In Gene Reviews; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Progeria Research Foundation. PRF by the Numbers. Available online: https://www.progeriaresearch.org/quick-facts (accessed on 24 January 2022).
- Harhouri, K.; Frankel, D.; Bartoli, C.; Roll, P.; Sandre-Giovannoli, A.d.; Lévy, N. An overview of treatment strategies for Hutchinson-Gilford Progeria syndrome. Nucleus 2018, 9, 246–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, S.; Comai, L. Lamin A, farnesylation and aging. Exp. Cell Res. 2012, 318, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Prokocimer, M.; Barkan, R.; Gruenbaum, Y. Hutchinson-Gilford progeria syndrome through the lens of transcription. Aging Cell 2013, 12, 533–543. [Google Scholar] [CrossRef]
- Koblan, L.W.; Erdos, M.R.; Wilson, C.; Cabral, W.A.; Levy, J.M.; Xiong, Z.-M.; Tavarez, U.L.; Davison, L.M.; Gete, Y.G.; Mao, X.; et al. In vivo base editing rescues Hutchinson-Gilford progeria syndrome in mice. Nature 2021, 589, 608–614. [Google Scholar] [CrossRef]
- Glynn, M.W.; Glover, T.W. Incomplete processing of mutant lamin A in Hutchinson-Gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibition. Hum. Mol. Genet. 2005, 14, 2959–2969. [Google Scholar] [CrossRef]
- Petersen, K.F.; Oral, E.A.; Dufour, S.; Befroy, D.; Ariyan, C.; Yu, C.; Cline, G.W.; DePaoli, A.M.; Taylor, S.I.; Gorden, P.; et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J. Clin. Investig. 2002, 109, 1345–1350. [Google Scholar] [CrossRef]
- Akinci, B.; Meral, R.; Oral, E.A. Phenotypic and Genetic Characteristics of Lipodystrophy: Pathophysiology, Metabolic Abnormalities, and Comorbidities. Curr. Diabetes Rep. 2018, 18, 143. [Google Scholar] [CrossRef]
- Hussain, I.; Garg, A. Lipodystrophy Syndromes. Endocrinol. Metab. Clin. N. Am. 2016, 45, 783–797. [Google Scholar] [CrossRef] [Green Version]
- Frigolet, M.E.; Gutiérrez-Aguilar, R. Los colores del tejido adiposo. Gac. Med. Mex. 2020, 156, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Oelkrug, R.; Polymeropoulos, E.T.; Jastroch, M. Brown adipose tissue: Physiological function and evolutionary significance. J. Comp. Physiol. B 2015, 185, 587–606. [Google Scholar] [CrossRef]
- Park, A.; Kim, W.K.; Bae, K.-H. Distinction of white, beige and brown adipocytes derived from mesenchymal stem cells. World J. Stem Cells 2014, 6, 33–42. [Google Scholar] [CrossRef]
- Boon, M.R.; van Marken Lichtenbelt, W.D. Brown adipose tissue: A Human perspective. In Metabolic Control; Herzig, S., Ed.; Springer International Publishing: Cham, Switzerland, 2016; Volume 233, pp. 301–319. [Google Scholar]
- Saely, C.H.; Geiger, K.; Drexel, H. Brown versus white adipose tissue: A mini-review. Gerontology 2012, 58, 15–23. [Google Scholar] [CrossRef]
- Ibrahim, M.M. Subcutaneous and visceral adipose tissue: Structural and functional differences. Obes. Rev. 2010, 11, 11–18. [Google Scholar] [CrossRef]
- Arner, P. Regional adipocity in man. J. Endocrinol. 1997, 155, 191–192. [Google Scholar] [CrossRef]
- Heinonen, S.; Jokinen, R.; Rissanen, A.; Pietiläinen, K.H. White adipose tissue mitochondrial metabolism in health and in obesity. Obes. Rev. 2020, 21, e12958. [Google Scholar] [CrossRef]
- Brown, R.J.; Araujo-Vilar, D.; Cheung, P.T.; Dunger, D.; Garg, A.; Jack, M.; Mungai, L.; Oral, E.A.; Patni, N.; Rother, K.I.; et al. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J. Clin. Endocrinol. Metab. 2016, 101, 4500–4511. [Google Scholar] [CrossRef]
- Hennekam, R.C.M. Hutchinson-Gilford progeria syndrome: Review of the phenotype. Am. J. Med. Genet. Part A 2006, 140, 2603–2624. [Google Scholar] [CrossRef] [Green Version]
- Friesen, M.; Cowan, C.A. FPLD2 LMNA mutation R482W dysregulates iPSC-derived adipocyte function and lipid metabolism. Biochem. Biophys. Res. Commun. 2018, 495, 254–260. [Google Scholar] [CrossRef]
- Hitzert, M.M.; van der Crabben, S.N.; Baldewsingh, G.; van Amstel, H.K.P.; van den Wijngaard, A.; van Ravenswaaij-Arts, C.M.A.; Zijlmans, C.W.R. Mandibuloacral dysplasia type B (MADB): A cohort of eight patients from Suriname with a homozygous founder mutation in ZMPSTE24 (FACE1), clinical diagnostic criteria and management guidelines. Orphanet J. Rare Dis. 2019, 14, 294. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, F.; Evangelisti, C.; Cenni, V.; Fazio, A.; Paganelli, F.; Martelli, A.M.; Lattanzi, G. The Cutting Edge: The Role of mTOR Signaling in Laminopathies. Int. J. Mol. Sci. 2019, 20, 847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gargiuli, C.; Schena, E.; Mattioli, E.; Columbaro, M.; D’Apice, M.R.; Novelli, G.; Greggi, T.; Lattanzi, G. Lamins and bone disorders: Current understanding and perspectives. Oncotarget 2018, 9, 22817–22831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Hegele, R.A. Nuclear lamin A/C R482Q mutation in canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 2000, 9, 109–112. [Google Scholar] [CrossRef] [Green Version]
- Capanni, C.; Mattioli, E.; Columbaro, M.; Lucarelli, E.; Parnaik, V.K.; Novelli, G.; Wehnert, M.; Cenni, V.; Maraldi, N.M.; Squarzoni, S.; et al. Altered pre-lamin A processing is a common mechanism leading to lipodystrophy. Hum. Mol. Genet. 2005, 14, 1489–1502. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, C.; Columbaro, M.; Schena, E.; Prencipe, S.; Andrenacci, D.; Iozzo, P.; Guzzardi, M.A.; Capanni, C.; Mattioli, E.; Loi, M.; et al. Altered adipocyte differentiation and unbalanced autophagy in type 2 Familial Partial Lipodystrophy: An in vitro and in vivo study of adipose tissue browning. Exp. Mol. Med. 2019, 51, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Navarro, C.L.; Esteves-Vieira, V.; Courrier, S.; Boyer, A.; Duong Nguyen, T.; Huong, L.T.T.; Meinke, P.; Schröder, W.; Cormier-Daire, V.; Sznajer, Y.; et al. New ZMPSTE24 (FACE1) mutations in patients affected with restrictive dermopathy or related progeroid syndromes and mutation update. Eur. J. Hum. Genet. 2014, 22, 1002–1011. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.S.; Ikram, S.; Bibi, N.; Mir, A. Hutchinson-Gilford Progeria Syndrome: A Premature Aging Disease. Mol. Neurobiol. 2018, 55, 4417–4427. [Google Scholar] [CrossRef]
- Gonzalo, S.; Kreienkamp, R.; Askjaer, P. Hutchinson-Gilford Progeria Syndrome: A premature aging disease caused by LMNA gene mutations. Ageing Res. Rev. 2017, 33, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Osorio, F.G.; Bárcena, C.; Soria-Valles, C.; Ramsay, A.J.; de Carlos, F.; Cobo, J.; Fueyo, A.; Freije, J.M.; López-Otín, C. Nuclear lamina defects cause ATM-dependent NF-κB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012, 26, 2311–2324. [Google Scholar] [CrossRef] [Green Version]
- Porter, L.J.; Holt, M.R.; Soong, D.; Shanahan, C.M.; Warren, D.T. Prelamin A Accumulation Attenuates Rac1 Activity and Increases the Intrinsic Migrational Persistence of Aged Vascular Smooth Muscle Cells. Cells 2016, 5, 41. [Google Scholar] [CrossRef] [Green Version]
- Regulski, M.J. Cellular Senescence: What, Why, and How. Wounds 2017, 29, 168–174. [Google Scholar]
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes. Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [Green Version]
- Uyar, B.; Palmer, D.; Kowald, A.; Murua Escobar, H.; Barrantes, I.; Möller, S.; Akalin, A.; Fuellen, G. Single-cell analyses of aging, inflammation and senescence. Ageing Res. Rev. 2020, 64, 101156. [Google Scholar] [CrossRef]
- Squarzoni, S.; Schena, E.; Sabatelli, P.; Mattioli, E.; Capanni, C.; Cenni, V.; D’Apice, M.R.; Andrenacci, D.; Sarli, G.; Pellegrino, V.; et al. Interleukin-6 neutralization ameliorates symptoms in prematurely aged mice. Aging Cell 2021, 20, e13285. [Google Scholar] [CrossRef]
- Revêchon, G.; Viceconte, N.; McKenna, T.; Sola Carvajal, A.; Vrtačnik, P.; Stenvinkel, P.; Lundgren, T.; Hultenby, K.; Franco, I.; Eriksson, M. Rare progerin-expressing preadipocytes and adipocytes contribute to tissue depletion over time. Sci. Rep. 2017, 7, 4405. [Google Scholar] [CrossRef] [Green Version]
- Osorio, F.G.; Navarro, C.L.; Cadiñanos, J.; López-Mejía, I.C.; Quirós, P.M.; Bartoli, C.; Rivera, J.; Tazi, J.; Guzmán, G.; Varela, I.; et al. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci. Transl. Med. 2011, 3, 106ra107. [Google Scholar] [CrossRef]
- Kang, S.M.; Yoon, M.H.; Ahn, J.; Kim, J.E.; Kim, S.Y.; Kang, S.Y.; Joo, J.; Park, S.; Cho, J.H.; Woo, T.G.; et al. Progerinin, an optimized progerin-lamin A binding inhibitor, ameliorates premature senescence phenotypes of Hutchinson-Gilford progeria syndrome. Commun. Biol. 2021, 4, 5. [Google Scholar] [CrossRef]
- Gordon, L.B.; Kleinman, M.E.; Miller, D.T.; Neuberg, D.S.; Giobbie-Hurder, A.; Gerhard-Herman, M.; Smoot, L.B.; Gordon, C.M.; Cleveland, R.; Snyder, B.D.; et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2012, 109, 16666–16671. [Google Scholar] [CrossRef] [Green Version]
- Capell, B.C.; Erdos, M.R.; Madigan, J.P.; Fiordalisi, J.J.; Varga, R.; Conneely, K.N.; Gordon, L.B.; Der, C.J.; Cox, A.D.; Collins, F.S. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 12879–12884. [Google Scholar] [CrossRef] [Green Version]
- Fong, L.G.; Frost, D.; Meta, M.; Qiao, X.; Yang, S.H.; Coffinier, C.; Young, S.G. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science 2006, 311, 1621–1623. [Google Scholar] [CrossRef]
- Dhillon, S. Lonafarnib: First Approval. Drugs 2021, 81, 283–289. [Google Scholar] [CrossRef]
- Arnold, R.; Vehns, E.; Randl, H.; Djabali, K. Baricitinib, a JAK-STAT Inhibitor, Reduces the Cellular Toxicity of the Farnesyltransferase Inhibitor Lonafarnib in Progeria Cells. Int. J. Mol. Sci. 2021, 22, 7474. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Arnold, R.; Henriques, G.; Djabali, K. Inhibition of JAK-STAT Signaling with Baricitinib Reduces Inflammation and Improves Cellular Homeostasis in Progeria Cells. Cells 2019, 8, 1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogul, A.; Corsi, K.; McAuliffe, L. Baricitinib: The Second FDA-Approved JAK Inhibitor for the Treatment of Rheumatoid Arthritis. Ann. Pharmacother. 2019, 53, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yang, L.; Zeng, Z.; Feng, Y.; Wang, X.; Wu, X.; Luo, H.; Zhang, J.; Zhang, M.; Pakvasa, M.; et al. Leptin Potentiates BMP9-Induced Osteogenic Differentiation of Mesenchymal Stem Cells Through the Activation of JAK/STAT Signaling. Stem Cells Dev. 2020, 29, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Burrell, J.A.; Boudreau, A.; Stephens, J.M. Latest advances in STAT signaling and function in adipocytes. Clin. Sci. 2020, 134, 629–639. [Google Scholar] [CrossRef] [Green Version]
- McGillicuddy, F.C.; Chiquoine, E.H.; Hinkle, C.C.; Kim, R.J.; Shah, R.; Roche, H.M.; Smyth, E.M.; Reilly, M.P. Interferon gamma attenuates insulin signaling, lipid storage, and differentiation in human adipocytes via activation of the JAK/STAT pathway. J. Biol. Chem. 2009, 284, 31936–31944. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-C.; Sia, K.-C.; Chang, J.-F.; Lin, C.-M.; Yang, C.-M.; Huang, K.-Y.; Lin, W.-N. Lipopolysaccharide promoted proliferation and adipogenesis of preadipocytes through JAK/STAT and AMPK-regulated cPLA2 expression. Int. J. Med. Sci. 2019, 16, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Kang, X.; Yang, M.-Y.; Shi, Y.-X.; Xie, M.-M.; Zhu, M.; Zheng, X.-L.; Zhang, C.-K.; Ge, Z.-L.; Bian, X.-T.; Lv, J.-T.; et al. Interleukin-15 facilitates muscle regeneration through modulation of fibro/adipogenic progenitors. Cell Commun. Signal. 2018, 16, 42. [Google Scholar] [CrossRef] [Green Version]
- Budel, L.; Djabali, K. Rapid isolation and expansion of skin-derived precursor cells from human primary fibroblast cultures. Biol. Open 2017, 6, 1745–1755. [Google Scholar] [CrossRef] [Green Version]
- Najdi, F.; Krüger, P.; Djabali, K. Impact of Progerin Expression on Adipogenesis in Hutchinson-Gilford Progeria Skin-Derived Precursor Cells. Cells 2021, 10, 1598. [Google Scholar] [CrossRef]
- Toma, J.G.; McKenzie, I.A.; Bagli, D.; Miller, F.D. Isolation and characterization of multipotent skin-derived precursors from human skin. Stem Cells 2005, 23, 727–737. [Google Scholar] [CrossRef]
- Wenzel, V.; Roedl, D.; Gabriel, D.; Gordon, L.B.; Herlyn, M.; Schneider, R.; Ring, J.; Djabali, K. Naïve adult stem cells from patients with Hutchinson-Gilford progeria syndrome express low levels of progerin in vivo. Biol. Open 2012, 1, 516–526. [Google Scholar] [CrossRef] [Green Version]
- Bataille, A.; Leschiera, R.; L’Hérondelle, K.; Pennec, J.-P.; Le Goux, N.; Mignen, O.; Sakka, M.; Plée-Gautier, E.; Brun, C.; Oddos, T.; et al. In Vitro Differentiation of Human Skin-Derived Cells into Functional Sensory Neurons-Like. Cells 2020, 9, 1000. [Google Scholar] [CrossRef] [Green Version]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]
- McClintock, D.; Ratner, D.; Lokuge, M.; Owens, D.M.; Gordon, L.B.; Collins, F.S.; Djabali, K. The mutant form of lamin A that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PLoS ONE 2007, 2, e1269. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- de Leeuw, R.; Gruenbaum, Y.; Medalia, O. Nuclear Lamins: Thin Filaments with Major Functions. Trends Cell Biol. 2018, 28, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, N.J.; Kieran, M.W.; Miller, D.T.; Gordon, L.B.; Cho, Y.J.; Silvera, V.M.; Giobbie-Hurder, A.; Neuberg, D.; Kleinman, M.E. Neurologic features of Hutchinson-Gilford progeria syndrome after lonafarnib treatment. Neurology 2013, 81, 427–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talukder, P.; Saha, A.; Roy, S.; Ghosh, G.; Dutta Roy, D.; Barua, S. Progeria-a Rare Genetic Condition with Accelerated Ageing Process. Appl. Biochem. Biotechnol. 2022, 195, 2587–2596. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Y.; Zhou, Z.; Wang, R.; Fu, H.; Khuri, F.R. The farnesyltransferase inhibitor Lonafarnib induces growth arrest or apoptosis of human lung cancer cells without downregulation of Akt. Cancer Biol. Ther. 2004, 3, 1092–1098; discussion 1099–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toth, J.I.; Yang, S.H.; Qiao, X.; Beigneux, A.P.; Gelb, M.H.; Moulson, C.L.; Miner, J.H.; Young, S.G.; Fong, L.G. Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc. Natl. Acad. Sci. USA 2005, 102, 12873–12878. [Google Scholar] [CrossRef] [Green Version]
- Verstraeten, V.L.; Peckham, L.A.; Olive, M.; Capell, B.C.; Collins, F.S.; Nabel, E.G.; Young, S.G.; Fong, L.G.; Lammerding, J. Protein farnesylation inhibitors cause donut-shaped cell nuclei attributable to a centrosome separation defect. Proc. Natl. Acad. Sci. USA 2011, 108, 4997–5002. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ostlund, C.; Choi, J.C.; Swayne, T.C.; Gundersen, G.G.; Worman, H.J. Blocking farnesylation of the prelamin A variant in Hutchinson-Gilford progeria syndrome alters the distribution of A-type lamins. Nucleus 2012, 3, 452–462. [Google Scholar] [CrossRef] [Green Version]
- Adam, S.A.; Butin-Israeli, V.; Cleland, M.M.; Shimi, T.; Goldman, R.D. Disruption of lamin B1 and lamin B2 processing and localization by farnesyltransferase inhibitors. Nucleus 2013, 4, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Blondel, S.; Egesipe, A.L.; Picardi, P.; Jaskowiak, A.L.; Notarnicola, M.; Ragot, J.; Tournois, J.; Le Corf, A.; Brinon, B.; Poydenot, P.; et al. Drug screening on Hutchinson Gilford progeria pluripotent stem cells reveals aminopyrimidines as new modulators of farnesylation. Cell Death Dis. 2016, 7, e2105. [Google Scholar] [CrossRef] [Green Version]
- Clements, C.S.; Bikkul, M.U.; Ofosu, W.; Eskiw, C.; Tree, D.; Makarov, E.; Kill, I.R.; Bridger, J.M. Presence and distribution of progerin in HGPS cells is ameliorated by drugs that impact on the mevalonate and mTOR pathways. Biogerontology 2019, 20, 337–358. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.Y.; Chen, X.Y.; Zhang, Z.J.; Kang, Y.; Liao, W.M.; Yu, W.H.; Xiang, A.P. Expression patterns of transcription factor PPARγ and C/EBP family members during in vitro adipogenesis of human bone marrow mesenchymal stem cells. Cell Biol. Int. 2015, 39, 457–465. [Google Scholar] [CrossRef]
- Rivas, D.; Akter, R.; Duque, G. Inhibition of Protein Farnesylation Arrests Adipogenesis and Affects PPARgamma Expression and Activation in Differentiating Mesenchymal Stem Cells. PPAR Res. 2007, 2007, 81654. [Google Scholar] [CrossRef] [Green Version]
- Ponnusamy, A.; Sinha, S.; Hyde, G.D.; Borland, S.J.; Taylor, R.F.; Pond, E.; Eyre, H.J.; Inkson, C.A.; Gilmore, A.; Ashton, N.; et al. FTI-277 inhibits smooth muscle cell calcification by up-regulating PI3K/Akt signaling and inhibiting apoptosis. PLoS ONE 2018, 13, e0196232. [Google Scholar] [CrossRef] [Green Version]
- Sebti, S.M.; Hamilton, A.D. Farnesyltransferase and geranylgeranyltransferase I inhibitors in cancer therapy: Important mechanistic and bench to bedside issues. Expert Opin. Investig. Drugs 2000, 9, 2767–2782. [Google Scholar] [CrossRef]
- Cox, A.D.; Der, C.J. Farnesyltransferase inhibitors and cancer treatment: Targeting simply Ras? Biochim. Biophys. Acta 1997, 1333, F51–F71. [Google Scholar] [CrossRef]
- Basso, A.D.; Mirza, A.; Liu, G.; Long, B.J.; Bishop, W.R.; Kirschmeier, P. The farnesyl transferase inhibitor (FTI) SCH66336 (lonafarnib) inhibits Rheb farnesylation and mTOR signaling. Role in FTI enhancement of taxane and tamoxifen anti-tumor activity. J. Biol. Chem. 2005, 280, 31101–31108. [Google Scholar] [CrossRef] [Green Version]
- Morgillo, F.; Lee, H.Y. Lonafarnib in cancer therapy. Expert Opin. Investig. Drugs 2006, 15, 709–719. [Google Scholar] [CrossRef]
- Roy, A.; Ghosh, A.; Jana, A.; Liu, X.; Brahmachari, S.; Gendelman, H.E.; Pahan, K. Sodium phenylbutyrate controls neuroinflammatory and antioxidant activities and protects dopaminergic neurons in mouse models of Parkinson’s disease. PLoS ONE 2012, 7, e38113. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; She, M.; Xu, Z.X.; Sun, L.; Yeung, S.C. Farnesyltransferase inhibitors induce DNA damage via reactive oxygen species in human cancer cells. Cancer Res. 2005, 65, 3671–3681. [Google Scholar] [CrossRef] [Green Version]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell. Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef]
- Gordon, L.B.; Massaro, J.; D’Agostino, R.B., Sr.; Campbell, S.E.; Brazier, J.; Brown, W.T.; Kleinman, M.E.; Kieran, M.W. Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation 2014, 130, 27–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caron, M.; Auclair, M.; Donadille, B.; Béréziat, V.; Guerci, B.; Laville, M.; Narbonne, H.; Bodemer, C.; Lascols, O.; Capeau, J.; et al. Human lipodystrophies linked to mutations in A-type lamins and to HIV protease inhibitor therapy are both associated with prelamin A accumulation, oxidative stress and premature cellular senescence. Cell Death Differ. 2007, 14, 1759–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maynard, S.; Hall, A.; Galanos, P.; Rizza, S.; Yamamoto, T.; Gram, H.H.; Munk, S.H.N.; Shoaib, M.; Sørensen, C.S.; Bohr, V.A.; et al. Lamin A/C impairments cause mitochondrial dysfunction by attenuating PGC1α and the NAMPT-NAD+ pathway. Nucleic Acids Res. 2022, 50, 9948–9965. [Google Scholar] [CrossRef] [PubMed]
- Peinado, J.R.; Quirós, P.M.; Pulido, M.R.; Mariño, G.; Martínez-Chantar, M.L.; Vázquez-Martínez, R.; Freije, J.M.; López-Otín, C.; Malagón, M.M. Proteomic profiling of adipose tissue from Zmpste24-/- mice, a model of lipodystrophy and premature aging, reveals major changes in mitochondrial function and vimentin processing. Mol. Cell. Proteom. 2011, 10, M111.008094. [Google Scholar] [CrossRef] [Green Version]
- Elouej, S.; Harhouri, K.; Le Mao, M.; Baujat, G.; Nampoothiri, S.; Kayserili, H.; Menabawy, N.A.; Selim, L.; Paneque, A.L.; Kubisch, C.; et al. Loss of MTX2 causes mandibuloacral dysplasia and links mitochondrial dysfunction to altered nuclear morphology. Nat. Commun. 2020, 11, 4589. [Google Scholar] [CrossRef]
- Heizer, P.J.; Yang, Y.; Tu, Y.; Kim, P.H.; Chen, N.Y.; Hu, Y.; Yoshinaga, Y.; de Jong, P.J.; Vergnes, L.; Morales, J.E.; et al. Deficiency in ZMPSTE24 and resulting farnesyl-prelamin A accumulation only modestly affect mouse adipose tissue stores. J. Lipid Res. 2020, 61, 413–421. [Google Scholar] [CrossRef]
- Vigouroux, C.; Auclair, M.; Dubosclard, E.; Pouchelet, M.; Capeau, J.; Courvalin, J.C.; Buendia, B. Nuclear envelope disorganization in fibroblasts from lipodystrophic patients with heterozygous R482Q/W mutations in the lamin A/C gene. J. Cell. Sci. 2001, 114, 4459–4468. [Google Scholar] [CrossRef]
- Lattanzi, G.; Columbaro, M.; Mattioli, E.; Cenni, V.; Camozzi, D.; Wehnert, M.; Santi, S.; Riccio, M.; Del Coco, R.; Maraldi, N.M.; et al. Pre-Lamin A processing is linked to heterochromatin organization. J. Cell. Biochem. 2007, 102, 1149–1159. [Google Scholar] [CrossRef]
- Osorio, F.G.; Varela, I.; Lara, E.; Puente, X.S.; Espada, J.; Santoro, R.; Freije, J.M.; Fraga, M.F.; López-Otín, C. Nuclear envelope alterations generate an aging-like epigenetic pattern in mice deficient in Zmpste24 metalloprotease. Aging Cell 2010, 9, 947–957. [Google Scholar] [CrossRef] [Green Version]
- Ragnauth, C.D.; Warren, D.T.; Liu, Y.; McNair, R.; Tajsic, T.; Figg, N.; Shroff, R.; Skepper, J.; Shanahan, C.M. Prelamin A acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation 2010, 121, 2200–2210. [Google Scholar] [CrossRef] [Green Version]
- Cenni, V.; Capanni, C.; Mattioli, E.; Columbaro, M.; Wehnert, M.; Ortolani, M.; Fini, M.; Novelli, G.; Bertacchini, J.; Maraldi, N.M.; et al. Rapamycin treatment of Mandibuloacral dysplasia cells rescues localization of chromatin-associated proteins and cell cycle dynamics. Aging 2014, 6, 755–770. [Google Scholar] [CrossRef] [Green Version]
- Dreesen, O.; Ong, P.F.; Chojnowski, A.; Colman, A. The contrasting roles of lamin B1 in cellular aging and human disease. Nucleus 2013, 4, 283–290. [Google Scholar] [CrossRef] [Green Version]
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; Hamanaka, R.B.; Goldman, A.E.; Lucas, C.A.; Shumaker, D.K.; Kosak, S.T.; Chandel, N.S.; Goldman, R.D. The role of nuclear lamin B1 in cell proliferation and senescence. Genes. Dev. 2011, 25, 2579–2593. [Google Scholar] [CrossRef] [Green Version]
- Turgay, Y.; Eibauer, M.; Goldman, A.E.; Shimi, T.; Khayat, M.; Ben-Harush, K.; Dubrovsky-Gaupp, A.; Sapra, K.T.; Goldman, R.D.; Medalia, O. The molecular architecture of lamins in somatic cells. Nature 2017, 543, 261–264. [Google Scholar] [CrossRef] [Green Version]
- Evangelisti, C.; Rusciano, I.; Mongiorgi, S.; Ramazzotti, G.; Lattanzi, G.; Manzoli, L.; Cocco, L.; Ratti, S. The wide and growing range of lamin B-related diseases: From laminopathies to cancer. Cell. Mol. Life Sci. 2022, 79, 126. [Google Scholar] [CrossRef]
- Janota, C.S.; Calero-Cuenca, F.J.; Gomes, E.R. The role of the cell nucleus in mechanotransduction. Curr. Opin. Cell Biol. 2020, 63, 204–211. [Google Scholar] [CrossRef]
- Donnaloja, F.; Carnevali, F.; Jacchetti, E.; Raimondi, M.T. Lamin A/C Mechanotransduction in Laminopathies. Cells 2020, 9, 1306. [Google Scholar] [CrossRef]
- Maraldi, N.M.; Capanni, C.; Cenni, V.; Fini, M.; Lattanzi, G. Laminopathies and lamin-associated signaling pathways. J. Cell. Biochem. 2011, 112, 979–992. [Google Scholar] [CrossRef]
- Lelliott, C.J.; Logie, L.; Sewter, C.P.; Berger, D.; Jani, P.; Blows, F.; O’Rahilly, S.; Vidal-Puig, A. Lamin expression in human adipose cells in relation to anatomical site and differentiation state. J. Clin. Endocrinol. Metab. 2002, 87, 728–734. [Google Scholar] [CrossRef]
- Camps, J.; Erdos, M.R.; Ried, T. The role of lamin B1 for the maintenance of nuclear structure and function. Nucleus 2015, 6, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Ivanovska, I.; Vashisth, M.; Discher, D.E. Nuclear mechanoprotection: From tissue atlases as blueprints to distinctive regulation of nuclear lamins. APL Bioeng. 2022, 6, 021504. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hartinger, R.; Lederer, E.-M.; Schena, E.; Lattanzi, G.; Djabali, K. Impact of Combined Baricitinib and FTI Treatment on Adipogenesis in Hutchinson–Gilford Progeria Syndrome and Other Lipodystrophic Laminopathies. Cells 2023, 12, 1350. https://doi.org/10.3390/cells12101350
Hartinger R, Lederer E-M, Schena E, Lattanzi G, Djabali K. Impact of Combined Baricitinib and FTI Treatment on Adipogenesis in Hutchinson–Gilford Progeria Syndrome and Other Lipodystrophic Laminopathies. Cells. 2023; 12(10):1350. https://doi.org/10.3390/cells12101350
Chicago/Turabian StyleHartinger, Ramona, Eva-Maria Lederer, Elisa Schena, Giovanna Lattanzi, and Karima Djabali. 2023. "Impact of Combined Baricitinib and FTI Treatment on Adipogenesis in Hutchinson–Gilford Progeria Syndrome and Other Lipodystrophic Laminopathies" Cells 12, no. 10: 1350. https://doi.org/10.3390/cells12101350
APA StyleHartinger, R., Lederer, E. -M., Schena, E., Lattanzi, G., & Djabali, K. (2023). Impact of Combined Baricitinib and FTI Treatment on Adipogenesis in Hutchinson–Gilford Progeria Syndrome and Other Lipodystrophic Laminopathies. Cells, 12(10), 1350. https://doi.org/10.3390/cells12101350