
Restoring Axonal Organelle Motility and Regeneration in Cultured FUS-ALS Motoneurons through Magnetic Field Stimulation Suggests an Alternative Therapeutic Approach

 , , , ,
, , , , 
Abstract
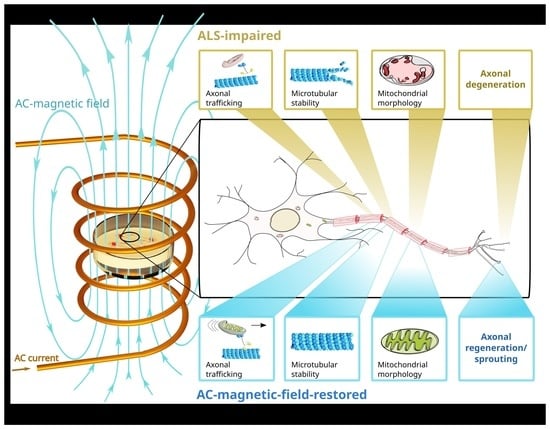
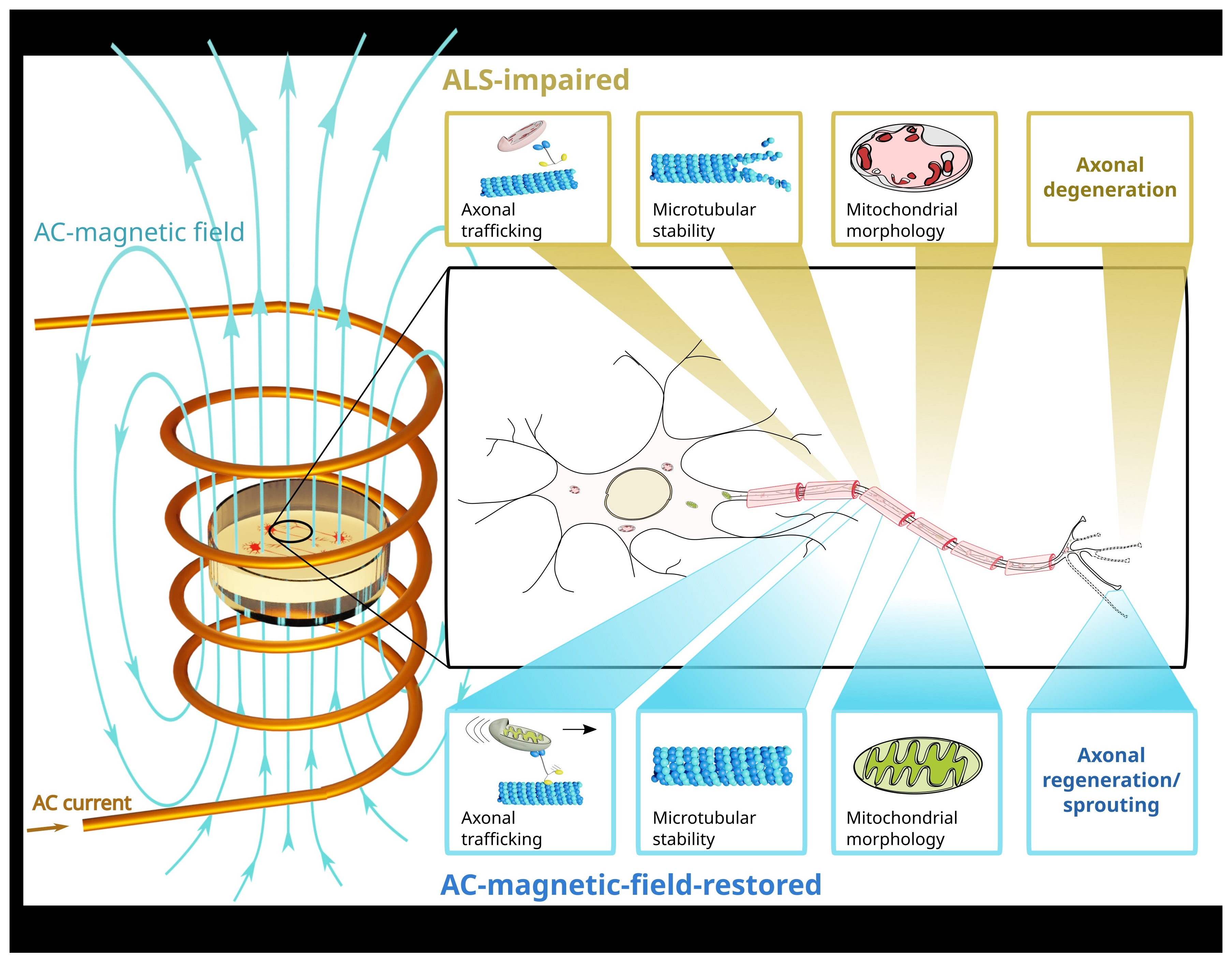
:
1. Introduction
2. Materials and Methods
2.1. Characteristics of Patients for iPSC Derivation
2.2. Genotyping
2.3. Mycoplasma Testing
2.4. Generating, Gene-Editing and Differentiation of Human iPSC Cell Lines to MNs in Microfluidic Chamber
2.5. Magnetic Field Stimulation (MS)
2.6. Immunofluorescent Stainings
2.7. Immunofluorescence Intensity Analysis
2.8. Axonal Live Cell Imaging of MNs in MFCs
2.9. Axonal Organelle Tracking and Shape Analysis of Live Imaging Movies
2.10. Multiparametric High Content (HC) Phenotypic Profiles
2.11. Live Cell Imaging of Growth Cones
2.12. Outgrowth Analysis of Advancing Growth Cone Velocity
- In FIJI, the macro “Growth cone characteristics” was loaded and executed. User input was then required to locate the parental folder containing all movie stacks.
- The pixel dimension calibrations from the microscope were automatically imported from the movie’s metadata and implemented for the analysis.
- For each movie, a rectangular ROI for the initial growth cone detection was manually drawn, which covered the entire migration throughout the whole movie stack. The ROI was carefully selected to eliminate erratic detections of objects other than growth cones. Some preliminary image optimizations were conducted in the ROI, as follows:
- ◦
- Contrast enhancement by allowing 0.1% saturated pixels and histogram normalization,
- ◦
- Background subtraction with an eroding rolling ball of 20 pixels in radius on light background,
- ◦
- Image segmentation with the thresholding function “Percentile” on a dark background,
- ◦
- Background setting to black,
- ◦
- Conversion of the obtained segmented images to masks.
- Growth cone tracking was performed on the black/white masks of all segmented objects. Given the dynamic nature of the growth cone’s morphology over consecutive frames, their tracking required a new determination of their center of mass for each frame. This was achieved by an iterative mask shrinking process starting from the borders of the selected ROI via decreasing circular masks following an intensity gradient that finally shrunk down to the intensity center of each recognized object to determine its current coordinates, thereby enabling its linking to consecutive frames. As a pragmatic approximation, it was assumed that the center of intensity equated to the center of mass. The radius was successively reduced until some selection remained. The remaining selection was then re-inflated back to its original size and the center-of-mass was calculated and highlighted in detected growth cones.
- In the case of several growth cones in the ROI, the user had to select a single growth cone of interest for further analysis.
- The selected growth cone (outlined in Figure 3b) was automatically analyzed by the macro with respect to its mean travel distance between consecutive frames over the entire movie stack (Figure 3b). To this end, the algorithm scanned an area surrounding the x-y coordinates of the cone’s center of mass deduced in the previous frame. In the case of multiple plausible positions, the algorithm chose the object with the closest distance to the previous frame as the new position. This process was repeated frame by frame.
2.13. Quantification and Statistics
3. Results
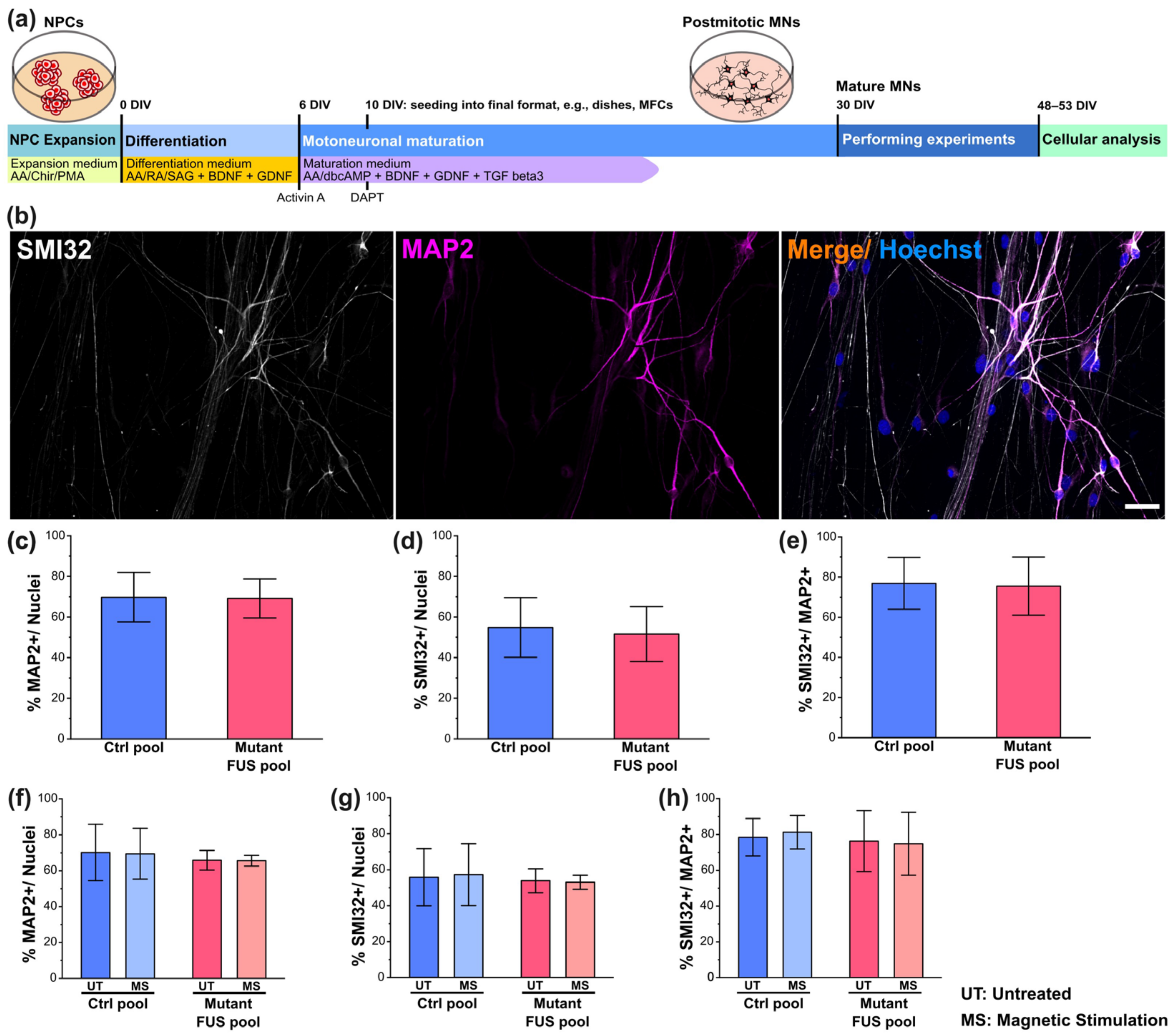
3.1. Neuronal Characterization and the Effect of Magnetic Stimulations on Neuronal Differentiation
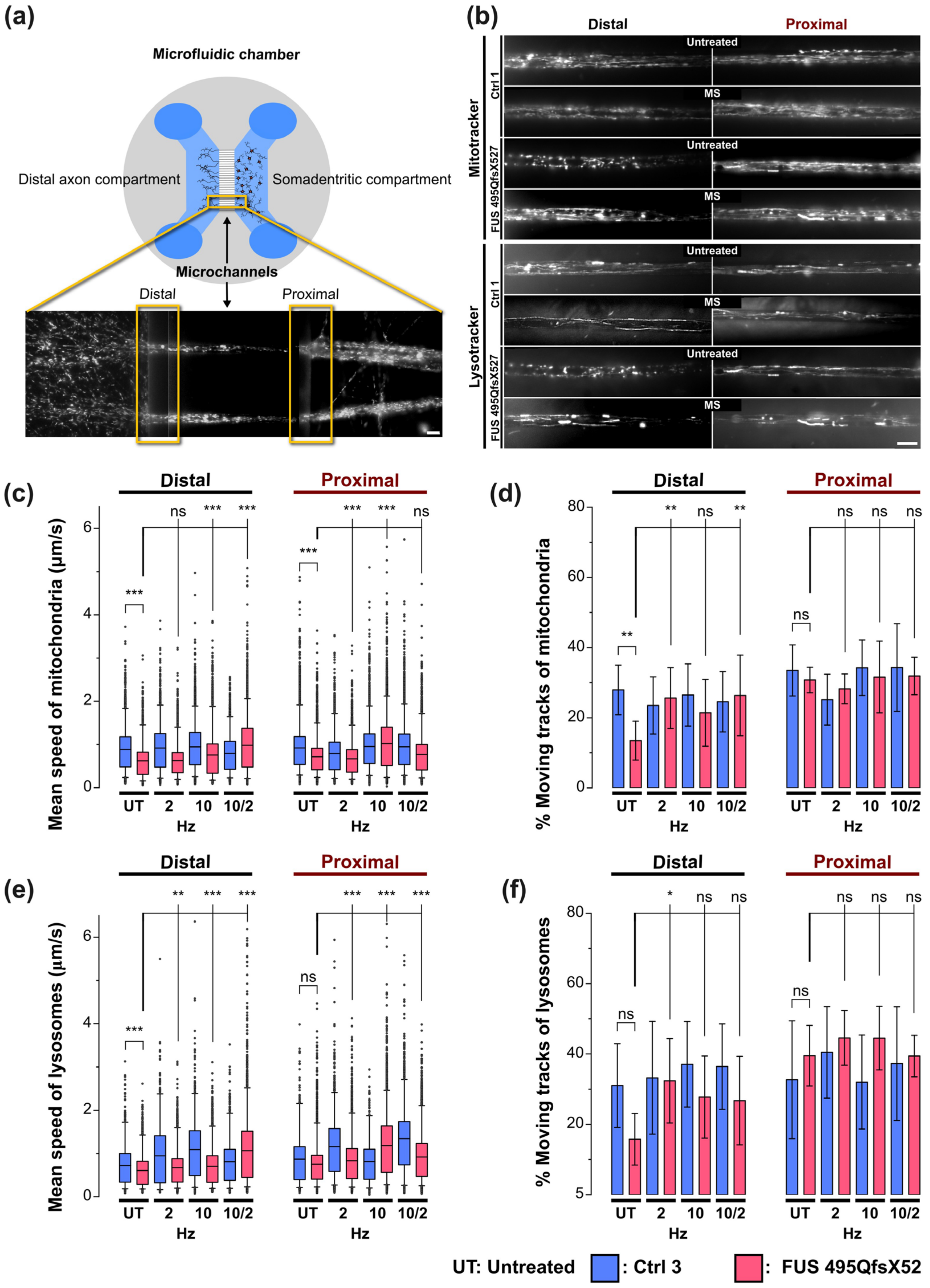
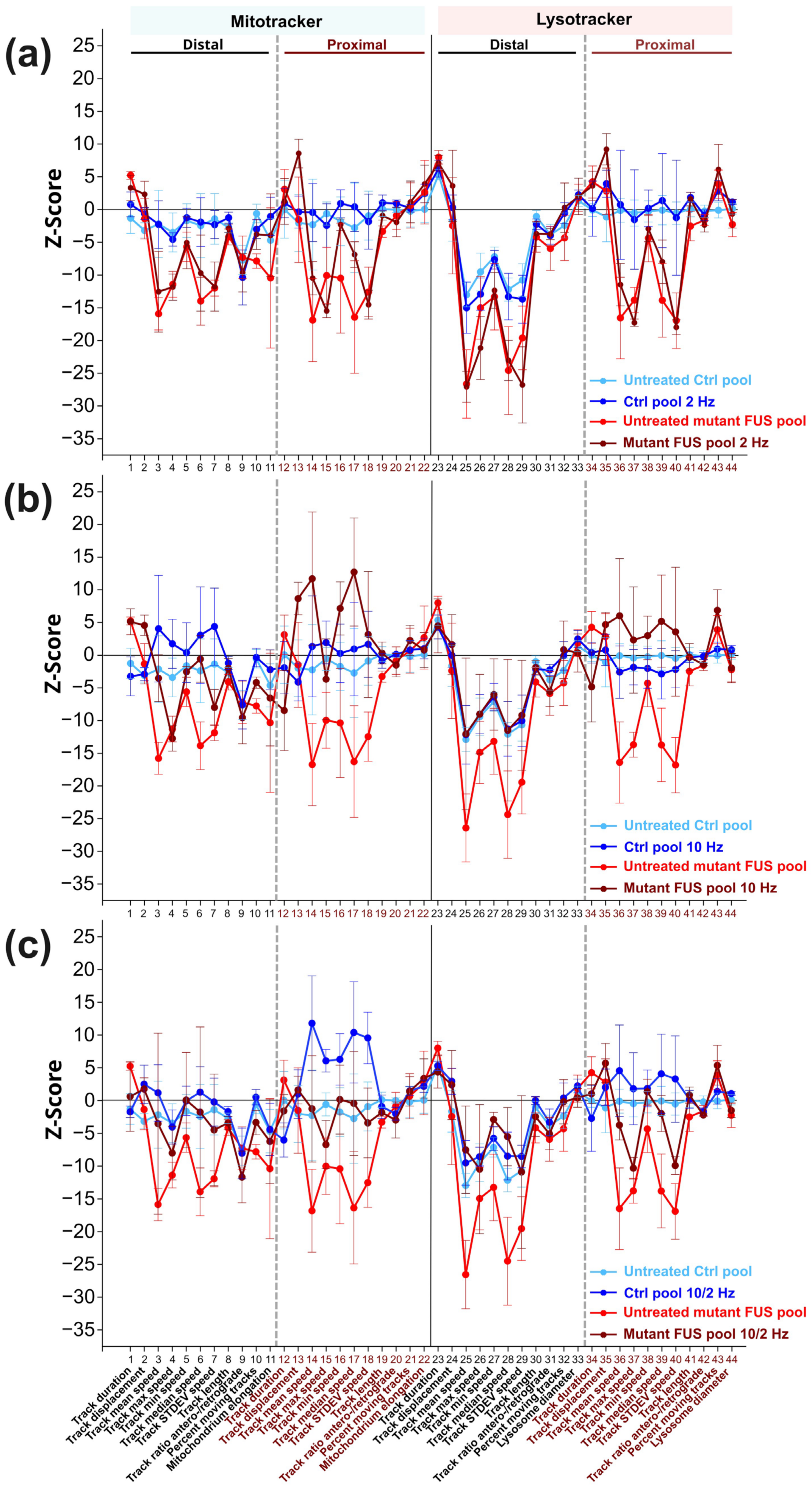
3.2. MS Restores Deficient Axonal Organelle Transport in FUS-ALS
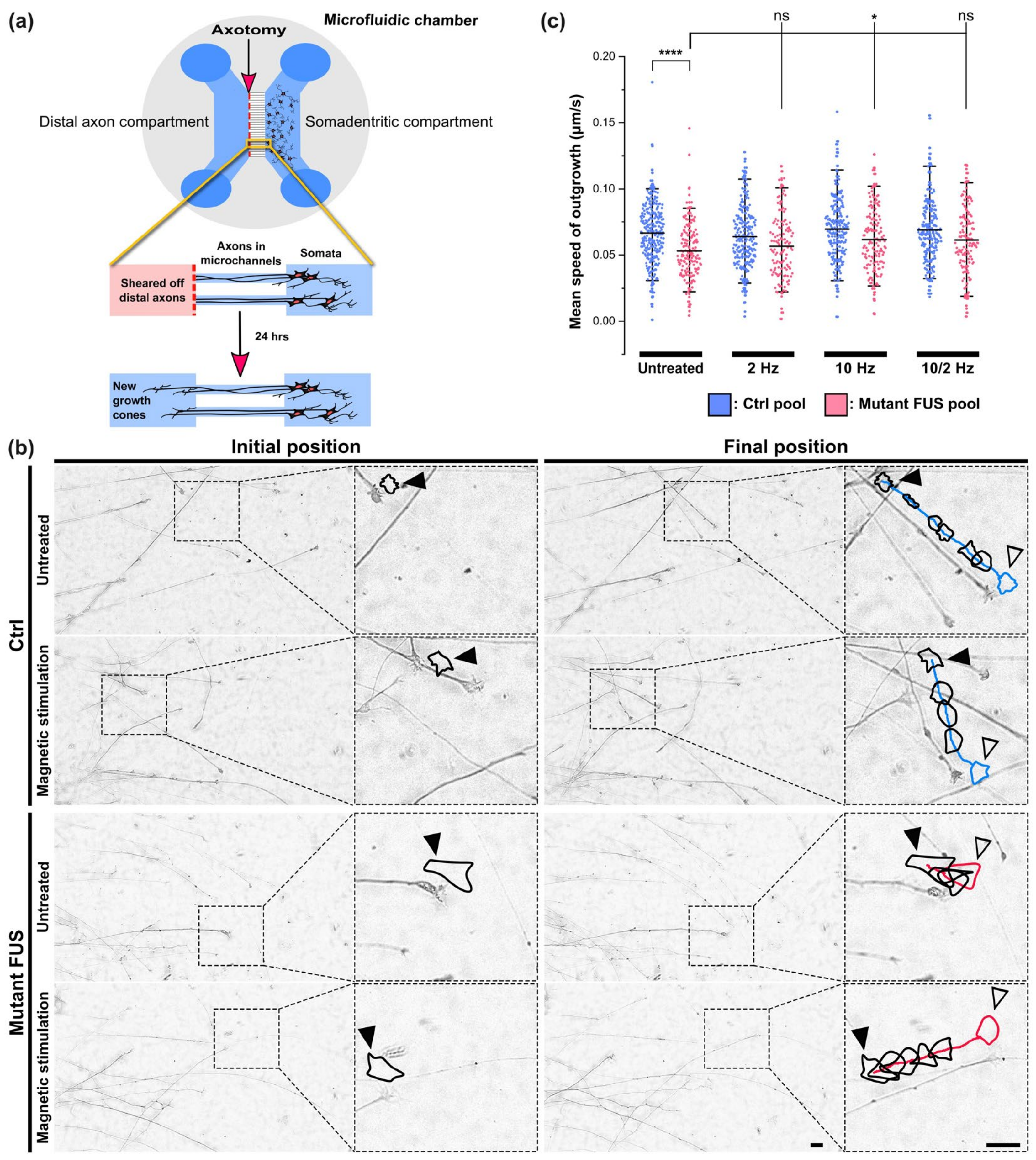
3.3. Rescue of Axonal Regeneration Defects in FUS-ALS through MS
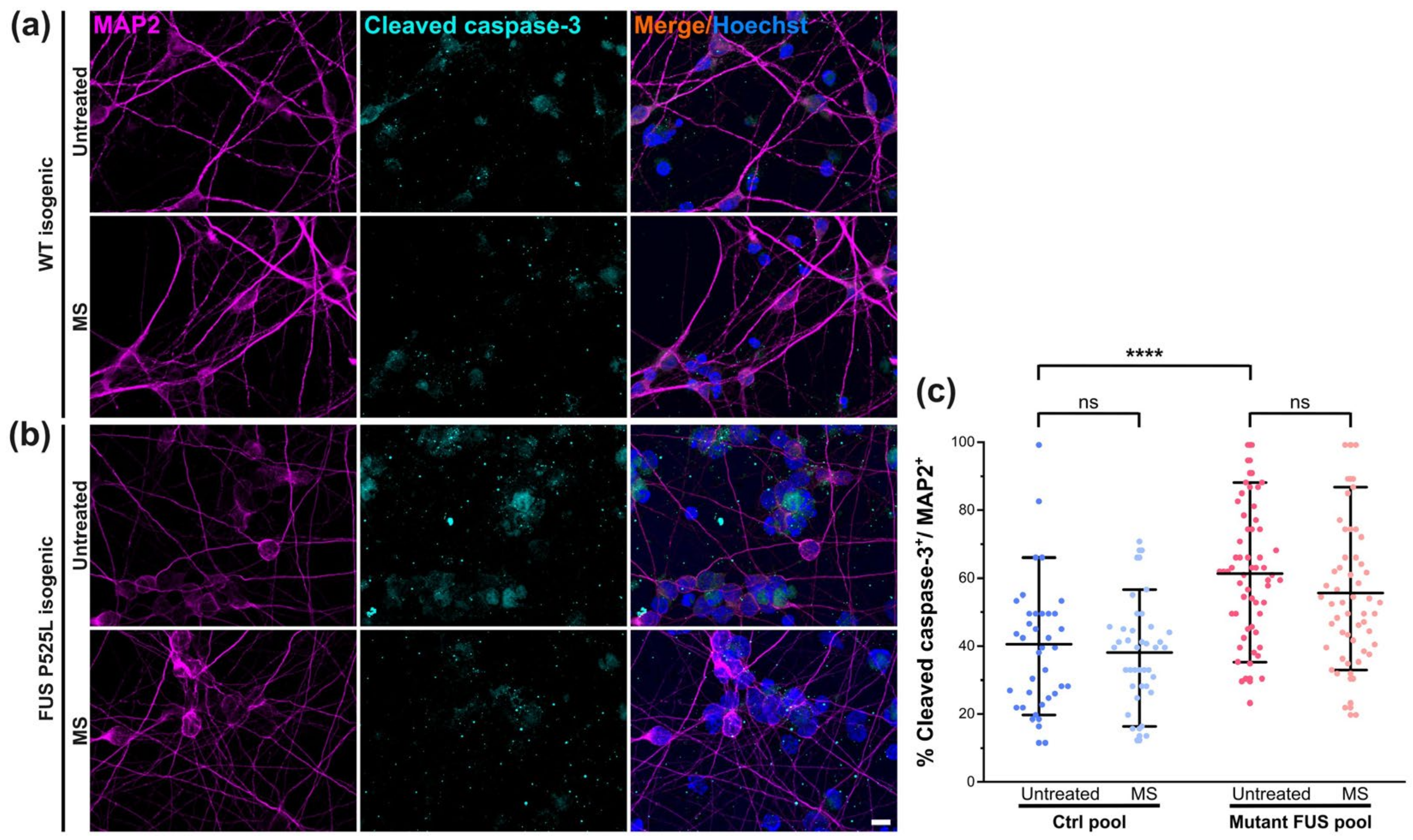
3.4. MS Did Not Alter Neuronal Survival
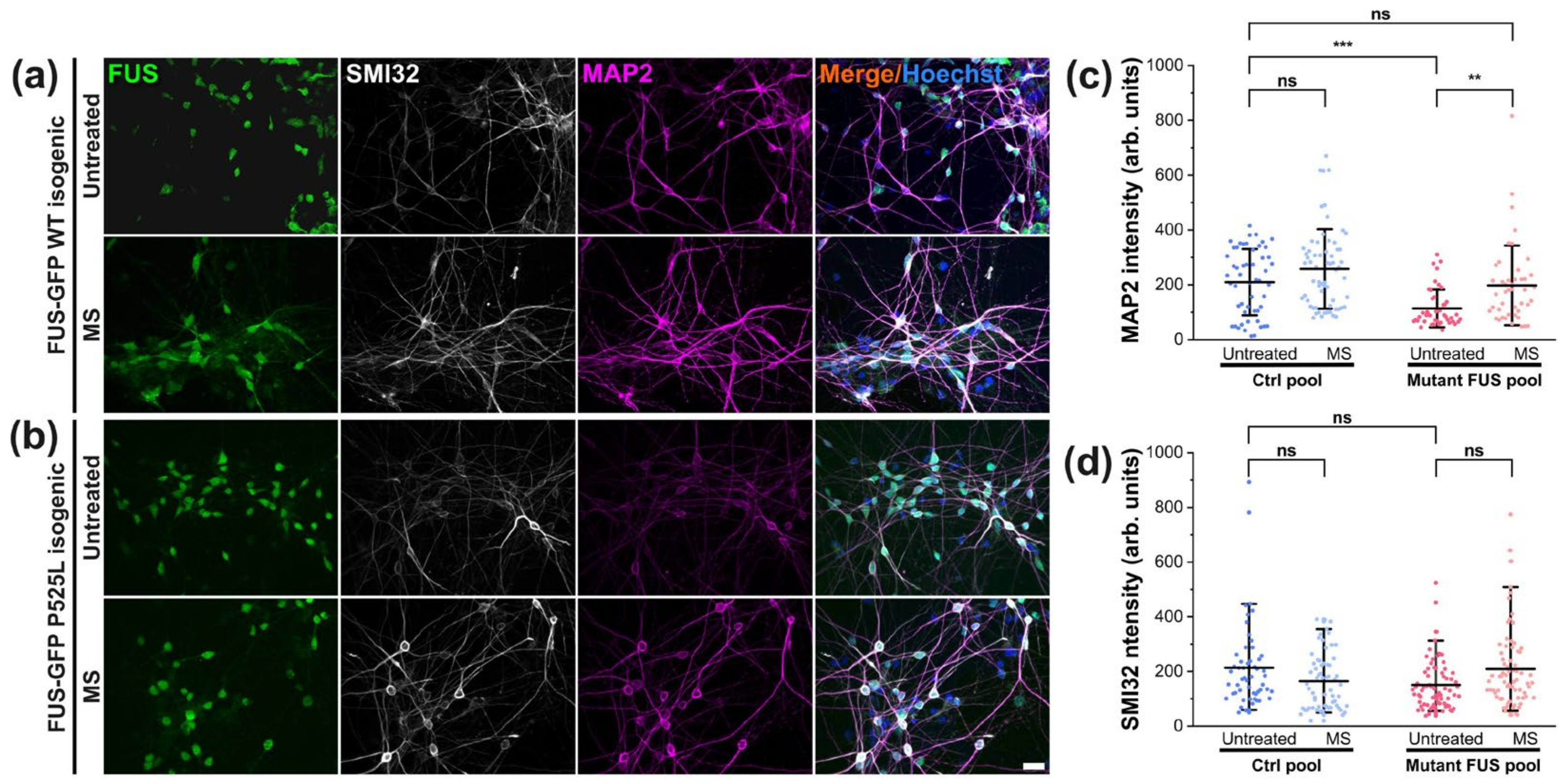
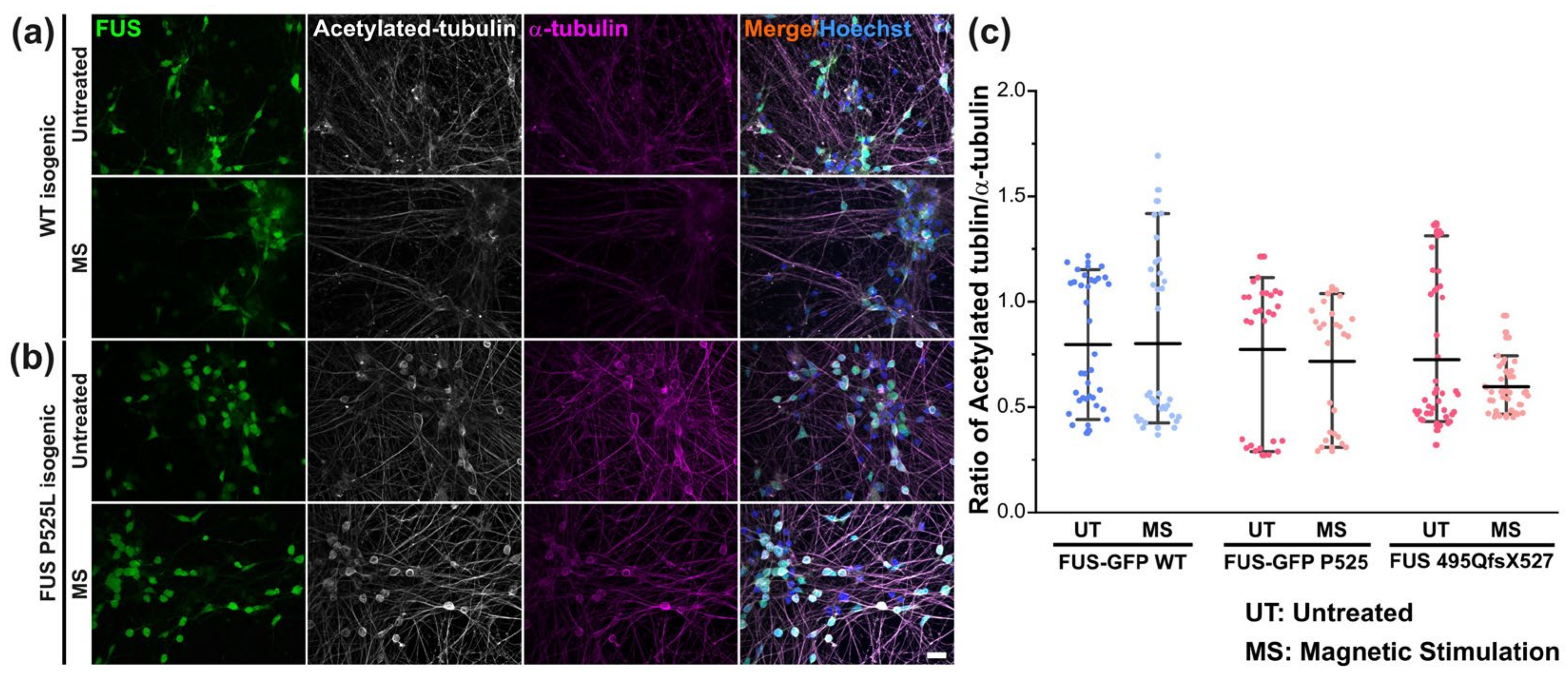
3.5. Magnetic Stimulations Modulated Cytoskeleton Integrity in MNs with FUS Mutations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cook, C.; Petrucelli, L. Genetic Convergence Brings Clarity to the Enigmatic Red Line in ALS. Neuron 2019, 101, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Peters, O.M.; Ghasemi, M.; Jr, R.H.B. Emerging Mechanisms of Molecular Pathology in ALS Find the Latest Version: Emerging Mechanisms of Molecular Pathology in ALS. J. Clin. Investig. 2015, 125, 1767–1779. [Google Scholar] [CrossRef] [PubMed]
- Baeumer, D.; Hilton, D.; Paine, S.M.L.; Turner, M.R.; Lowe, J.; Talbot, K.; Ansorge, O. Juvenile ALS with Basophilic Inclusions Is a FUS Proteinopathy with FUS Mutations. Neurology 2010, 75, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Conte, A.; Lattante, S.; Zollino, M.; Marangi, G.; Luigetti, M.; Del Grande, A.; Servidei, S.; Trombetta, F.; Sabatelli, M. P525L FUS Mutation Is Consistently Associated with a Severe Form of Juvenile Amyotrophic Lateral Sclerosis. Neuromuscul. Disord. 2012, 22, 73–75. [Google Scholar] [CrossRef]
- Naumann, M.; Peikert, K.; Günther, R.; van der Kooi, A.J.; Aronica, E.; Hübers, A.; Danel, V.; Corcia, P.; Pan-Montojo, F.; Cirak, S.; et al. Phenotypes and Malignancy Risk of Different FUS Mutations in Genetic Amyotrophic Lateral Sclerosis. Ann. Clin. Transl. Neurol. 2019, 6, 2384–2394. [Google Scholar] [CrossRef]
- Grassano, M.; Brodini, G.; Marco, G.D.; Casale, F.; Fuda, G.; Salamone, P.; Brunetti, M.; Sbaiz, L.; Gallone, S.; Cugnasco, P.; et al. Phenotype Analysis of Fused in Sarcoma Mutations in Amyotrophic Lateral Sclerosis. Neurol. Genet. 2022, 8, e200011. [Google Scholar] [CrossRef]
- Giancarlo, L.; Marco, P.; Benoît, M.; Emma, N.; Foad, A.-A.; Ahmed, A.; Fares, A.; Weldegebreal, A.S.; Ashish, A.; Yazan, C.; et al. Global, Regional, and National Burden of Motor Neuron Diseases 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2016, 12, 1083–1097. [Google Scholar] [CrossRef]
- Miller, R.G.; Mitchell, J.D.; Lyon, M.; Moore, D.H. Riluzole for Amyotrophic Lateral Sclerosis (ALS)/Motor Neuron Disease (MND). Cochrance Database Syst. Rev. 2002, 4, 191–206. [Google Scholar] [CrossRef]
- Abe, K.; Aoki, M.; Tsuji, S.; Itoyama, Y.; Sobue, G.; Togo, M.; Hamada, C.; Tanaka, M.; Akimoto, M.; Nakamura, K.; et al. Safety and Efficacy of Edaravone in Well Defined Patients with Amyotrophic Lateral Sclerosis: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Witzel, S.; Maier, A.; Steinbach, R.; Grosskreutz, J.; Koch, J.C.; Sarikidi, A.; Petri, S.; Günther, R.; Wolf, J.; Hermann, A.; et al. Safety and Effectiveness of Long-Term Intravenous Administration of Edaravone for Treatment of Patients With Amyotrophic Lateral Sclerosis. JAMA Neurol. 2022, 79, 121–130. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H., Jr.; Don, W. Decoding ALS: From Genes to Mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Gao, K.; Jankovic, J. The Role of FUS Gene Variants in Neurodegenerative Diseases. Nat. Rev. Neurol. 2014, 10, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Naumann, M.; Pal, A.; Goswami, A.; Lojewski, X.; Japtok, J.; Vehlow, A.; Naujock, M.; Günther, R.; Jin, M.; Stanslowsky, N.; et al. Impaired DNA Damage Response Signaling by FUS- NLS Mutations Leads to Neurodegeneration and FUS Aggregate Formation. Nat. Commun. 2018, 9, 335. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovás, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 Inhibition Reverses Axonal Transport Defects in Motor Neurons Derived from FUS-ALS Patients. Nat. Commun. 2017, 8, 861. [Google Scholar] [CrossRef] [PubMed]
- Alhindi, A.; Boehm, I.; Chaytow, H. Small Junction, Big Problems: Neuromuscular Junction Pathology in Mouse Models of Amyotrophic Lateral Sclerosis (ALS). J. Anat. 2021, 241, 1089–1107. [Google Scholar] [CrossRef] [PubMed]
- Hallett, M. Transcranial Magnetic Stimulation and the Human Brain. Nature 2000, 406, 147–150. [Google Scholar] [CrossRef]
- Petersen, N.T.; Pyndt, H.S.; Nielsen, J.B. Investigating Human Motor Control by Transcranial Magnetic Stimulation. Exp. Brain Res. 2003, 152, 1–16. [Google Scholar] [CrossRef]
- Fregni, F.; Pascual-leone, A. Technology Insight: Noninvasive Brain Stimulation in Neurology—Perspectives on the Therapeutic Potential of RTMS and TDCS. Nat. Publ. Group 2007, 3, 383–393. [Google Scholar] [CrossRef]
- Stölting, M.N.L.; Arnold, A.S.; Haralampieva, D.; Handschin, C.; Sulser, T.; Eberli, D. Magnetic Stimulation Supports Muscle and Nerve Regeneration after Trauma in Mice. Muscle Nerve 2016, 53, 598–607. [Google Scholar] [CrossRef]
- Musarò, A.; Dobrowolny, G.; Cambieri, C.; Onesti, E.; Ceccanti, M.; Frasca, V.; Pisano, A.; Cerbelli, B.; Lepore, E.; Ruffolo, G.; et al. Neuromuscular Magnetic Stimulation Counteracts Muscle Decline in ALS Patients: Results of a Randomized, Double-Blind, Controlled Study. Sci. Rep. 2019, 9, 2837. [Google Scholar] [CrossRef]
- Pal, A.; Kretner, B.; Abo-Rady, M.; Glab, H.; Dash, B.P.; Naumann, M.; Japtok, J.; Kreiter, N.; Dhingra, A.; Heutink, P.; et al. Concomitant Gain and Loss of Function Pathomechanisms in C9ORF72 Amyotrophic Lateral Sclerosis. Life Sci. Alliance 2021, 4, 1–26. [Google Scholar] [CrossRef]
- Bruneteau, G.; Bauché, S.; de Aguilar, J.L.G.; Brochier, G.; Mandjee, N.; Tanguy, M.-L.; Hussain, G.; Behin, A.; Khiami, F.; Sariali, E.; et al. Endplate Denervation Correlates with Nogo-A Muscle Expression in Amyotrophic Lateral Sclerosis Patients. Ann. Clin. Transl. Neurol. 2015, 2, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Japtok, J.; Lojewksi, X.; Naumann, M.; Klingenstein, M.; Reinhardt, P.; Sterneckert, J.; Putz, S.; Demestre, M.; Boeckers, T.M.; Ludolph, A.C.; et al. Stepwise Acquirement of Hallmark Neuropathology in FUS-ALS IPSC Models Depends on Mutation Type and Neuronal Aging. Neurobiol. Dis. 2015, 82, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Higelin, J.; Demestre, M.; Putz, S.; Delling, J.P.; Jacob, C.; Lutz, A.K.; Bausinger, J.; Huber, A.K.; Klingenstein, M.; Barbi, G.; et al. FUS Mislocalization and Vulnerability to DNA Damage in ALS Patients Derived HiPSCs and Aging Motoneurons. Front. Cell. Neurosci. 2016, 10, 290. [Google Scholar] [CrossRef] [PubMed]
- Naujock, M.; Stanslowsky, N.; Bufler, S.; Naumann, M.; Reinhardt, P.; Sterneckert, J.; Ekaterini, K.; Carola, K.; Franziska, B.; Lojewski, X.; et al. 4-Aminopyridine Induced Activity Rescues Hypoexcitable Motor Neurons from Amyotrophic Lateral Sclerosis Patient-Derived Induced Pluripotent Stem Cells. Stem Cells 2016, 34, 1563–1575. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, P.; Glatza, M.; Hemmer, K.; Tsytsyura, Y.; Thiel, C.S.; Höing, S.; Moritz, S.; Parga, J.A.; Wagner, L.; Bruder, J.M.; et al. Derivation and Expansion Using Only Small Molecules of Human Neural Progenitors for Neurodegenerative Disease Modeling. PLoS ONE 2013, 8, e59252. [Google Scholar] [CrossRef]
- Pal, A.; Glaß, H.; Naumann, M.; Kreiter, N.; Japtok, J.; Sczech, R.; Hermann, A. High Content Organelle Trafficking Enables Disease State Profiling as Powerful Tool for Disease Modelling. Sci. Data 2018, 5, 180241. [Google Scholar] [CrossRef]
- Taylor, A.M.; Blurton-Jones, M.; Rhee, S.W.; Cribbs, D.H.; Cotman, C.W.; Jeon, N.L. A Microfluidic Culture Platform for CNS Axonal Injury, Regeneration and Transport. Nat. Methods 2005, 2, 599–605. [Google Scholar] [CrossRef]
- Tsantoulas, C.; Farmer, C.; Machado, P.; Baba, K.; McMahon, S.B.; Raouf, R. Probing Functional Properties of Nociceptive Axons Using a Microfluidic Culture System. PLoS ONE 2013, 8, e80722. [Google Scholar] [CrossRef]
- Vysokov, N.; McMahon, S.B.; Raouf, R. The Role of NaV Channels in Synaptic Transmission after Axotomy in a Microfluidic Culture Platform. Sci. Rep. 2019, 9, 12915. [Google Scholar] [CrossRef]
- Piacentini, R.; Ripoli, C.; Mezzogori, D.; Azzena, G.B.; Grassi, C. Extremely Low-Frequency Electromagnetic Fields Promote In Vitro Neurogenesis Via Upregulation of Ca v 1-Channel Activity. J. Cell. Physiol. 2008, 251, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Maeder, C.I.; Shen, K.; Hoogenraad, C.C. Axon and Dendritic Trafficking. Curr. Opin. Neurobiol. 2014, 27, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Pierre, U. Axonal Transport Deficits and Neurodegenerative Diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [Google Scholar] [CrossRef]
- Glaß, H.; Neumann, P.; Pal, A.; Reinhardt, P.; Storch, A.; Sterneckert, J.; Hermann, A. Combined Dendritic and Axonal Deterioration Are Responsible for Motoneuronopathy in Patient-Derived Neuronal Cell Models of Chorea-Acanthocytosis. Int. J. Mol. Sci. 2020, 21, 1797. [Google Scholar] [CrossRef]
- Mateos-aparicio, P.; Rodríguez-moreno, A. The Impact of Studying Brain Plasticity. Front. Cell. Neurosci. 2019, 13, 66. [Google Scholar] [CrossRef]
- Burke, S.N.; Barnes, C.A. Neural Plasticity in the Ageing Brain. Nature 2006, 7, 30–40. [Google Scholar] [CrossRef]
- Bradke, F.; Fawcett, J.W.; Spira, M.E. Assembly of a New Growth Cone after Axotomy: The Precursor to Axon Regeneration. Nat. Rev. Neurosci. 2012, 13, 183–193. [Google Scholar] [CrossRef]
- Alvarez, F.J.; Rotterman, T.M.; Akhter, E.T.; Lane, A.R.; English, A.W.; Cope, T.C. Synaptic Plasticity on Motoneurons After Axotomy: A Necessary Change in Paradigm. Front. Mol. Neurosci. 2020, 13, 68. [Google Scholar] [CrossRef]
- Groen, E.J.N.; Fumoto, K.; Blokhuis, A.M.; Engelen-lee, J.; Heuvel, D.M.A.V.D.; Koppers, M.; Diggelen, F.V.; Heest, J.V.; Demmers, J.A.A.; Kirby, J.; et al. ALS-Associated Mutations in FUS Disrupt the Axonal Distribution and Function of SMN. Hum. Mol. Genet. 2013, 22, 3690–3704. [Google Scholar] [CrossRef]
- Chapin, S.J.; Bulinski, J.C.; Gundersen, G. Microtubule Bundling in Cells. Nature 1991, 349, 24. [Google Scholar] [CrossRef]
- Xu, Z.; Schaedel, L.; Portran, D.; Aguilar, A.; Gaillard, J.; Marinkovich, M.P.; Théry, M.; Nachury, M.V. Microtubules Acquire Resistance from Mechanical Breakage through Intralumenal Acetylation. Science 2017, 356, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Janke, C.; Montagnac, G. Causes and Consequences of Microtubule Acetylation. Curr. Biol. 2017, 27, R1287–R1292. [Google Scholar] [CrossRef] [PubMed]
- Bozzo, F.; Mirra, A.; Carrì, M.T. Oxidative Stress and Mitochondrial Damage in the Pathogenesis of ALS: New Perspectives. Neurosci. Lett. 2017, 636, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Burk, K.; Pasterkamp, R.J. Disrupted Neuronal Trafficking in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2019, 137, 859–877. [Google Scholar] [CrossRef]
- Kreiter, N.; Pal, A.; Lojewski, X.; Corcia, P.; Naujock, M.; Reinhardt, P.; Sterneckert, J.; Petri, S.; Wegner, F.; Storch, A.; et al. Age-Dependent Neurodegeneration and Organelle Transport Deficiencies in Mutant TDP43 Patient-Derived Neurons Are Independent of TDP43 Aggregation. Neurobiol. Dis. 2018, 115, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Dis, V.V.; Kuijpers, M.; Haasdijk, E.D.; Teuling, E.; Oakes, S.A.; Hoogenraad, C.C.; Jaarsma, D. Golgi Fragmentation Precedes Neuromuscular Denervation and Is Associated with Endosome Abnormalities in SOD1-ALS Mouse Motor Neurons. Acta Neuropathol. Commun. 2014, 2, 38. [Google Scholar] [CrossRef] [PubMed]
- Workman, M.J.; Lim, R.G.; Wu, J.; Frank, A.; Ornelas, L.; Panther, L.; Galvez, E.; Perez, D.; Meepe, I.; Lei, S.; et al. Large-Scale Differentiation of IPSC-Derived Motor Neurons from ALS and Control Subjects. Neuron 2023, 111, 1191–1204.e5. [Google Scholar] [CrossRef]
- Campanari, M.; Ciura, S. Neuromuscular Junction Impairment in Amyotrophic Lateral Sclerosis: Reassessing the Role of Acetylcholinesterase. Front. Mol. Neurosci. 2016, 9, 160. [Google Scholar] [CrossRef]
- Sivadasan, R.; Hornburg, D.; Drepper, C.; Frank, N.; Jablonka, S.; Hansel, A.; Lojewski, X.; Sterneckert, J.; Hermann, A.; Shaw, P.J.; et al. C9ORF72 Interaction with Cofilin Modulates Actin Dynamics in Motor Neurons. Nat. Neurosci. 2016, 19, 1610–1618. [Google Scholar] [CrossRef]
- So, E.; Mitchell, J.C.; Memmi, C.; Chennell, G.; Vizcay-barrena, G.; Allison, L.; Shaw, C.E.; Vance, C. Mitochondrial Abnormalities and Disruption of the Neuromuscular Junction Precede the Clinical Phenotype and Motor Neuron Loss in HFUS WT Transgenic Mice. Hum. Mol. Genet. 2018, 27, 463–474. [Google Scholar] [CrossRef]
- Deshpande, D.; Higelin, J.; Schoen, M.; Vomhof, T.; Boeckers, T.M.; Demestre, M.; Michaelis, J.; Vance, C. Synaptic FUS Localization During Motoneuron Development and Its Accumulation in Human ALS Synapses. Front. Cell. Neurosci. 2019, 13, 256. [Google Scholar] [CrossRef]
- Zholudeva, L.V.; Qiang, L.; Marchenko, V.; Dougherty, K.J.; Sakiyama-elbert, S.E.; Lane, M.A.; Stop, C. The Neuroplastic and Therapeutic Potential of Spinal Interneurons in the Injured Spinal Cord. Trends Neurosci. 2018, 41, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Sengottuvel, V.; Leibinger, M.; Pfreimer, M.; Andreadaki, A.; Fischer, D. Taxol Facilitates Axon Regeneration in the Mature CNS. J. Neurosci. 2011, 31, 2688–2699. [Google Scholar] [CrossRef] [PubMed]
- Rivieccio, M.A.; Brochier, C.; Willis, D.E.; Walker, B.A.; D’Annibale, M.A.; McLaughlin, K.; Siddiq, A.; Kozikowski, A.P.; Jaffrey, S.R.; Twiss, J.L.; et al. HDAC6 Is a Target for Protection and Regeneration Following Injury in the Nervous System. Proc. Natl. Acad. Sci. USA 2009, 106, 19599–19604. [Google Scholar] [CrossRef]
- Guo, W.; Ludo, V.D.B. Therapeutic Potential of HDAC6 in Amyotrophic Lateral Sclerosis. Cell Stress 2017, 2, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Binder, L.I.; Rosenbaum, J.L. The Periodic Association of MAP2 with Brain Microtubules in Vitro. J. Cell Biol. 1979, 80, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Balabanian, L.; Berger, C.L.; Hendricks, A.G. Acetylated Microtubules Are Preferentially Bundled Leading to Enhanced Kinesin-1 Motility. Biophys. J. 2017, 113, 1551–1560. [Google Scholar] [CrossRef]
- Garone, M.G.; Birsa, N.; Rosito, M.; Salaris, F.; Mochi, M.; de Turris, V.; Nair, R.R.; Cunningham, T.J.; Fisher, E.M.C.; Morlando, M.; et al. ALS-Related FUS Mutations Alter Axon Growth in Motoneurons and Affect HuD/ELAVL4 and FMRP Activity. Commun. Biol. 2021, 4, 1025. [Google Scholar] [CrossRef]
- Akiyama, T.; Suzuki, N.; Ishikawa, M.; Fujimori, K.; Sone, T.; Kawada, J.; Funayama, R.; Fujishima, F.; Mitsuzawa, S.; Ikeda, K.; et al. Aberrant Axon Branching via Fos-B Dysregulation in FUS-ALS Motor Neurons. eBioMedicine 2019, 45, 362–378. [Google Scholar] [CrossRef]
- Osking, Z.; Ayers, J.I.; Hildebrandt, R.; Skruber, K.; Brown, H.; Ryu, D.; Eukovich, A.R.; Golde, T.E.; Borchelt, D.R.; Read, T.-A.; et al. ALS-Linked SOD1 Mutants Enhance Neurite Outgrowth and Branching in Adult Motor Neurons. iScience 2019, 11, 294–304. [Google Scholar] [CrossRef]
- Marshall, K.L.; Rajbhandari, L.; Venkatesan, A.; Maragakis, N.J.; Farah, M.H. Enhanced Axonal Regeneration of ALS Patient IPSC-Derived Motor Neurons Harboring SOD1(A4V) Mutation. Sci. Rep. 2023, 13, 5597. [Google Scholar] [CrossRef]
- McWhorter, M.L.; Monani, U.R.; Burghes, A.H.M.; Beattie, C.E. Knockdown of the Survival Motor Neuron (Smn) Protein in Zebrafish Causes Defects in Motor Axon Outgrowth and Pathfinding. J. Cell Biol. 2003, 162, 919–931. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gender | Age of Biopsy | Written as | Mutation | Clinical Features | Previously Characterized in | |
|---|---|---|---|---|---|---|
| Control | Male | 34 | Ctrl1 | - | - | [13] |
| Female | n.d. | Ctrl2 | - | - | [13] | |
| Female | 53 | Ctrl3 | - | - | [13] | |
| ALS-FUS | Female | 58 | FUS R521C | R521Chet | Spinal | [13] |
| Isogenic control of R521C | Same as above | FUS WT | WT-EGFP | n/a | [13] | |
| Isogenic variant of R521C | Same as above | FUS P525L | P525L-EGFP | n/a | [13] | |
| Female | 65 | FUS R521L | R521Lhet | Spinal | [13] | |
| Male | 29 | FUS 495QfsX527 | R495QfsX527het | Spinal | [13] |
| Bonferroni’s Multiple Comparison Test | Significant Different | Adjusted p-Value | Summary |
|---|---|---|---|
| Ctrl untreated vs. 2 Hz | No | ˃0.9999 | ns |
| Ctrl untreated vs. 10 Hz | No | ˃0.9999 | ns |
| Ctrl untreated vs. 10/2 Hz | Yes | 0.0063 | ** |
| mutant FUS untreated vs. Ctrl untreated | Yes | ˂0.0001 | **** |
| mutant FUS untreated vs. 2 Hz | No | 0.9708 | ns |
| mutant FUS untreated vs. 10 Hz | Yes | ˂0.0001 | **** |
| mutant FUS untreated vs. 10/2 Hz | Yes | ˂0.0001 | **** |
| mutant FUS 2 Hz vs. Ctrl untreated | Yes | ˂0.0001 | **** |
| mutant FUS 10 Hz vs. Ctrl untreated | No | 0.9482 | ns |
| mutant FUS 10/2 Hz vs. Ctrl untreated | No | ˃0.9999 | ns |
| mutant FUS 2 Hz vs. Ctrl 2 Hz | Yes | ˂0.0001 | **** |
| mutant FUS 10 Hz vs. Ctrl 10 Hz | No | ˃0.9999 | ns |
| mutant FUS 10/2 Hz vs. Ctrl 10/2Hz | Yes | 0.0084 | ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kandhavivorn, W.; Glaß, H.; Herrmannsdörfer, T.; Böckers, T.M.; Uhlarz, M.; Gronemann, J.; Funk, R.H.W.; Pietzsch, J.; Pal, A.; Hermann, A. Restoring Axonal Organelle Motility and Regeneration in Cultured FUS-ALS Motoneurons through Magnetic Field Stimulation Suggests an Alternative Therapeutic Approach. Cells 2023, 12, 1502. https://doi.org/10.3390/cells12111502
Kandhavivorn W, Glaß H, Herrmannsdörfer T, Böckers TM, Uhlarz M, Gronemann J, Funk RHW, Pietzsch J, Pal A, Hermann A. Restoring Axonal Organelle Motility and Regeneration in Cultured FUS-ALS Motoneurons through Magnetic Field Stimulation Suggests an Alternative Therapeutic Approach. Cells. 2023; 12(11):1502. https://doi.org/10.3390/cells12111502
Chicago/Turabian StyleKandhavivorn, Wonphorn, Hannes Glaß, Thomas Herrmannsdörfer, Tobias M. Böckers, Marc Uhlarz, Jonas Gronemann, Richard H. W. Funk, Jens Pietzsch, Arun Pal, and Andreas Hermann. 2023. "Restoring Axonal Organelle Motility and Regeneration in Cultured FUS-ALS Motoneurons through Magnetic Field Stimulation Suggests an Alternative Therapeutic Approach" Cells 12, no. 11: 1502. https://doi.org/10.3390/cells12111502
APA StyleKandhavivorn, W., Glaß, H., Herrmannsdörfer, T., Böckers, T. M., Uhlarz, M., Gronemann, J., Funk, R. H. W., Pietzsch, J., Pal, A., & Hermann, A. (2023). Restoring Axonal Organelle Motility and Regeneration in Cultured FUS-ALS Motoneurons through Magnetic Field Stimulation Suggests an Alternative Therapeutic Approach. Cells, 12(11), 1502. https://doi.org/10.3390/cells12111502