

Co-Stimulation of AGEs and LPS Induces Inflammatory Mediators through PLCγ1/JNK/NF-κB Pathway in MC3T3-E1 Cells

 ,
,
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Culture
2.2. Preparation of AGEs
2.3. Real-Time Polymerase Chain Reaction (Real-Time PCR)
2.4. Enzyme-Linked Immune-Sorbent Assay (ELISA)
2.5. Western Blotting
2.6. Immunofluorescence Localization of NF-κB
2.7. Short Interfering (si)RNA
2.8. Statistical Analysis
3. Results
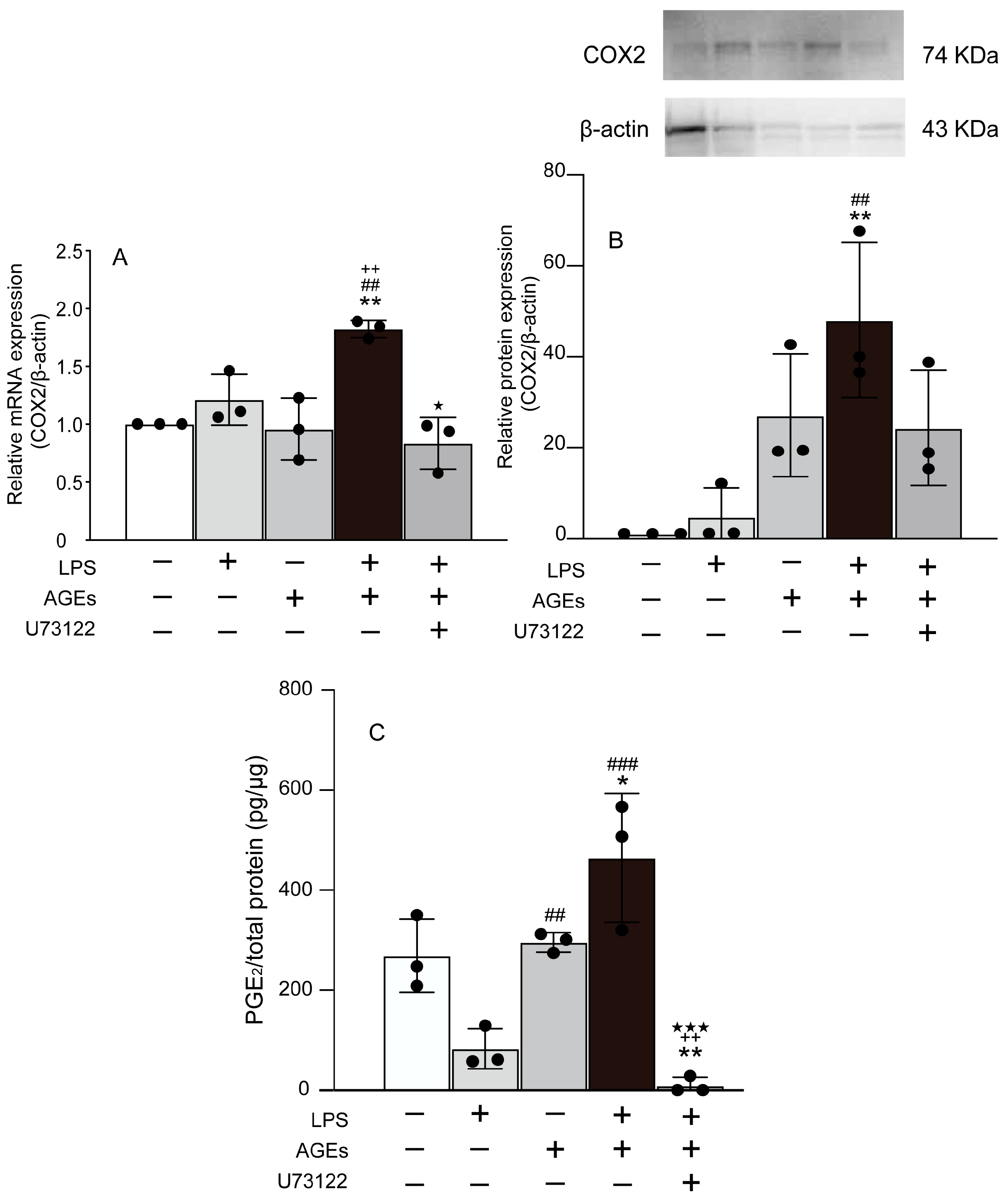
3.1. AGEs+LPS Increased the Expression of COX2 and PGE2 in MC3T3-E1 Cells
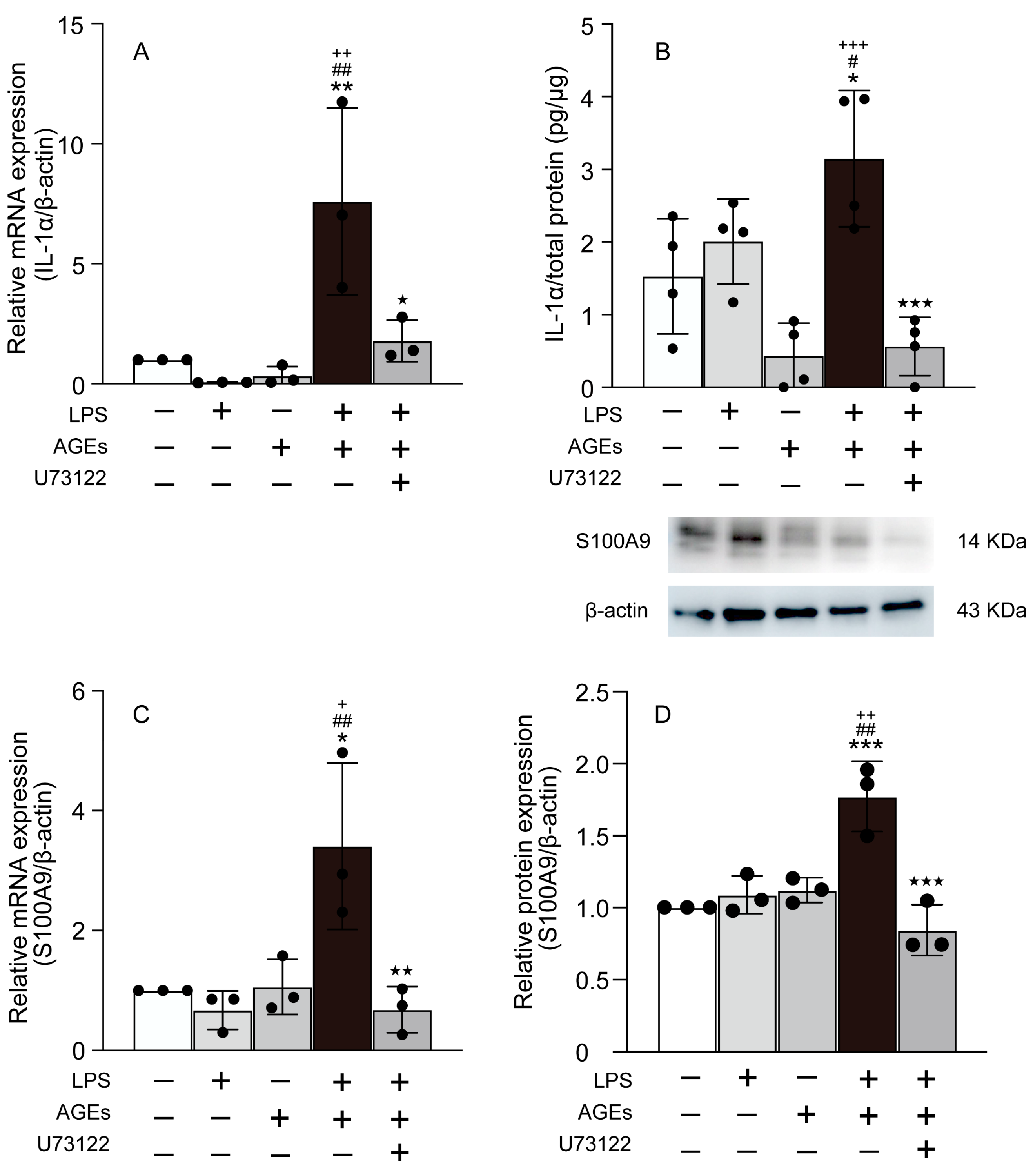
3.2. AGEs+LPS Increased the Expression of IL-1α, and S100A9 in MC3T3-E1 Cells
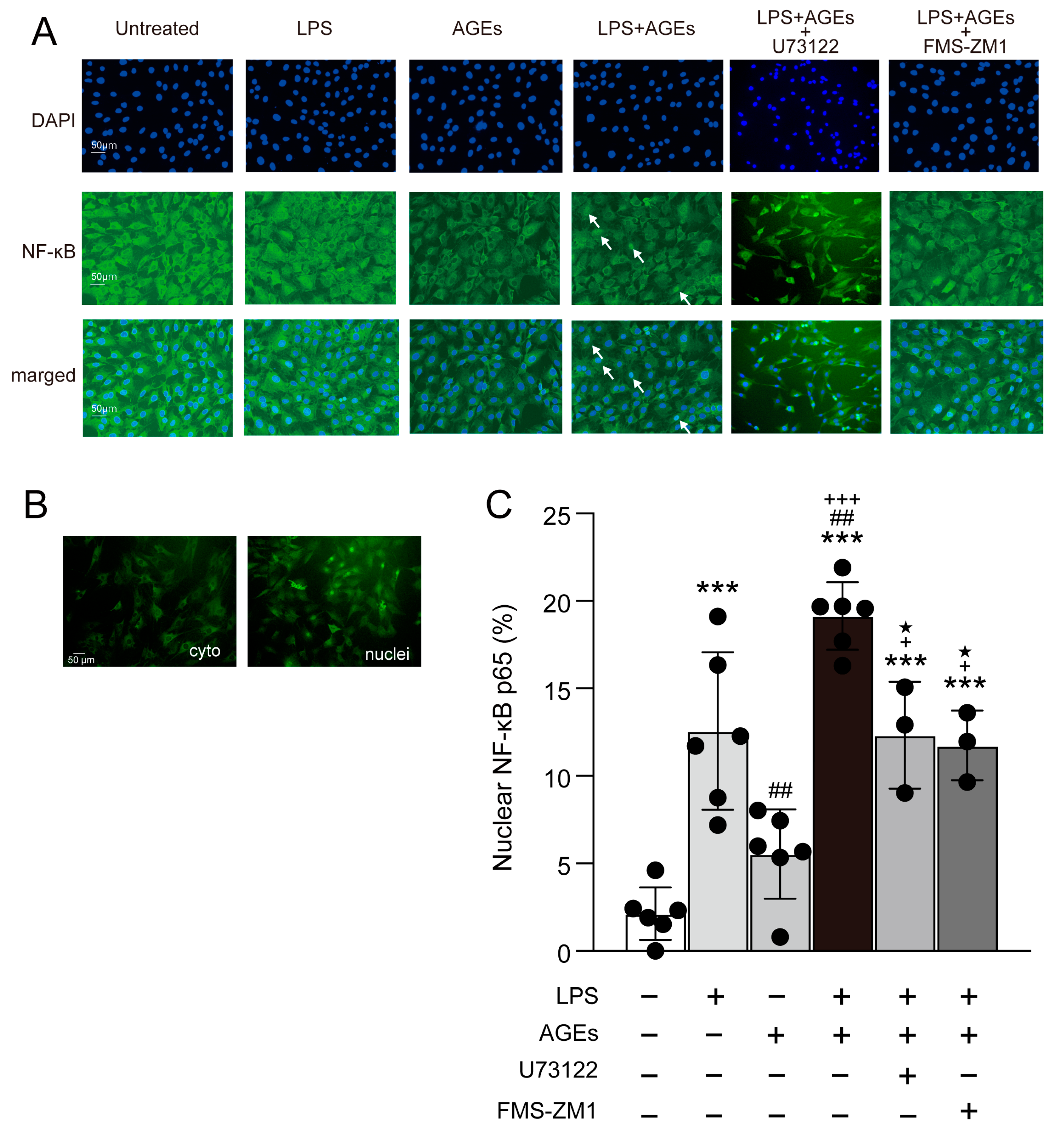
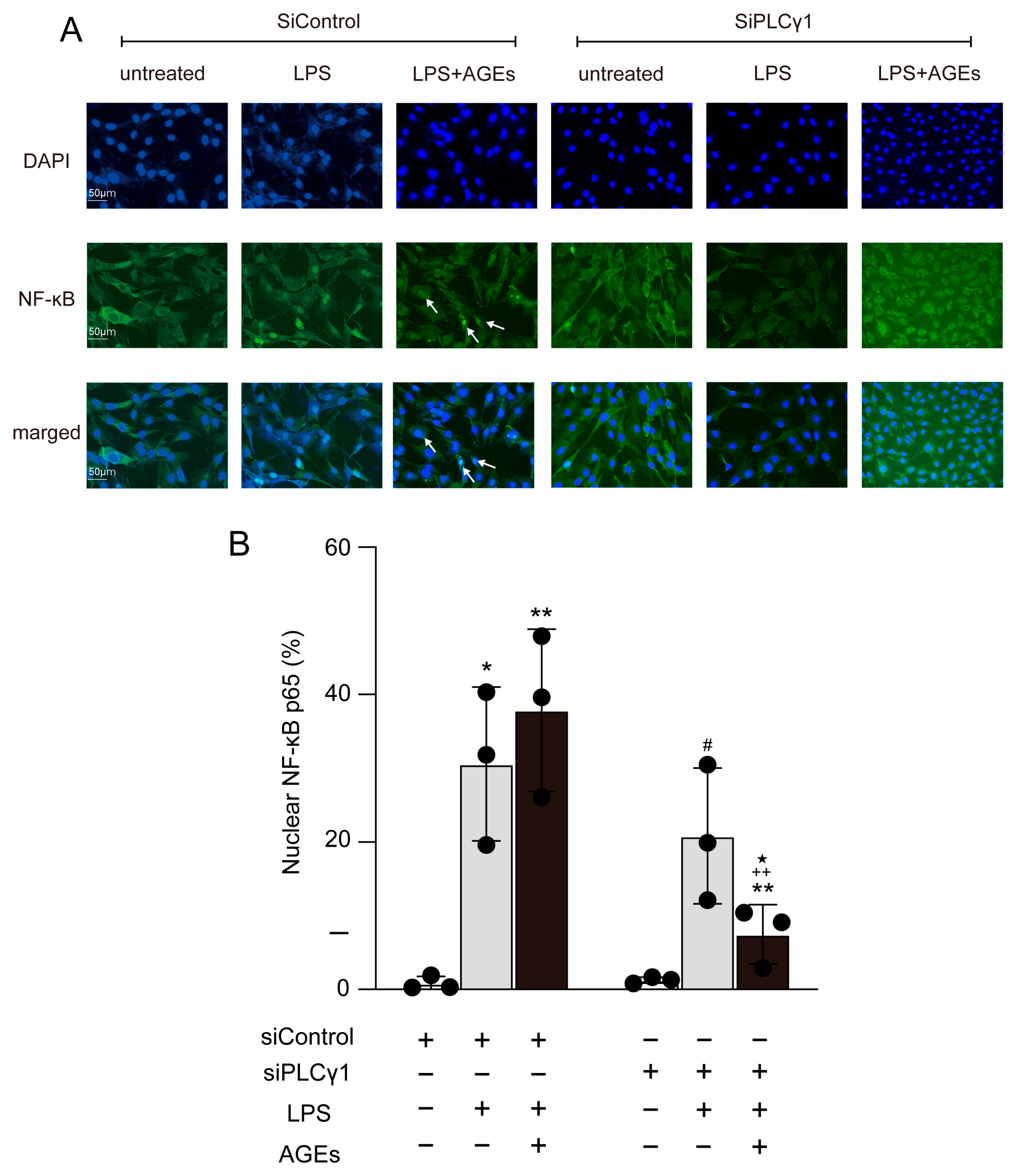
3.3. LPS+AGEs Increase NF-kB Nuclear Localization
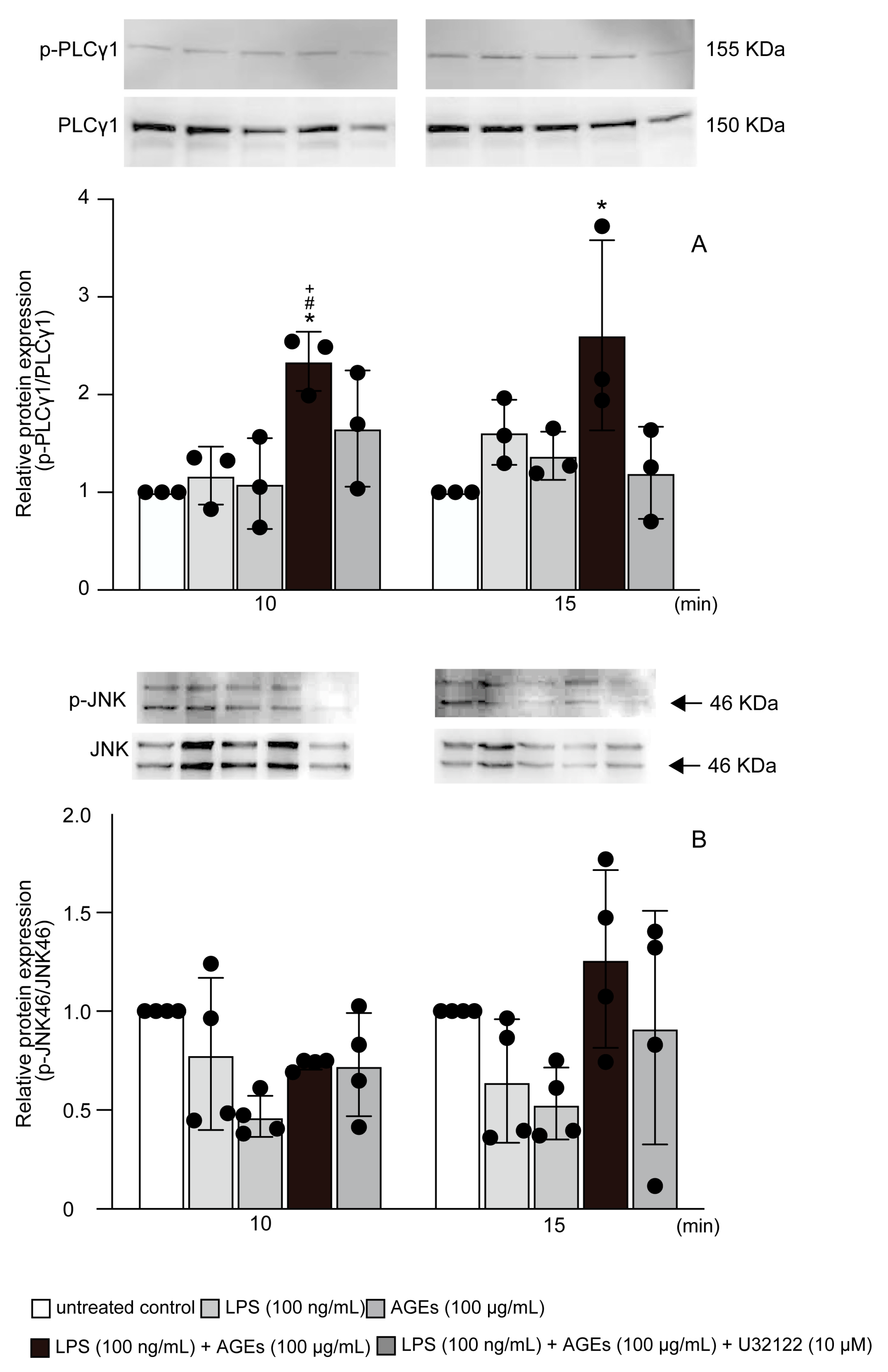
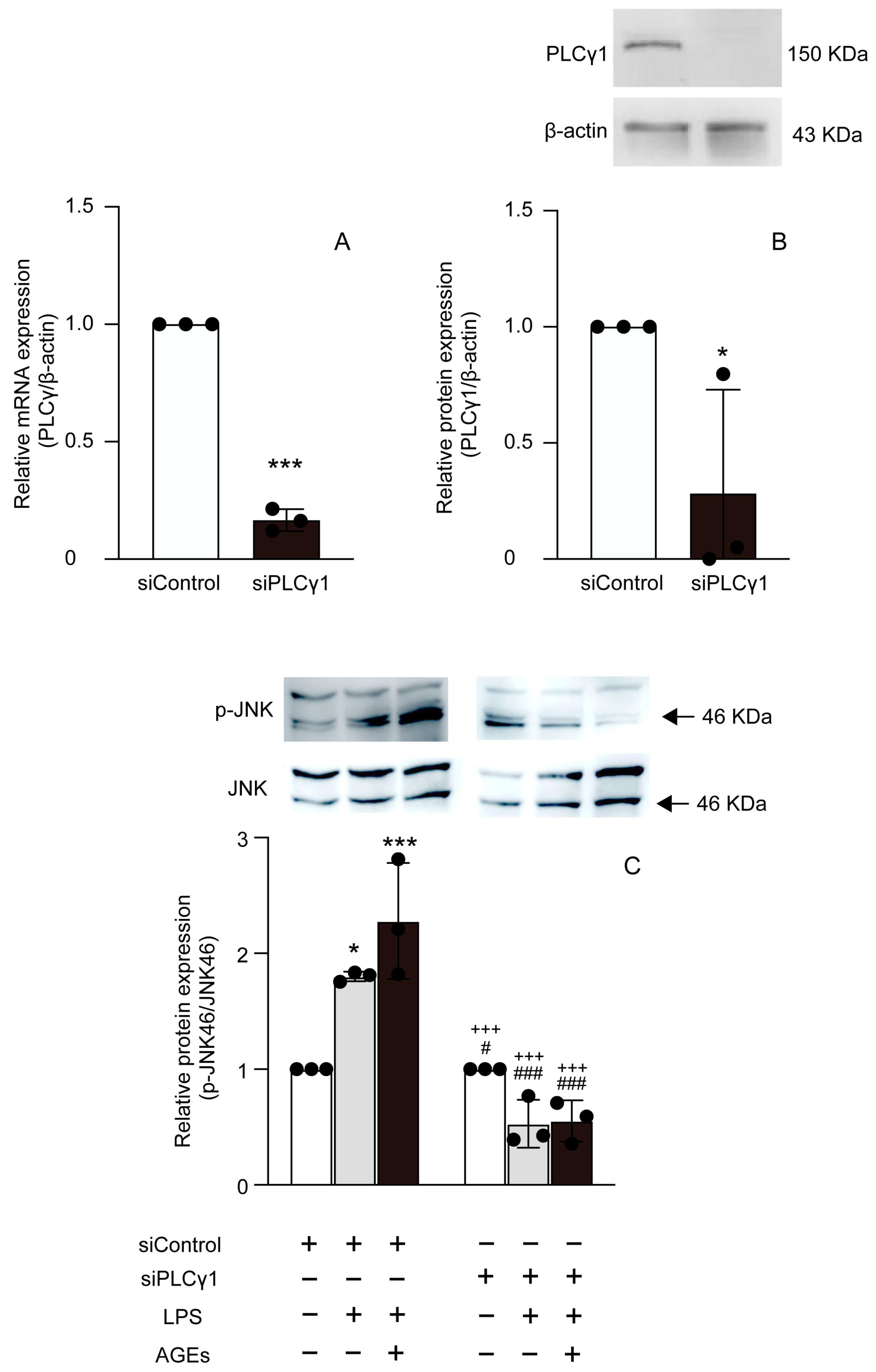
3.4. LPS+AGEs Increased p-PLCγ1 and p-JNK46 in MC3T3-E1 Cells
3.5. AGEs+LPS Increased the Nuclear Translocation of NF-κB Nuclear Localization through the PLCγ1-JNK Pathway
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AGEs | advanced glycation end-products |
| RAGE | receptor for advanced glycation end-products |
| DAMPs | damage-associated molecular pattern molecules |
| HMGB1 | high mobility group 1 or amphoterin |
| RA | rheumatoid arthritis |
| IL-1 | interleukin-1 |
| IL-6 | interleukin-6 |
| IL-8 | interleukin-8 |
| IL-17 | interleukin-17 |
| TNFα | tumor necrosis factor-α |
| RANKL | receptor activator of nuclear factor-kappa B ligand |
| LPS | lipopolysaccharide |
| RA | rheumatoid arthritis |
| PGE2 | prostaglandin E2 |
| COX2 | cyclooxygenase 2 |
| ELISA | enzyme-linked immuno-sorbent assay |
| PLC | phospholipase C |
| p-PLCγ1 | phosphorylated Phospholipase Cγ1 |
| NF-κB | nuclear factor-kappa B |
| JNK: | c-Jun N-terminal kinase |
| p-JNK | phosphorylated c-Jun N-terminal kinase |
| S100A9 | S100 calcium binding protein A9 |
| TLR | toll-like receptor |
| EGF | epidermal growth factor |
References
- Twarda-clapa, A.; Olczak, A.; Białkowska, A.M.; Koziołkiewicz, M. Advanced Glycation End-Products (AGEs): Formation, Chemistry, Classification, Receptors, and Diseases Related to AGEs. Cells 2022, 11, 1312. [Google Scholar] [CrossRef] [PubMed]
- Khalid, M.; Petroianu, G.; Adem, A. Advanced Glycation End Products and Diabetes Mellitus: Mechanisms and Perspectives. Biomolecules 2022, 12, 542. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Iwaki, M.; Shimogaito, N.; Wu, X.; Yamagishi, S.; Takeuchi, M. TAGE (Toxic AGEs) Theory in Diabetic Complications. Curr. Mol. Med. 2006, 6, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Miura, J.; Yamagishi, S.; Uchigata, Y.; Takeichi, M.; Yamamoto, H.; Makita, Z.; Iwamoto, Y. Serum Levels of Non-carboxymethyllysine Advanced Glycation Endproducts are Correlated to Severity of Microvascular Complications in Patients with Type 1 Diabetes. J. Diabetes Complicat. 2003, 17, 16–21. [Google Scholar] [CrossRef]
- Dariya, B.; Nagaraju, G.P. Advanced Glycation End Products in Diabetes, Cancer and Phytochemical Therapy. Drug Discov. Today 2020, 25, 1614–1623. [Google Scholar] [CrossRef]
- Kierdorf, K.; Fritz, G. RAGE Regulation and Signaling in Inflammation and Beyond. J. Leukoc. Biol. 2013, 94, 55–68. [Google Scholar] [CrossRef]
- Hudson, B.I.; Lippman, M.E. Targeting RAGE Signaling in Inflammatory Disease. Annu. Rev. Med. 2018, 69, 349–364. [Google Scholar] [CrossRef]
- Plemmenos, G.; Piperi, C. Pathogenic Molecular Mechanisms in Periodontitis and Peri-Implantitis: Role of Advanced Glycation End Products. Life 2022, 12, 218. [Google Scholar] [CrossRef] [PubMed]
- Keeting, P.E.; Rifas, L.; Harris, S.A.; Colvard, D.S.; Spelsberg, T.C.; Peck, W.A.; Riggs, B.L. Evidence for Interleukin-1β Production by Cultured Normal Human Osteoblast-like Cells. J. Bone Miner. Res. 1991, 6, 827–833. [Google Scholar] [CrossRef]
- Shoji, M.; Tanabe, N.; Mitsui, N.; Tanaka, H.; Suzuki, N.; Takeichi, O.; Sugaya, A.; Maeno, M. Lipopolysaccharide stimulates the production of prostaglandin E2 and the receptor Ep4 in osteoblasts. Life Sci. 2006, 78, 2012–2018. [Google Scholar] [CrossRef]
- Page, R.C. The Role of Inflammatory Mediators in the Pathogenesis of Periodontal Disease. J. Periodontal Res. 1991, 26, 230–242. [Google Scholar] [CrossRef]
- Nagao, M.; Tanabe, N.; Manaka, S.; Naito, M.; Sekino, J.; Takayama, T.; Kawato, T.; Torigoe, G.; Kato, S.; Tsukune, N.; et al. LIPUS Suppressed LPS-Induced IL-1α through the Inhibition of NF-κB Nuclear Translocation via AT1-PLCβ Pathway in MC3T3-E1 Cells. J. Cell. Physiol. 2017, 232, 3337–3346. [Google Scholar] [CrossRef]
- Monu; Agnihotri, P.; Biswas, S. AGE/Non-AGE Glycation: An Important Event in Rheumatoid Arthritis Pathophysiology. Inflammation 2022, 45, 477–496. [Google Scholar] [CrossRef]
- Takeda, M.; Ojima, M.; Yoshioka, H.; Inaba, H.; Kogo, M.; Shizukuishi, S.; Nomura, M.; Amano, A. Relationship of Serum Advanced Glycation End Products with Deterioration of Periodontitis in Type 2 Diabetes Patients. J. Periodontol. 2006, 77, 15–20. [Google Scholar] [CrossRef]
- Akram, Z.; Alqahtani, F.; Alqahtani, M.; Al-Kheraif, A.A.; Javed, F. Levels of Advanced Glycation End Products in Gingival Crevicular Fluid of Chronic Periodontitis Patients with and without Type-2 Diabetes Mellitus. J. Periodontol. 2020, 91, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, K.; Yamaguchi, T.; Tanaka, K.; Notsu, M.; Ogawa, N.; Yano, S.; Sugimoto, T. Advanced Glycation End Products (AGEs), but Not High Glucose, Inhibit the Osteoblastic Differentiation of Mouse Stromal ST2 Cells Through the Suppression of Osterix Expression, and Inhibit Cell Growth and Increasing Cell Apoptosis. Calcif. Tissue Int. 2012, 91, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Cekici, A.; Kantarci, A.; Hasturk, H.; Van Dyke, T.E. Inflammatory and Immune Pathways in the Pathogenesis of Periodontal Disease. Periodontol. 2000 2014, 64, 57–80. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Wang, M.; Wang, P.; Wen, J.; Wang, J.; Cha, S.; Xiao, X.; He, Y.; Shu, R.; Bai, D. LncRNA NEAT1 Ameliorates LPS-induced Inflammation in MG63 Cells by Activating Autophagy and Suppressing the NLRP3 Inflammasome. Int. J. Mol. Med. 2021, 47, 607–620. [Google Scholar] [CrossRef]
- Rhee, S.H. Lipopolysaccharide: Basic Biochemistry, Intracellular Signaling, and Physiological Impacts in the Gut. Intest. Res. 2014, 12, 90–95. [Google Scholar] [CrossRef]
- Smith, W.L. The eicosanoids and their biochemical mechanisms of action. Biochem. J. 1989, 259, 315–324. [Google Scholar] [CrossRef]
- Berenbaum, F. Proinflammatory cytokines, prostaglandins, and the chondrocyte: Mechanisms of intracellular activation. Jt. Bone Spine 2000, 67, 561–564. [Google Scholar] [CrossRef]
- Funk, C.D. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef]
- Park, J.Y.; Pillinger, M.H.; Abramson, S.B. Prostaglandin E2 synthesis and secretion: The role of PGE2 synthases. Clin. Immunol. 2006, 119, 229–240. [Google Scholar] [CrossRef]
- Liu, X.H.; Kirschenbaum, A.; Yao, S.; Levine, A.C. Cross-talk between the interleukin-6 and prostaglandin E2 signaling systems results in enhancement of osteoclastogenesis through effects on the osteoprotegerin/receptor activator of nuclear factor-κB (RANK) ligand/RANK system. Endocrinology 2005, 146, 1991–1998. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C. Overview of the IL-1 Family in Innate Inflammation and Acquired Immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef]
- Hiroshima, Y.; Sakamoto, E.; Yoshida, K.; Abe, K.; Naruishi, K.; Yamamoto, T.; Shinohara, Y.; Kido, J.I.; Geczy, C.L. Advanced Glycation End-Products and Porphyromonas Gingivalis Lipopolysaccharide Increase Calprotectin Expression in Human Gingival Epithelial Cells. J. Cell. Biochem. 2018, 119, 1591–1603. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Su, Q.; Wen, J.; Zhu, K.; Tan, J.; Fu, Q.; Sun, G. S100A9 Induces Nucleus Pulposus Cell Degeneration through Activation of the NF-κB Signaling Pathway. J. Cell. Mol. Med. 2021, 25, 4709–4720. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Firestein, G.S. NF-ΚB: A Key Role in Inflammatory Diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Ben-Neriah, Y. Phosphorylation Meets Ubiquitination: The Control of NF-κB Activity. Annu. Rev. Immunol. 2003, 18, 621–663. [Google Scholar] [CrossRef]
- Mak, T.W.; Yeh, W.C. Signaling for Survival and Apoptosis in the Immune System. Arthritis Res. 2002, 4 (Suppl. S3), S243–S252. [Google Scholar] [CrossRef] [PubMed]
- Komarova, S.V.; Pilkington, M.F.; Weidema, A.F.; Dixon, S.J.; Sims, S.M. RANK Ligand-Induced Elevation of Cytosolic Ca2+ Accelerates Nuclear Translocation of Nuclear Factor Kappa B in Osteoclasts. J. Biol. Chem. 2003, 278, 8286–8293. [Google Scholar] [CrossRef]
- Li, X.; Hua, L.; Deng, F.; Bai, X.; Zeng, W.; Lu, D.; Su, Y.; Luo, S. NF-ΚB and Hsp70 Are Involved in the Phospholipase Cγ1 Signaling Pathway in Colorectal Cancer Cells. Life Sci. 2005, 77, 2794–2803. [Google Scholar] [CrossRef]
- Zeng, X.Z.; Zhang, Y.Y.; Yang, Q.; Wang, S.; Zou, B.H.; Tan, Y.H.; Zou, M.; Liu, S.W.; Li, X.J. Artesunate Attenuates LPS-Induced Osteoclastogenesis by Suppressing TLR4/TRAF6 and PLCγ1-Ca2+-NFATc1 Signaling pathway. Acta Pharmacol. Sin. 2020, 41, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Wang, M.-Q.; Ni, S.-H.; Liu, L.-Y.; You, H.-Y.; Wu, X.-H.; Wang, Y.-J.; Lu, L.; Wei, L.-B. Salidroside Ameliorates Endothelial Inflammation and Oxidative Stress by Regulating the AMPK/NF-κB/NLRP3 Signaling Pathway in AGEs-Induced HUVECs. Eur. J. Pharmacol. 2020, 867, 172797. [Google Scholar] [CrossRef]
- Li, J.-S.; Ji, T.; Su, S.-L.; Zhu, Y.; Chen, X.-L.; Shang, E.-X.; Guo, S.; Qian, D.-W.; Duan, J.-A. Mulberry Leaves Ameliorate Diabetes via Regulating Metabolic Profiling and AGEs/RAGE and P38 MAPK/NF-κB Pathway. J. Ethnopharmacol. 2022, 283, 114713. [Google Scholar] [CrossRef]
- Lai, J.-L.; Liu, Y.-H.; Liu, C.; Qi, M.-P.; Liu, R.-N.; Zhu, X.-F.; Zhou, Q.-G.; Chen, Y.-Y.; Guo, A.-Z.; Hu, C.-M. Indirubin Inhibits LPS-Induced Inflammation via TLR4 Abrogation Mediated by the NF-κB and MAPK Signaling Pathways. Inflammation 2017, 40, 1–12. [Google Scholar] [CrossRef]
- Wu, X.; Mi, Y.; Yang, H.; Hu, A.; Zhang, Q.; Shang, C. The Activation of HMGB1 as a Progression Factor on Inflammation Response in Normal Human Bronchial Epithelial Cells through RAGE/JNK/NF-κB Pathway. Mol. Cell. Biochem. 2013, 380, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Young, D.W.; Gusovsky, F.; Chow, J.C. Cellular Events Mediated by Lipopolysaccharide-Stimulated Toll-like Receptor 4. MD-2 Is Required for Activation of Mitogen-Activated Protein Kinases and Elk-1. J. Biol. Chem. 2000, 275, 20861–20866. [Google Scholar] [CrossRef]
- Cario, E.; Rosenberg, I.M.; Brandwein, S.L.; Beck, P.L.; Reinecker, H.-C.; Podolsky, D.K. Lipopolysaccharide Activates Distinct Signaling Pathways in Intestinal Epithelial Cell Lines Expressing Toll-Like Receptors. J. Immunol. 2000, 164, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.X.; Kirschning, C.J.; Mancinelli, R.; Xu, X.P.; Jin, Y.; Faure, E.; Mantovani, A.; Rothe, M.; Muzio, M.; Arditi, M. Bacterial Lipopolysaccharide Activates Nuclear Factor-KappaB through Interleukin-1 Signaling Mediators in Cultured Human Dermal Endothelial Cells and Mononuclear Phagocytes. J. Biol. Chem. 1999, 274, 7611–7614. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Primers | GenBank Acc. |
|---|---|---|
| IL-1α | 5′-TGGTTAAATGACCTGCAACAGGAA-3′ 5′-AGGTCGGTCTCACTACCTTGTGATG-3′ | NM_010554.4 |
| COX2 | 5′-GCCAGGCTGAACTTCGAAACA-3′ 5′-GCTCACGAGGCCACTGATACCTA-3′ | NM_011198.5 |
| S100A9 | 5′-ACCACCATCATCGACACCTTC-3′ 5′-AAAGGTTGCCAACTGTGCTTC-3′ | NM_009114.3 |
| PLCγ | 5′-CGTCAACGTGGAGGACAAGA-3′ 5′-ATCACCGAAGGACAGCTTGG-3′ | NM_172285.2 |
| β-actin | 5′-CATCCGTAAAGACCTCTATGCCAAC-3′ 5′-ATGGAGCCACCGATCCACA-3′ | NM_007393.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanabe, N.; Tomita, K.; Manaka, S.; Ichikawa, R.; Takayama, T.; Kawato, T.; Ono, M.; Masai, Y.; Utsu, A.; Suzuki, N.; et al. Co-Stimulation of AGEs and LPS Induces Inflammatory Mediators through PLCγ1/JNK/NF-κB Pathway in MC3T3-E1 Cells. Cells 2023, 12, 1383. https://doi.org/10.3390/cells12101383
Tanabe N, Tomita K, Manaka S, Ichikawa R, Takayama T, Kawato T, Ono M, Masai Y, Utsu A, Suzuki N, et al. Co-Stimulation of AGEs and LPS Induces Inflammatory Mediators through PLCγ1/JNK/NF-κB Pathway in MC3T3-E1 Cells. Cells. 2023; 12(10):1383. https://doi.org/10.3390/cells12101383
Chicago/Turabian StyleTanabe, Natsuko, Keiko Tomita, Soichiro Manaka, Risa Ichikawa, Tadahiro Takayama, Takayuki Kawato, Misae Ono, Yuma Masai, Akihisa Utsu, Naoto Suzuki, and et al. 2023. "Co-Stimulation of AGEs and LPS Induces Inflammatory Mediators through PLCγ1/JNK/NF-κB Pathway in MC3T3-E1 Cells" Cells 12, no. 10: 1383. https://doi.org/10.3390/cells12101383
APA StyleTanabe, N., Tomita, K., Manaka, S., Ichikawa, R., Takayama, T., Kawato, T., Ono, M., Masai, Y., Utsu, A., Suzuki, N., & Sato, S. (2023). Co-Stimulation of AGEs and LPS Induces Inflammatory Mediators through PLCγ1/JNK/NF-κB Pathway in MC3T3-E1 Cells. Cells, 12(10), 1383. https://doi.org/10.3390/cells12101383