

Resistance to Gemcitabine in Pancreatic Cancer Is Connected to Methylglyoxal Stress and Heat Shock Response

 , ,
, ,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines, Culturing, and Chemicals
2.2. Clinical Tumor Samples
2.3. Immunohistochemistry (IHC)
2.4. Gemcitabine Inhibitory Concentration 50 (IC50) Determination
2.5. Extracellular Flux Analysis
2.6. Western Blotting
2.7. Cell Proliferation Assay
2.8. GLO1 Activity
2.9. L-Lactate Production
2.10. RNA Isolation and Quantitative Reverse Transcription PCR (qRT-PCR)
2.11. Oncogenic Mutation Analysis
2.12. In Silico Analysis
2.13. Statistical Analysis
3. Results
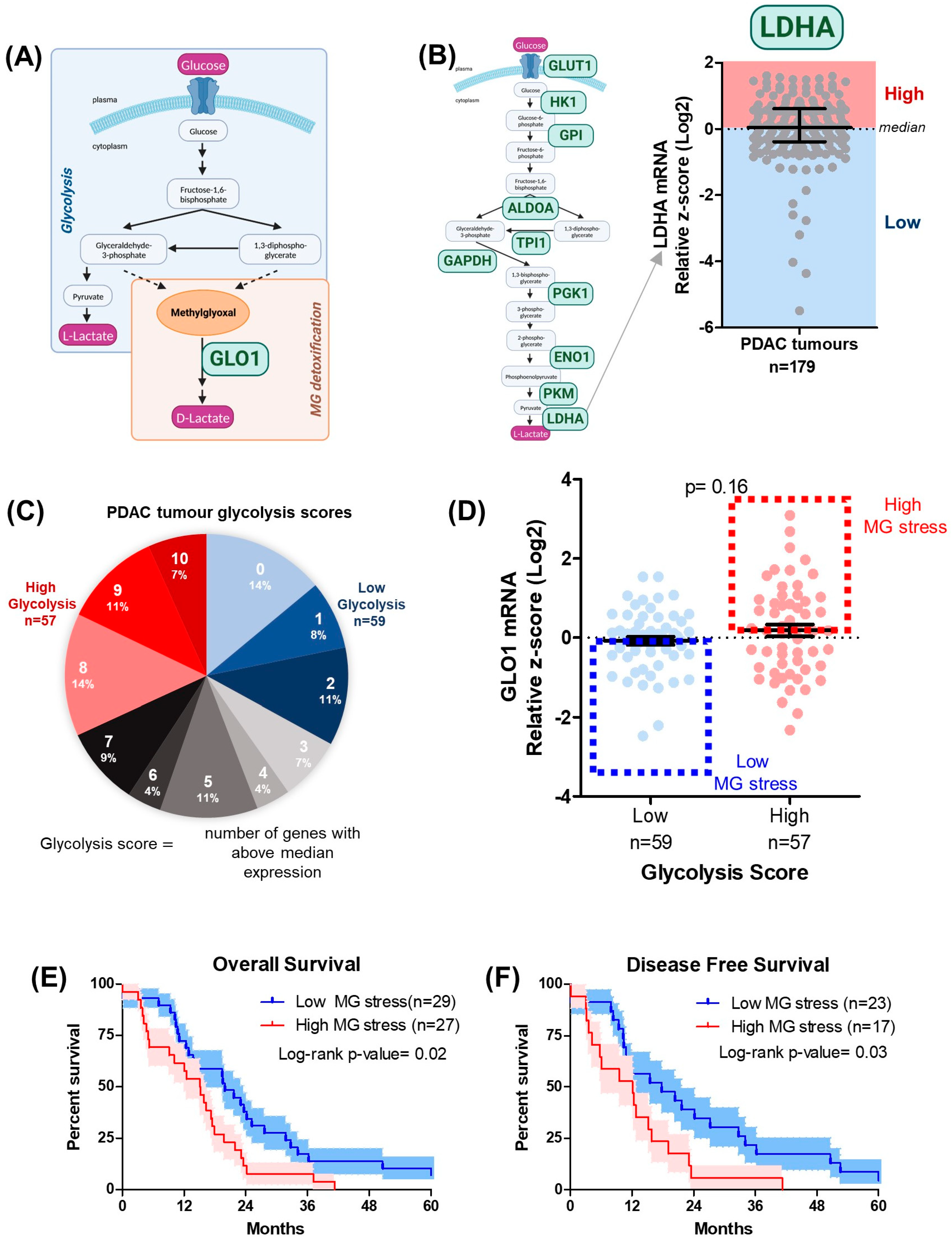
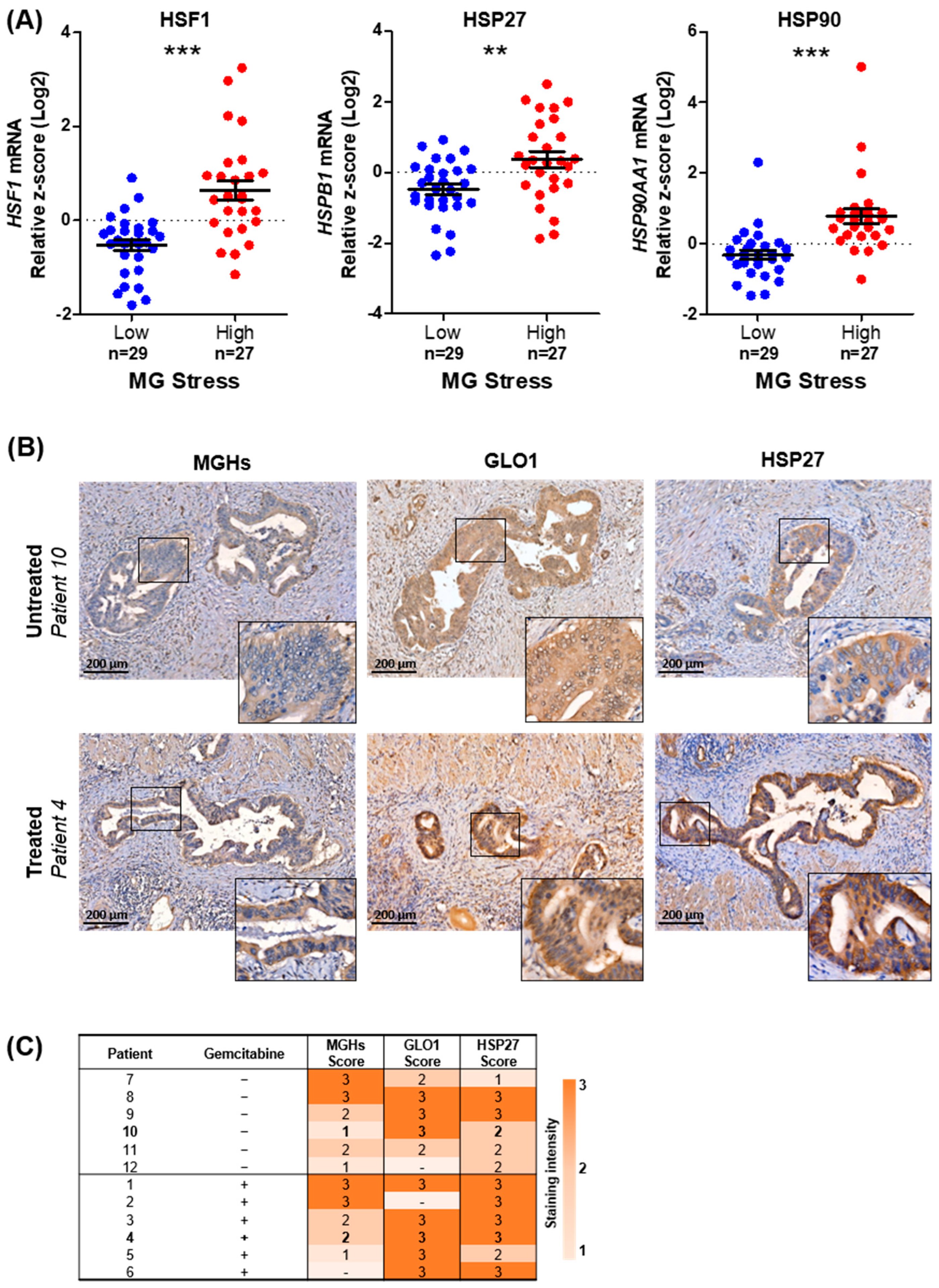
3.1. MG Stress Is Associated with Poor Patient Outcome in PDAC
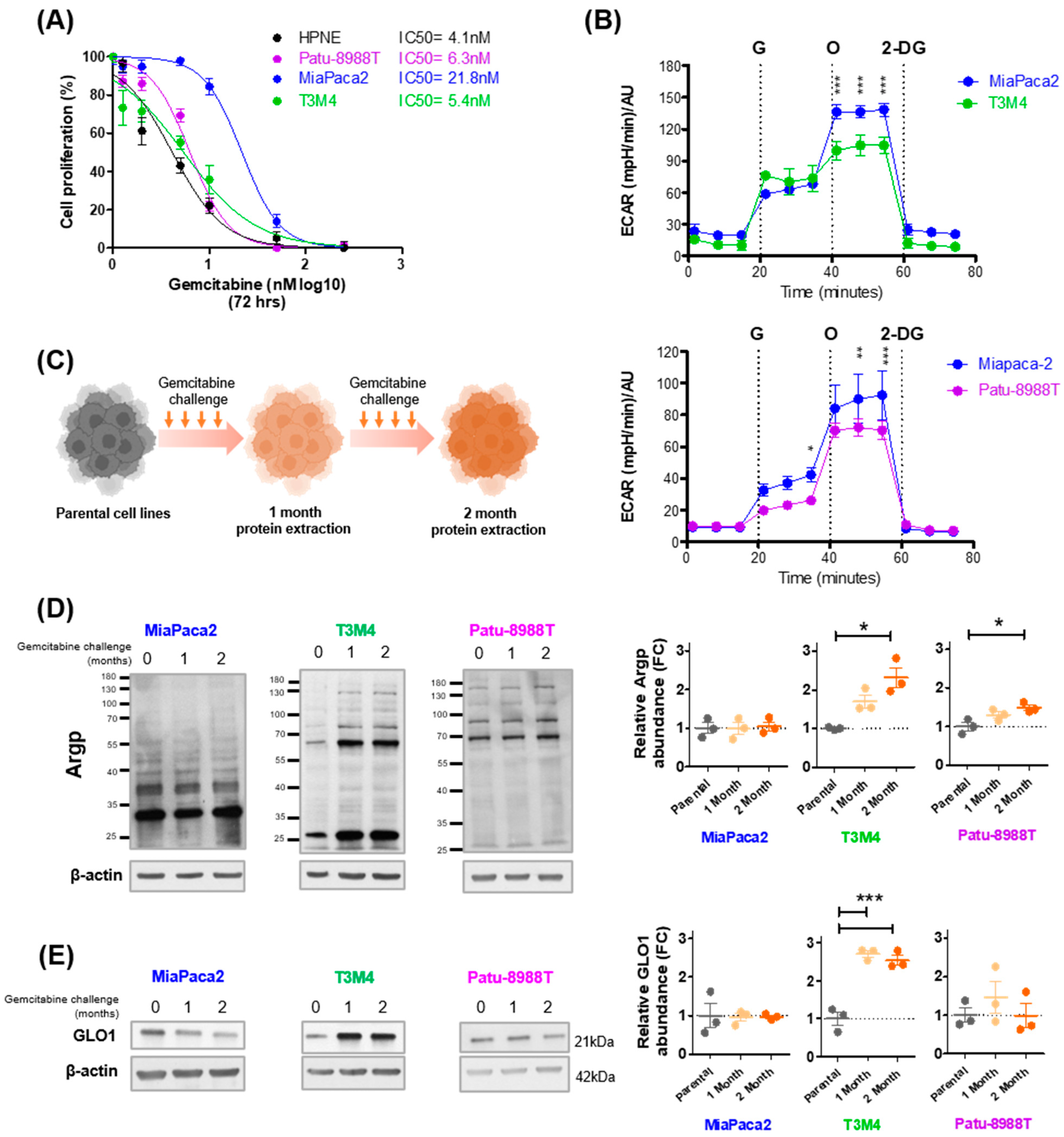
3.2. Gemcitabine Increases MG Stress in Gemcitabine-Sensitive Pancreatic Cancer Cells
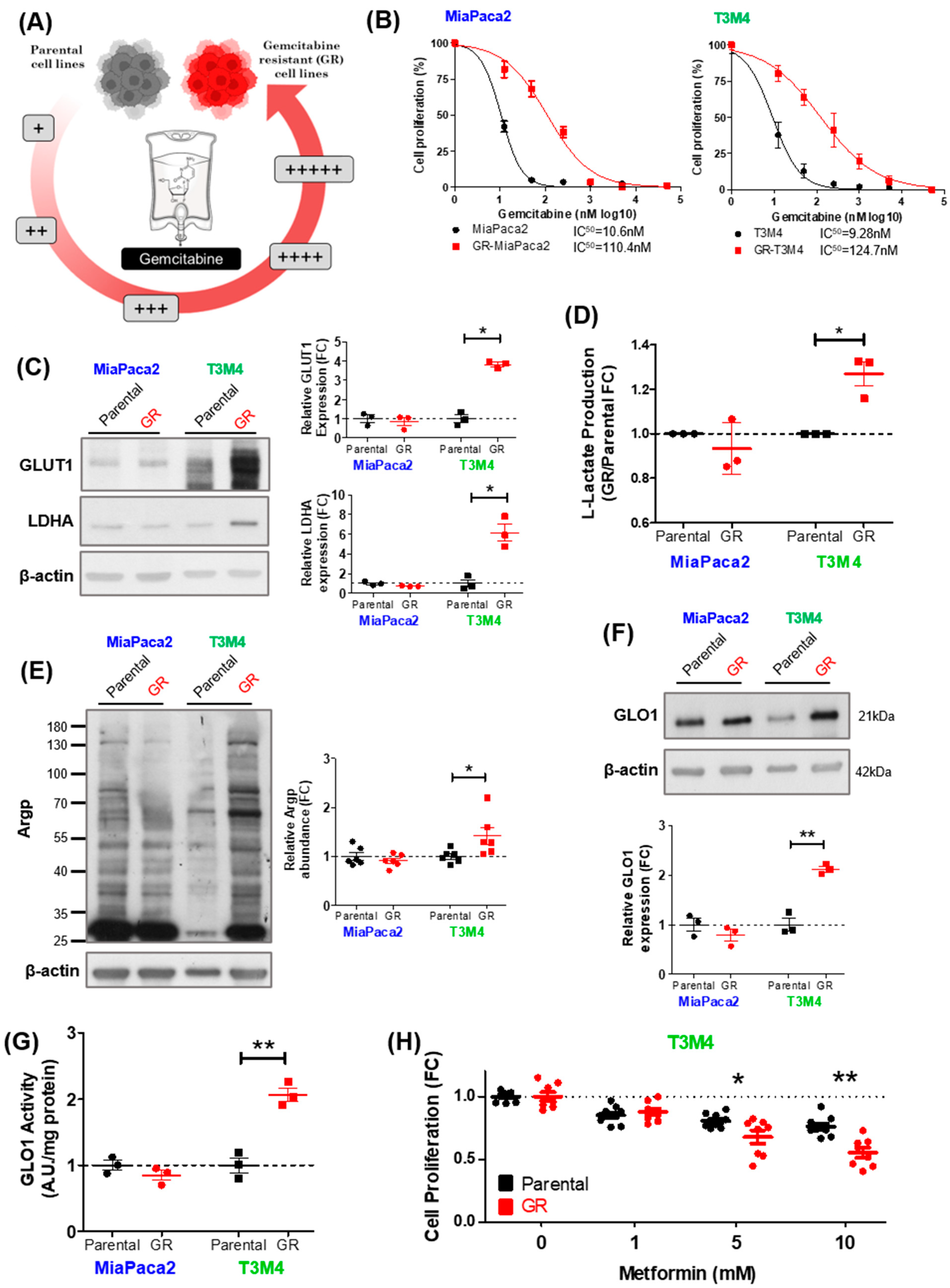
3.3. MG Stress Is Associated with Gemcitabine Resistance in PDAC
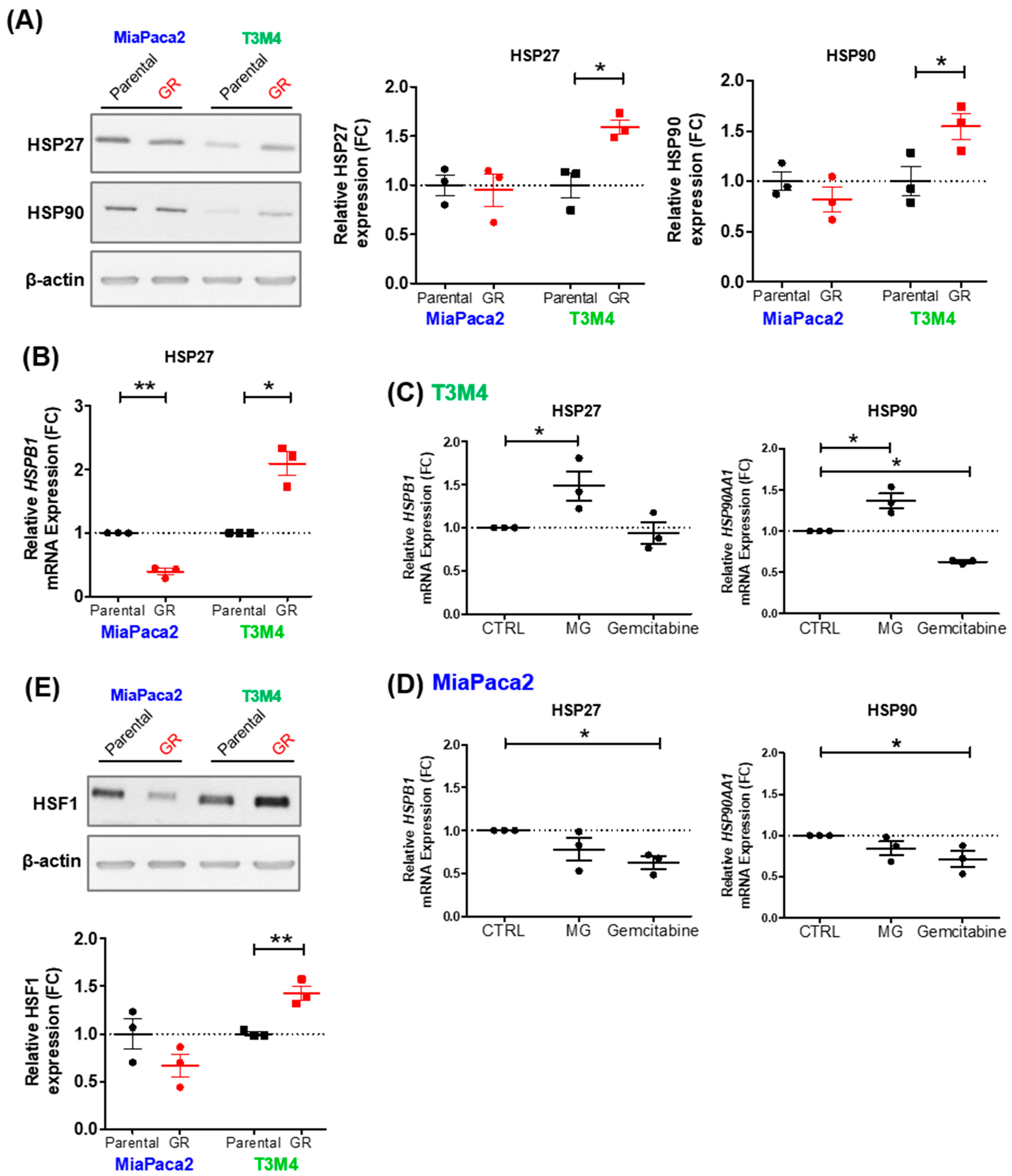
3.4. MG Stress Mediates Gemcitabine Resistance Acquisition through Regulation of the Heat Shock Response
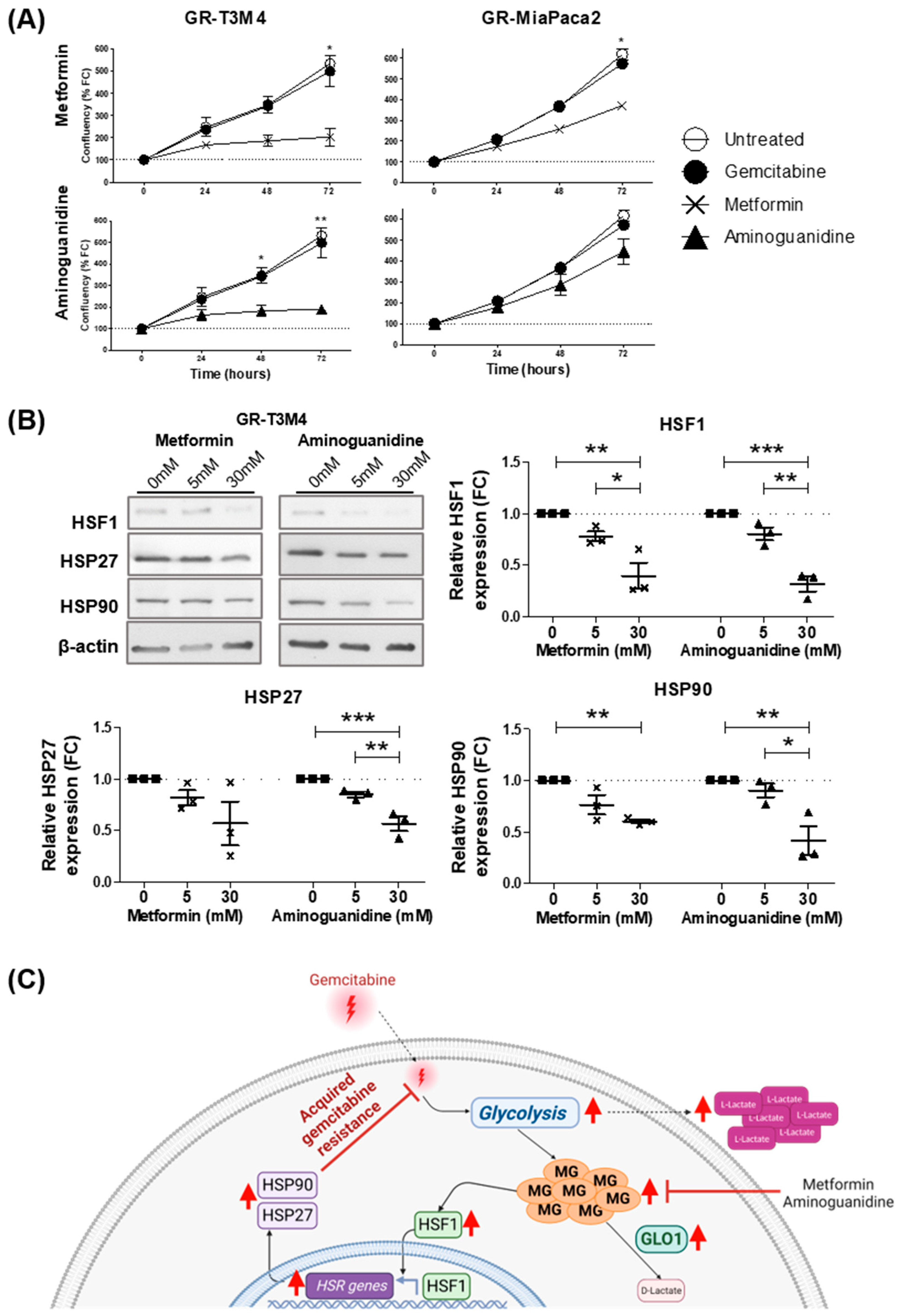
3.5. MG Scavenging Inhibits Gemcitabine-Resistant PDAC Cell Proliferation and Expression of Heat Shock Response Proteins
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Partensky, C.; Bray, F. More deaths from pancreatic cancer than breast cancer in the EU by 2017. Acta Oncol. 2016, 55, 1158–1160. [Google Scholar] [CrossRef]
- Lambert, A.; Schwarz, L.; Borbath, I.; Henry, A.; Van Laethem, J.L.; Malka, D.; Ducreux, M.; Conroy, T. An update on treatment options for pancreatic adenocarcinoma. Adv. Med. Oncol. 2019, 11, 1758835919875568. [Google Scholar] [CrossRef]
- Binenbaum, Y.; Na’ara, S.; Gil, Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug. Resist. Updat. 2015, 23, 55–68. [Google Scholar] [CrossRef]
- Maréchal, R.; Bachet, J.B.; Mackey, J.R.; Dalban, C.; Demetter, P.; Graham, K.; Couvelard, A.; Svrcek, M.; Bardier-Dupas, A.; Hammel, P.; et al. Levels of gemcitabine transport and metabolism proteins predict survival times of patients treated with gemcitabine for pancreatic adenocarcinoma. Gastroenterology 2012, 143, 664–674.e666. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.K.; Purohit, V.; Mehla, K.; Gunda, V.; Chaika, N.V.; Vernucci, E.; King, R.J.; Abrego, J.; Goode, G.D.; Dasgupta, A.; et al. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 2017, 32, 71–87.e77. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, K.; Fidler, I.J. Hypoxia increases resistance of human pancreatic cancer cells to apoptosis induced by gemcitabine. Clin. Cancer Res. 2004, 10, 2299–2306. [Google Scholar] [CrossRef]
- Nicolle, R.; Gayet, O.; Duconseil, P.; Vanbrugghe, C.; Roques, J.; Bigonnet, M.; Blum, Y.; Elarouci, N.; Armenoult, L.; Ayadi, M.; et al. A transcriptomic signature to predict adjuvant gemcitabine sensitivity in pancreatic adenocarcinoma. Ann. Oncol. 2021, 32, 250–260. [Google Scholar] [CrossRef]
- Peng, X.; Chen, Z.; Farshidfar, F.; Xu, X.; Lorenzi, P.L.; Wang, Y.; Cheng, F.; Tan, L.; Mojumdar, K.; Du, D.; et al. Molecular Characterization and Clinical Relevance of Metabolic Expression Subtypes in Human Cancers. Cell Rep. 2018, 23, 255–269.e254. [Google Scholar] [CrossRef]
- Xi, Y.; Yuan, P.; Li, T.; Zhang, M.; Liu, M.F.; Li, B. hENT1 reverses chemoresistance by regulating glycolysis in pancreatic cancer. Cancer Lett. 2020, 479, 112–122. [Google Scholar] [CrossRef]
- Dai, S.; Peng, Y.; Zhu, Y.; Xu, D.; Zhu, F.; Xu, W.; Chen, Q.; Zhu, X.; Liu, T.; Hou, C.; et al. Glycolysis promotes the progression of pancreatic cancer and reduces cancer cell sensitivity to gemcitabine. Biomed. Pharmacother. 2020, 121, 109521. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Guillaumond, F.; Leca, J.; Olivares, O.; Lavaut, M.N.; Vidal, N.; Berthezène, P.; Dusetti, N.J.; Loncle, C.; Calvo, E.; Turrini, O.; et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 3919–3924. [Google Scholar] [CrossRef]
- Phillips, S.A.; Thornalley, P.J. The formation of methylglyoxal from triose phosphates. Investigation using a specific assay for methylglyoxal. Eur. J. Biochem. FEBS 1993, 212, 101–105. [Google Scholar] [CrossRef]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef]
- Chiavarina, B.; Nokin, M.J.; Bellier, J.; Durieux, F.; Bletard, N.; Sherer, F.; Lovinfosse, P.; Peulen, O.; Verset, L.; Dehon, R.; et al. Methylglyoxal-Mediated Stress Correlates with High Metabolic Activity and Promotes Tumor Growth in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 213. [Google Scholar] [CrossRef]
- Chiavarina, B.; Nokin, M.J.; Durieux, F.; Bianchi, E.; Turtoi, A.; Peulen, O.; Peixoto, P.; Irigaray, P.; Uchida, K.; Belpomme, D.; et al. Triple negative tumors accumulate significantly less methylglyoxal specific adducts than other human breast cancer subtypes. Oncotarget 2014, 5, 5472–5482. [Google Scholar] [CrossRef]
- van Heijst, J.W.; Niessen, H.W.; Hoekman, K.; Schalkwijk, C.G. Advanced glycation end products in human cancer tissues: Detection of Nepsilon-(carboxymethyl)lysine and argpyrimidine. Ann. N. Y. Acad. Sci. 2005, 1043, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Nokin, M.J.; Durieux, F.; Peixoto, P.; Chiavarina, B.; Peulen, O.; Blomme, A.; Turtoi, A.; Costanza, B.; Smargiasso, N.; Baiwir, D.; et al. Methylglyoxal, a glycolysis side-product, induces Hsp90 glycation and YAP-mediated tumor growth and metastasis. eLife 2016, 5, e19375. [Google Scholar] [CrossRef] [PubMed]
- Nokin, M.J.; Bellier, J.; Durieux, F.; Peulen, O.; Rademaker, G.; Gabriel, M.; Monseur, C.; Charloteaux, B.; Verbeke, L.; van Laere, S.; et al. Methylglyoxal, a glycolysis metabolite, triggers metastasis through MEK/ERK/SMAD1 pathway activation in breast cancer. Breast Cancer Res. 2019, 21, 11. [Google Scholar] [CrossRef] [PubMed]
- Beisswenger, P.J.; Howell, S.K.; Touchette, A.D.; Lal, S.; Szwergold, B.S. Metformin reduces systemic methylglyoxal levels in type 2 diabetes. Diabetes 1999, 48, 198–202. [Google Scholar] [CrossRef]
- Kinsky, O.R.; Hargraves, T.L.; Anumol, T.; Jacobsen, N.E.; Dai, J.; Snyder, S.A.; Monks, T.J.; Lau, S.S. Metformin Scavenges Methylglyoxal to Form a Novel Imidazolinone Metabolite in Humans. Chem. Res. Toxicol. 2016, 29, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Lo, T.W.; Selwood, T.; Thornalley, P.J. The reaction of methylglyoxal with aminoguanidine under physiological conditions and prevention of methylglyoxal binding to plasma proteins. Biochem. Pharmacol. 1994, 48, 1865–1870. [Google Scholar] [CrossRef]
- Bellier, J.; Nokin, M.J.; Caprasse, M.; Tiamiou, A.; Blomme, A.; Scheijen, J.L.; Koopmansch, B.; MacKay, G.M.; Chiavarina, B.; Costanza, B.; et al. Methylglyoxal Scavengers Resensitize KRAS-Mutated Colorectal Tumors to Cetuximab. Cell Rep. 2020, 30, 1400–1416.e1406. [Google Scholar] [CrossRef] [PubMed]
- Nokin, M.J.; Durieux, F.; Bellier, J.; Peulen, O.; Uchida, K.; Spiegel, D.A.; Cochrane, J.R.; Hutton, C.A.; Castronovo, V.; Bellahcene, A. Hormetic potential of methylglyoxal, a side-product of glycolysis, in switching tumours from growth to death. Sci. Rep. 2017, 7, 11722. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Gui, D.Y.; Vander Heiden, M.G. Altered metabolite levels in cancer: Implications for tumour biology and cancer therapy. Nat. Rev. Cancer 2016, 16, 680–693. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, C. Oncometabolites in Cancer: Current Understanding and Challenges. Cancer Res. 2021, 81, 2820–2823. [Google Scholar] [CrossRef]
- Bommer, G.T.; Van Schaftingen, E.; Veiga-da-Cunha, M. Metabolite Repair Enzymes Control Metabolic Damage in Glycolysis. Trends Biochem. Sci. 2020, 45, 228–243. [Google Scholar] [CrossRef]
- Alasady, M.J.; Mendillo, M.L. The Multifaceted Role of HSF1 in Tumorigenesis. Adv. Exp. Med. Biol. 2020, 1243, 69–85. [Google Scholar] [CrossRef]
- Bento, C.F.; Marques, F.; Fernandes, R.; Pereira, P. Methylglyoxal alters the function and stability of critical components of the protein quality control. PLoS ONE 2010, 5, e13007. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Mashima, T.; Yamamoto, K.; Tsuruo, T. Modulation of heat-shock protein 27 (Hsp27) anti-apoptotic activity by methylglyoxal modification. J. Biol. Chem. 2002, 277, 45770–45775. [Google Scholar] [CrossRef] [PubMed]
- van Heijst, J.W.; Niessen, H.W.; Musters, R.J.; van Hinsbergh, V.W.; Hoekman, K.; Schalkwijk, C.G. Argpyrimidine-modified Heat shock protein 27 in human non-small cell lung cancer: A possible mechanism for evasion of apoptosis. Cancer Lett. 2006, 241, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Chen, K.; Li, J.; Qian, W.; Xiao, Y.; Wu, E.; Ma, J.; Chen, Z.; Wang, Z.; Ma, Q.; et al. Heat shock factor 1 inhibition sensitizes pancreatic cancer to gemcitabine via the suppression of cancer stem cell-like properties. Biomed. Pharmacother. 2022, 148, 112713. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Lim, D.H.; Jang, K.T.; Lim, T.; Lee, J.; Choi, Y.L.; Jang, H.L.; Yi, J.H.; Baek, K.K.; Park, S.H.; et al. Impact of KRAS mutations on clinical outcomes in pancreatic cancer patients treated with first-line gemcitabine-based chemotherapy. Mol. Cancer Ther. 2011, 10, 1993–1999. [Google Scholar] [CrossRef]
- Oya-Ito, T.; Naito, Y.; Takagi, T.; Handa, O.; Matsui, H.; Yamada, M.; Shima, K.; Yoshikawa, T. Heat-shock protein 27 (Hsp27) as a target of methylglyoxal in gastrointestinal cancer. Biochim. Biophys. Acta 2011, 1812, 769–781. [Google Scholar] [CrossRef]
- Rong, Y.; Wu, W.; Ni, X.; Kuang, T.; Jin, D.; Wang, D.; Lou, W. Lactate dehydrogenase A is overexpressed in pancreatic cancer and promotes the growth of pancreatic cancer cells. Tumour Biol. 2013, 34, 1523–1530. [Google Scholar] [CrossRef]
- Wang, Y.; Kuramitsu, Y.; Ueno, T.; Suzuki, N.; Yoshino, S.; Iizuka, N.; Akada, J.; Kitagawa, T.; Oka, M.; Nakamura, K. Glyoxalase I (GLO1) is up-regulated in pancreatic cancerous tissues compared with related non-cancerous tissues. Anticancer Res. 2012, 32, 3219–3222. [Google Scholar]
- Acunzo, J.; Andrieu, C.; Baylot, V.; So, A.; Rocchi, P. Hsp27 as a therapeutic target in cancers. Curr. Drug Targets 2014, 15, 423–431. [Google Scholar] [CrossRef]
- Wang, X.; Chen, M.; Zhou, J.; Zhang, X. HSP27, 70 and 90, anti-apoptotic proteins, in clinical cancer therapy. Int. J. Oncol. 2014, 45, 18–30. [Google Scholar] [CrossRef]
- Mori-Iwamoto, S.; Kuramitsu, Y.; Ryozawa, S.; Taba, K.; Fujimoto, M.; Okita, K.; Nakamura, K.; Sakaida, I. A proteomic profiling of gemcitabine resistance in pancreatic cancer cell lines. Mol. Med. Rep. 2008, 1, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Baylot, V.; Andrieu, C.; Katsogiannou, M.; Taieb, D.; Garcia, S.; Giusiano, S.; Acunzo, J.; Iovanna, J.; Gleave, M.; Garrido, C.; et al. OGX-427 inhibits tumor progression and enhances gemcitabine chemotherapy in pancreatic cancer. Cell Death Dis. 2011, 2, e221. [Google Scholar] [CrossRef] [PubMed]
- Mori-Iwamoto, S.; Kuramitsu, Y.; Ryozawa, S.; Mikuria, K.; Fujimoto, M.; Maehara, S.; Maehara, Y.; Okita, K.; Nakamura, K.; Sakaida, I. Proteomics finding heat shock protein 27 as a biomarker for resistance of pancreatic cancer cells to gemcitabine. Int. J. Oncol. 2007, 31, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Umar, H.I.; Ajayi, A.T.; Mukerjee, N.; Aborode, A.T.; Hasan, M.M.; Maitra, S.; Bello, R.O.; Alabere, H.O.; Sanusi, A.A.; Awolaja, O.O.; et al. Discovery of Novel HSP27 Inhibitors as Prospective Anti-Cancer Agents Utilizing Computer-Assisted Therapeutic Discovery Approaches. Cells 2022, 11, 2412. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.K.; Kam, H.; Kim, K.Y.; Park, S.I.; Lee, Y.S. Targeting Heat Shock Protein 27 in Cancer: A Druggable Target for Cancer Treatment? Cancers 2019, 11, 1195. [Google Scholar] [CrossRef]
- Hansen, R.K.; Parra, I.; Lemieux, P.; Oesterreich, S.; Hilsenbeck, S.G.; Fuqua, S.A. Hsp27 overexpression inhibits doxorubicin-induced apoptosis in human breast cancer cells. Breast Cancer Res. Treat. 1999, 56, 187–196. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDAC Patient | Neo-Adj Chemo | Sex | Age (Diag) | TNM Stage (at Diag) | Total # of Neo-Adj Cycles | Survival since Diag (Months) | Mets (Y/N) | KRAS Variant |
|---|---|---|---|---|---|---|---|---|
| 1 | Gem | F | 69.3 | IIB | 3 | 5 | Yes | G12V |
| 2 | Gem | M | 65.2 | IIB | 3 | 29 | Yes | - |
| 3 | Gem | M | 59 | IIB | 3 | 74.9 | Yes | - |
| 4 | Gem | M | 70.2 | IB | 3 | 19.9 | Yes | - |
| 5 | Gem | F | 87.7 | IIB | 4 | 16.8 | - | - |
| 6 | Gem/Nab-P | M | 71.7 | III | 3 | 48.1 | No | - |
| 7 | F | 77.3 | IIB | - | 7.6 | Yes | - | |
| 8 | F | 50 | IA | - | 97.8 | Yes | wt | |
| 9 | M | 76.7 | III | - | 9.4 | Yes | Q61H | |
| 10 | M | 74 | IB | - | 15 | Yes | G12D | |
| 11 | F | 62.9 | IA | - | 99.2 | Yes | Q61H | |
| 12 | F | 82.9 | IB | - | 36 | No | wt |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crake, R.; Gasmi, I.; Dehaye, J.; Lardinois, F.; Peiffer, R.; Maloujahmoum, N.; Agirman, F.; Koopmansch, B.; D’Haene, N.; Azurmendi Senar, O.; et al. Resistance to Gemcitabine in Pancreatic Cancer Is Connected to Methylglyoxal Stress and Heat Shock Response. Cells 2023, 12, 1414. https://doi.org/10.3390/cells12101414
Crake R, Gasmi I, Dehaye J, Lardinois F, Peiffer R, Maloujahmoum N, Agirman F, Koopmansch B, D’Haene N, Azurmendi Senar O, et al. Resistance to Gemcitabine in Pancreatic Cancer Is Connected to Methylglyoxal Stress and Heat Shock Response. Cells. 2023; 12(10):1414. https://doi.org/10.3390/cells12101414
Chicago/Turabian StyleCrake, Rebekah, Imène Gasmi, Jordan Dehaye, Fanny Lardinois, Raphaël Peiffer, Naïma Maloujahmoum, Ferman Agirman, Benjamin Koopmansch, Nicky D’Haene, Oier Azurmendi Senar, and et al. 2023. "Resistance to Gemcitabine in Pancreatic Cancer Is Connected to Methylglyoxal Stress and Heat Shock Response" Cells 12, no. 10: 1414. https://doi.org/10.3390/cells12101414
APA StyleCrake, R., Gasmi, I., Dehaye, J., Lardinois, F., Peiffer, R., Maloujahmoum, N., Agirman, F., Koopmansch, B., D’Haene, N., Azurmendi Senar, O., Arsenijevic, T., Lambert, F., Peulen, O., Van Laethem, J. -L., & Bellahcène, A. (2023). Resistance to Gemcitabine in Pancreatic Cancer Is Connected to Methylglyoxal Stress and Heat Shock Response. Cells, 12(10), 1414. https://doi.org/10.3390/cells12101414