
Current State and Future Directions in the Therapy of ALS



Abstract
:1. Introduction
2. Current Treatment Options
2.1. Medical Treatment
2.1.1. Riluzole
2.1.2. Edaravone
2.1.3. Sodium Phenylbutyrate and Taurursodiol
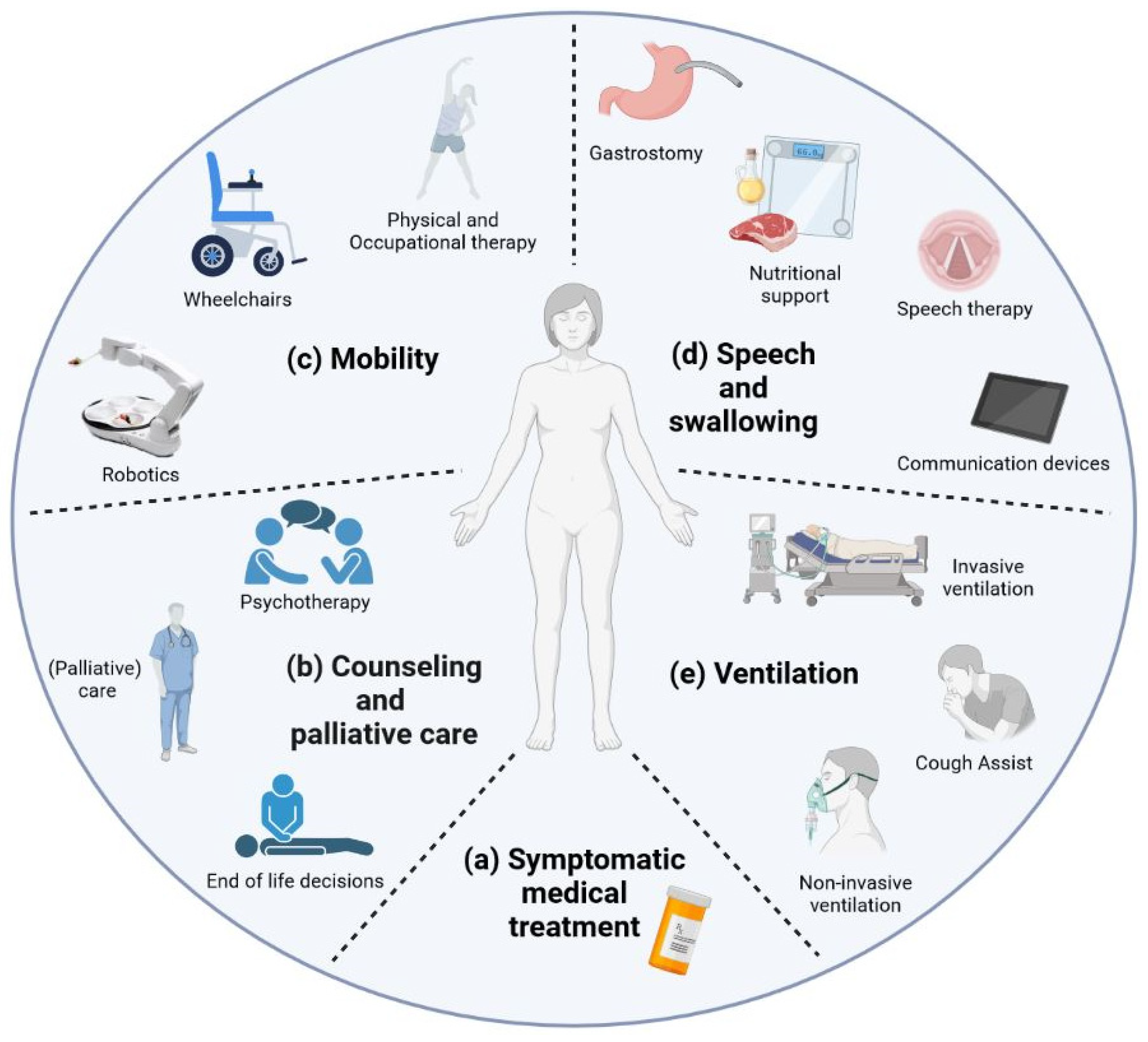
2.2. Supportive Therapy
2.2.1. Mobility
Physical and Occupational Therapy
Robotics
2.2.2. Speech and Swallowing
Speech Therapy and Communication Devices
Nutritional Therapy
2.2.3. Ventilation
3. On-Going Development and Future Perspectives in the Field
3.1. Therapies Currently Being Tested
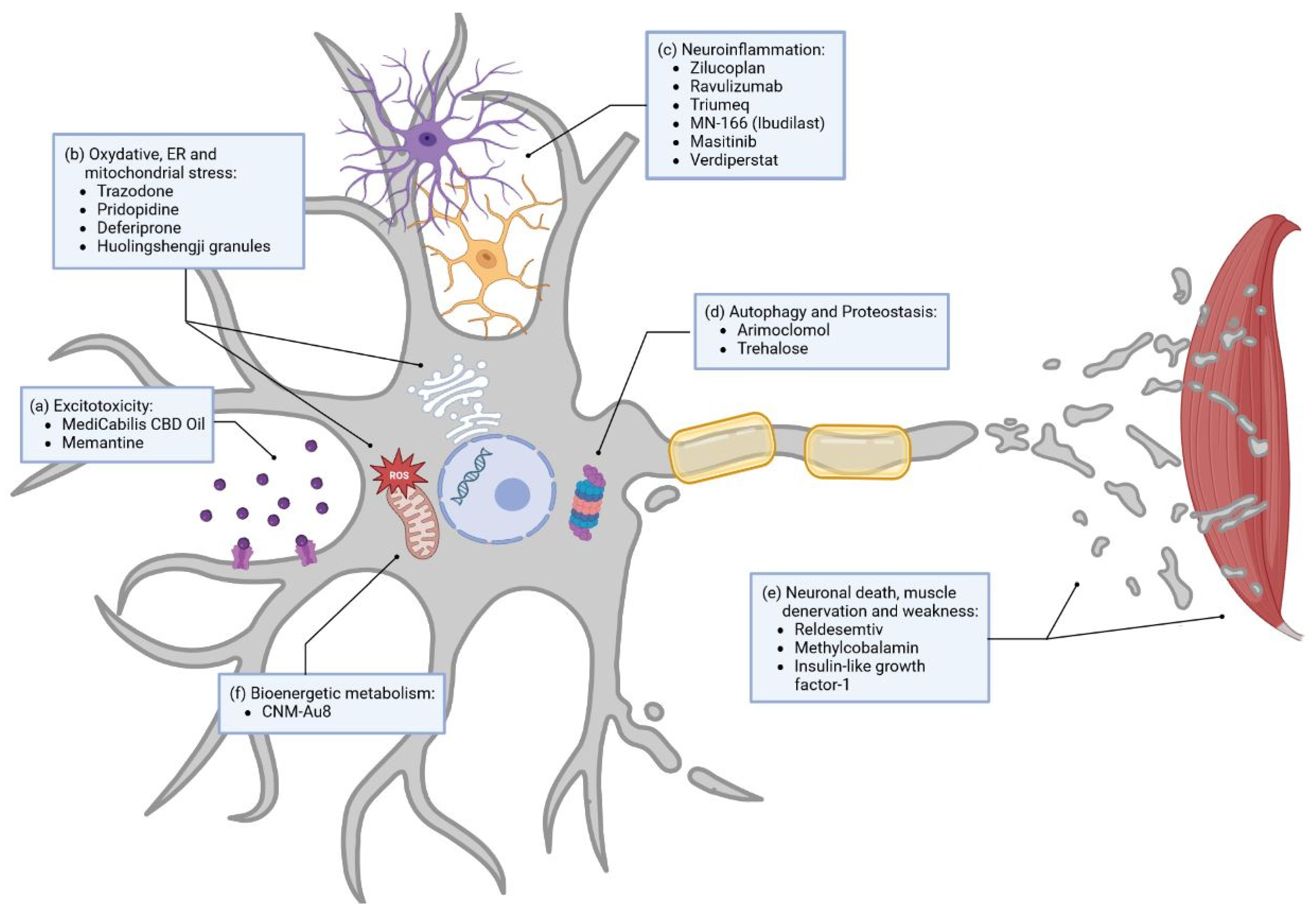
3.1.1. Small Molecules
Excitotoxicity
Oxidative Stress
Neuroinflammation
Others
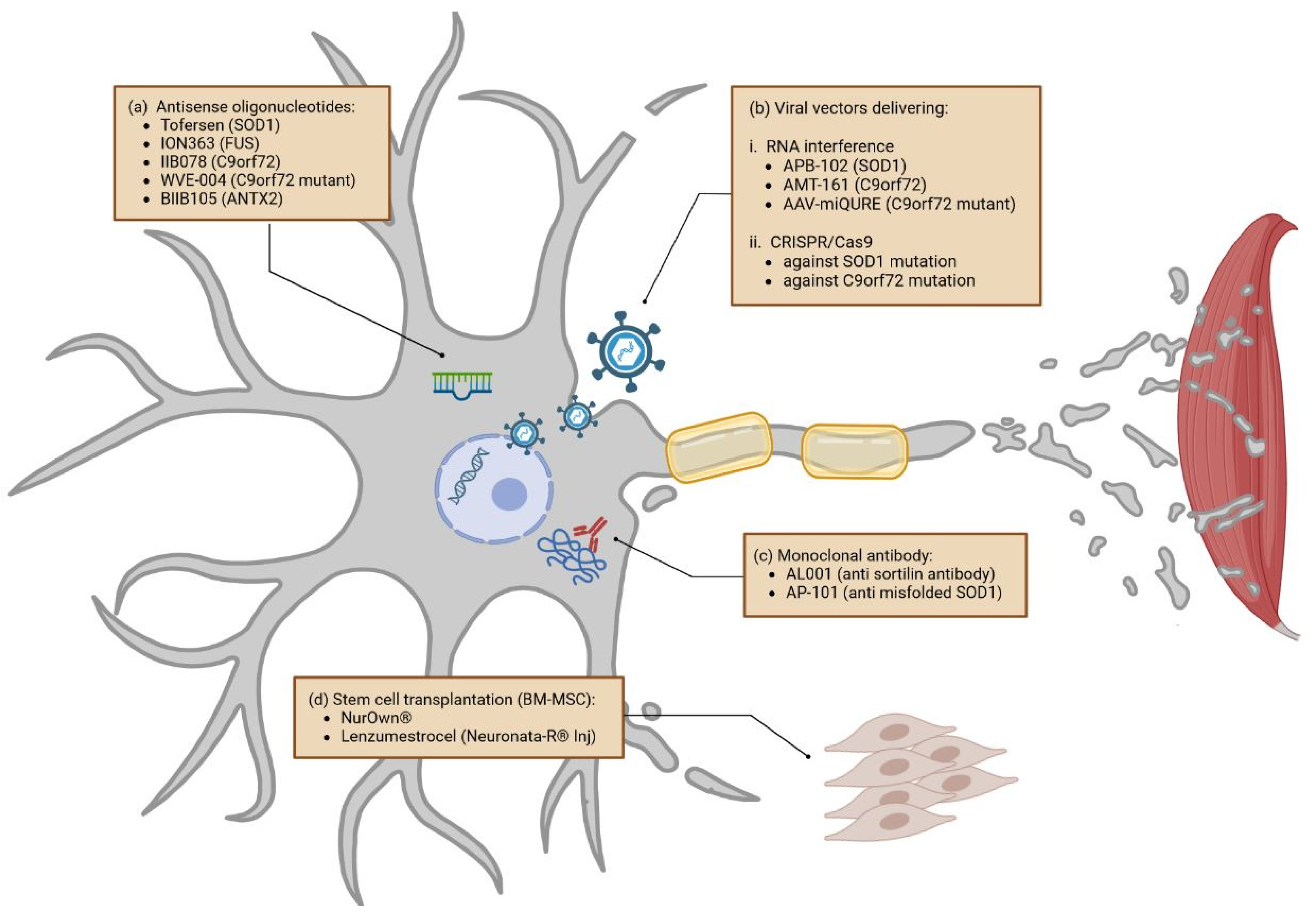
3.1.2. Gene-Specific Therapies
Antisense Oligonucleotides
Viral Vectors Delivering RNA Interference and CRISPR/Cas9
3.1.3. Monoclonal Antibodies
3.1.4. Stem Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Approved Drug | Pathway | Phase |
| Riluzole | Excitotoxicity | Approved |
| Edaravone | Oxidative stress | Approved |
| Sodium phenylbutyrate/Taurursodiol | Oxidative stress | Approved |
| Therapeutic being tested (selection) | Pathway/Approach | Phase |
| CBD oil (NCT03690791) | Excitotoxicity | Phase III |
| Memantine (NCT04302870) | Excitotoxicity | Phase II/III |
| Trazodone (NCT04302870) | Oxidative stress | Phase III |
| Huolingshengji granules (NCT04950933) | Oxidative stress | Phase III |
| Pridopidine (NCT04615923) | Oxidative stress | Phase II/III |
| Deferiprone (NCT03293069) | Oxidative stress | Phase II/III |
| Triumeq (NCT05193994) | Neuroinflammation | Phase III |
| Masitinib (NCT03127267) | Neuroinflammation | Phase III |
| MN-166/Ibudilast (NCT04057898) | Neuroinflammation | Phase II/III |
| Verdiperstat (NCT04436510) | Neuroinflammation | Phase II/III |
| Ravulizumab (NCT04248465) | Neuroinflammation | Stopped in January 2023 |
| Zilucoplan (NCT04436497) | Neuroinflammation | Stopped in June 2020 |
| Methylcobalamin (NCT03548311) | OTHERS Reduces denervation and muscle weakness | Phase III |
| Insulin-like growth factor (NCT00035815) | OTHERS Blockage of cell death pathways | Phase III |
| Trehalose (NCT05136885) | OTHERS Enhances autophagy, decreases SOD1 aggregates | Phase II/III |
| CNM-Au8 (NCT04615923) | OTHERS Energy Metabolism | Phase II/III |
| Reldesemtiv (NCT04944784) | OTHERS Slows calcium release | Stopped in March 2023 |
| Arimoclomol (NCT03836716) | OTHERS (Increased heat shock protein expression, enhancement of autophagy) | Stopped |
| Tofersen/BIIB067 (NCT04972487) | Gene specific–ASO, SOD1-mutations | Phase III |
| Jacifusen/ION363 (NCT04768972) | Gene specific–ASO, FUS-mutations | Phase III |
| WVE-004 (NCT04931862) | Gene specific–ASO, mutant C9orf72 | Phase I/II |
| BIIB105 (NCT04494256) | Gene specific–ASO, ATXN2 | Phase I |
| IIB078 (NCT03626012B) | Gene specific–ASO, C9orf72 | Stopped |
| APB-102 | SOD1 microRNA | Phase I/II (begin date mid–end 2023) |
| AMT-161 | C9orf72 microRNA | Substance in development |
| AAV-miQURE | Mutant C9orf72 microRNA | Substance in development |
| AL001 (NCT05053035) | Monoclonal AB, C9orf72 | Phase II |
| AP-101 (NCT05039099) | Monoclonal AB, SOD1 | Phase II |
| ANX-005 (NCT04569435) | Monoclonal AB | Phase II |
| AT-1501 (NCT04322149) | Monoclonal AB | Phase II |
| Tocilizumab (NCT02469896) | Monoclonal AB | Phase II |
| NurOwn (NCT04681118) | Stem cells | Phase III |
| Lenzumestrocel (NCT04745299) | Stem cells | Phase III |
| Blood-Brain Barrier Opening Using MR-Guided Focused Ultrasound (NCT03321487) | Blood-Brain Barrier Opening | Not Applicable |
| Fasudil (NCT03792490) | Post-translational modifications | Phase IIa |
| BIIB100 (NCT03945279) | Nucleocytoplasmic transport | Stopped |
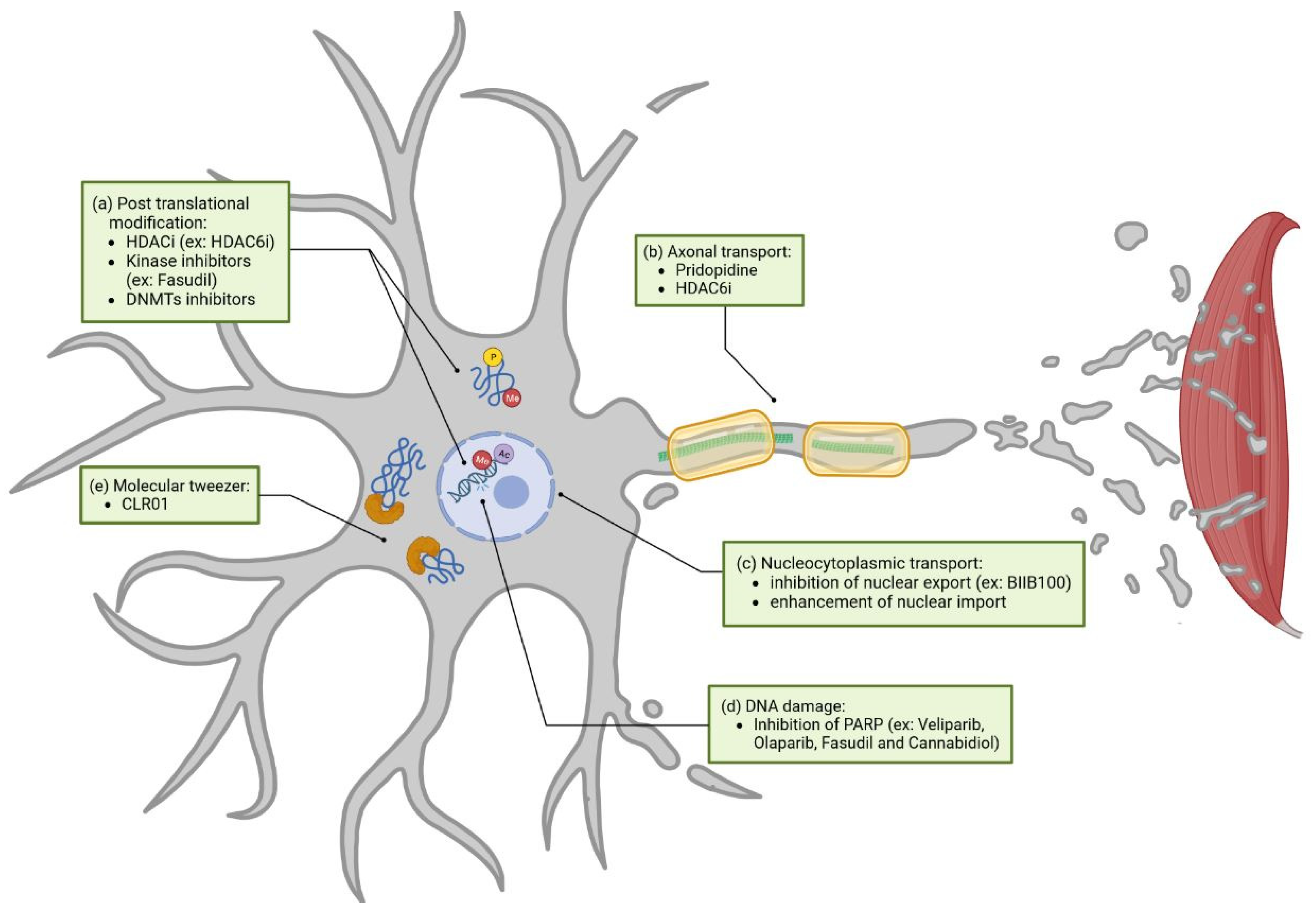
3.2. Novel Therapeutic Approaches (Unaddressed or Poorly Addressed Yet)
3.2.1. Post-Translational Modification
3.2.2. Axonal Transport
3.2.3. Nucleocytoplasmic Transport
3.2.4. DNA Damage
3.2.5. Molecular Tweezer
4. Stratification towards Personalized Medicine
4.1. Clinical Characteristics
4.2. Genetic Classification
4.3. The Power of Biomarkers
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AAVs | Adeno-associated viruses |
| ADP | Adenosine diphosphate |
| ALS | Amyotrophic Lateral Sclerosis |
| ALSFRS-R | Revised ALS Functional Rating Scale |
| AMPA | Aminomethylphosphonic acid |
| ASO | Antisense oligonucleotide |
| ATXN2 | Ataxin-2 |
| BMI | Body mass index |
| CBD | Cannabidiol |
| C9orf72 | Chromosome 9 open reading frame 72 |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| DNMTs | DNA methyltransferases |
| dsRNAs | Double strand RNA |
| EAAT2 | Excitatory amino acid transporter 2 |
| EEG | Electroencephalographic |
| EMG | Electromyographic |
| ER | Endoplasmic reticulum |
| fALS | Familial ALS |
| FTD | Frontotemporal Dementia |
| FUS | Fused in sarcoma |
| FVC | Forced vital capacity |
| GABA | Gamma-aminobutyric acid |
| HDACi | Histone deacetylase inhibitors |
| iPSC | Induced pluripotent stem cells |
| miRNA | micro RNA |
| MME | motor-assisted movement exercisers |
| Nf | Neurofilaments |
| NfL | Neurofilament light chain |
| NGT | Nasogastric tube |
| NIV | Non-invasive ventilation |
| NMDA | N-methyl-D-aspartate |
| PAR | Poly Adenosine diphosphate (ADP)-ribose |
| PARP | Poly ADP-ribose polymerase |
| PEG | Percutaneous gastrostomy |
| PERK | Protein kinase Ribonucleic acid (RNA)-like endoplasmic reticulum kinase |
| PGRN | Progranulin |
| PTMs | Post-translational modifications |
| RIG | Radiologically inserted gastrostomy |
| RISC | RNA-induced silencing complex |
| RNA | Ribonucleic acid |
| sALS | Sporadic ALS |
| SCA2 | Spinocerebellar Ataxia Type 2 |
| siRNA | Small interfering RNA |
| SNP | Sniff nasal pressure |
| SOD1 | Superoxide dismutase 1 |
| TDP-43 | Transactive response DNA binding protein 43 kDa |
| VC | Vital capacity |
References
- Swinnen, B.; Robberecht, W. The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Paganoni, S.; Macklin, E.A.; Hendrix, S.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; Quick, A.; et al. Trial of Sodium Phenylbutyrate-Taurursodiol for Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2020, 383, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Bensimon, G.; Lacomblez, L.; Meininger, V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Itoyama, Y.; Sobue, G.; Tsuji, S.; Aoki, M.; Doyu, M.; Hamada, C.; Kondo, K.; Yoneoka, T.; Akimoto, M.; et al. Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 610–617. [Google Scholar] [CrossRef]
- Cedarbaum, J.M.; Stambler, N.; Malta, E.; Fuller, C.; Hilt, D.; Thurmond, B.; Nakanishi, A. The ALSFRS-R: A revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J. Neurol. Sci. 1999, 169, 13–21. [Google Scholar] [CrossRef]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef]
- Georgoulopoulou, E.; Fini, N.; Vinceti, M.; Monelli, M.; Vacondio, P.; Bianconi, G.; Sola, P.; Nichelli, P.; Mandrioli, J. The impact of clinical factors, riluzole and therapeutic interventions on ALS survival: A population based study in Modena, Italy. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 338–345. [Google Scholar] [CrossRef]
- Hinchcliffe, M.; Smith, A. Riluzole: Real-world evidence supports significant extension of median survival times in patients with amyotrophic lateral sclerosis. Degener. Neurol. Neuromuscul. Dis. 2017, 7, 61–70. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef]
- Mead, R.J.; Shan, N.; Reiser, H.J.; Marshall, F.; Shaw, P.J. Amyotrophic lateral sclerosis: A neurodegenerative disorder poised for successful therapeutic translation. Nat. Rev. Drug Discov. 2023, 22, 185–212. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Halliday, G.M.; Kril, J.J.; Götz, J.; Hodges, J.R.; Kiernan, M.C. FTD and ALS--translating mouse studies into clinical trials. Nat. Rev. Neurol. 2015, 11, 360–366. [Google Scholar] [CrossRef]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment? Front. Aging Neurosci. 2017, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Shefner, J.M.; Bedlack, R.; Andrews, J.A.; Berry, J.D.; Bowser, R.; Brown, R.; Glass, J.D.; Maragakis, N.J.; Miller, T.M.; Rothstein, J.D.; et al. Amyotrophic Lateral Sclerosis Clinical Trials and Interpretation of Functional End Points and Fluid Biomarkers: A Review. JAMA Neurol. 2022, 79, 1312–1318. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.A.; Fang, T.; De Marchi, F.; Neel, D.; Van Weehaeghe, D.; Berry, J.D.; Paganoni, S. Pharmacotherapy for Amyotrophic Lateral Sclerosis: A Review of Approved and Upcoming Agents. Drugs 2022, 82, 1367–1388. [Google Scholar] [CrossRef]
- Ortega-Hombrados, L.; Molina-Torres, G.; Galán-Mercant, A.; Sánchez-Guerrero, E.; González-Sánchez, M.; Ruiz-Muñoz, M. Systematic Review of Therapeutic Physical Exercise in Patients with Amyotrophic Lateral Sclerosis over Time. Int. J. Environ. Res. Public Health 2021, 18, 1074. [Google Scholar] [CrossRef]
- Hwang, C.-S.; Weng, H.-H.; Wang, L.-F.; Tsai, C.-H.; Chang, H.-T. An eye-tracking assistive device improves the quality of life for ALS patients and reduces the caregivers’ burden. J. Mot. Behav. 2014, 46, 233–238. [Google Scholar] [CrossRef]
- Rosa Silva, J.P.; Santiago Júnior, J.B.; Dos Santos, E.L.; de Carvalho, F.O.; de França Costa, I.M.P.; de Mendonça, D.M.F. Quality of life and functional independence in amyotrophic lateral sclerosis: A systematic review. Neurosci. Biobehav. Rev. 2020, 111, 1–11. [Google Scholar] [CrossRef]
- EFNS Task Force on Diagnosis and Management of Amyotrophic Lateral Sclerosis; Andersen, P.M.; Abrahams, S.; Borasio, G.D.; de Carvalho, M.; Chio, A.; Van Damme, P.; Hardiman, O.; Kollewe, K.; Morrison, K.E.; et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)--revised report of an EFNS task force. Eur. J. Neurol. 2012, 19, 360–375. [Google Scholar] [CrossRef]
- Ludolph, A.C.; Dorst, J.; Dreyhaupt, J.; Weishaupt, J.H.; Kassubek, J.; Weiland, U.; Meyer, T.; Petri, S.; Hermann, A.; Emmer, A.; et al. Effect of High-Caloric Nutrition on Survival in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2020, 87, 206–216. [Google Scholar] [CrossRef]
- Bourke, S.C.; Tomlinson, M.; Williams, T.L.; Bullock, R.E.; Shaw, P.J.; Gibson, G.J. Effects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: A randomised controlled trial. Lancet Neurol. 2006, 5, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.A.; Lally, C.; Kupelian, V.; Flanders, W.D. Estimated Prevalence and Incidence of Amyotrophic Lateral Sclerosis and SOD1 and C9orf72 Genetic Variants. Neuroepidemiology 2021, 55, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Talbott, E.O.; Malek, A.M.; Lacomis, D. The epidemiology of amyotrophic lateral sclerosis. Handb. Clin. Neurol. 2016, 138, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Rosenbohm, A.; Peter, R.S.; Erhardt, S.; Lulé, D.; Rothenbacher, D.; Ludolph, A.C.; Nagel, G.; ALS Registry Study Group. Epidemiology of amyotrophic lateral sclerosis in Southern Germany. J. Neurol. 2017, 264, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Shen, D.; Yang, X.; Cui, B.; Tai, H.; Wang, Z.; Liu, S.; Zhang, K.; Liu, M.; Cui, L. Early onset but long survival and other prognostic factors in Chinese sporadic amyotrophic lateral sclerosis. J. Clin. Neurosci. 2019, 69, 74–80. [Google Scholar] [CrossRef]
- Gregory, J.M.; Fagegaltier, D.; Phatnani, H.; Harms, M.B. Genetics of Amyotrophic Lateral Sclerosis. Curr. Genet. Med. Rep. 2020, 8, 121–131. [Google Scholar] [CrossRef]
- Albo, F.; Pieri, M.; Zona, C. Modulation of AMPA receptors in spinal motor neurons by the neuroprotective agent riluzole. J. Neurosci. Res. 2004, 78, 200–207. [Google Scholar] [CrossRef]
- Kretschmer, B.D.; Kratzer, U.; Schmidt, W.J. Riluzole, a glutamate release inhibitor, and motor behavior. Naunyn-Schmiedeberg’s Arch. Pharm. 1998, 358, 181–190. [Google Scholar] [CrossRef]
- Fumagalli, E.; Funicello, M.; Rauen, T.; Gobbi, M.; Mennini, T. Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur. J. Pharm. 2008, 578, 171–176. [Google Scholar] [CrossRef]
- Theile, J.W.; Cummins, T.R. Inhibition of Navβ4 peptide-mediated resurgent sodium currents in Nav1.7 channels by carbamazepine, riluzole, and anandamide. Mol. Pharm. 2011, 80, 724–734. [Google Scholar] [CrossRef]
- He, Y.; Benz, A.; Fu, T.; Wang, M.; Covey, D.F.; Zorumski, C.F.; Mennerick, S. Neuroprotective agent riluzole potentiates postsynaptic GABA(A) receptor function. Neuropharmacology 2002, 42, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Lacomblez, L.; Bensimon, G.; Leigh, P.N.; Guillet, P.; Powe, L.; Durrleman, S.; Delumeau, J.C.; Meininger, V. A confirmatory dose-ranging study of riluzole in ALS. ALS/Riluzole Study Group-II. Neurology 1996, 47, S242–S250. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.A.; Jackson, C.E.; Heiman-Patterson, T.D.; Bettica, P.; Brooks, B.R.; Pioro, E.P. Real-world evidence of riluzole effectiveness in treating amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 509–518. [Google Scholar] [CrossRef]
- Brooks, B.R.; Bettica, P.; Cazzaniga, S. Riluzole Oral Suspension: Bioavailability Following Percutaneous Gastrostomy Tube-modeled Administration Versus Direct Oral Administration. Clin. Ther. 2019, 41, 2490–2499. [Google Scholar] [CrossRef]
- Dyer, A.M.; Smith, A. Riluzole 5 mg/mL oral suspension: For optimized drug delivery in amyotrophic lateral sclerosis. Drug Des. Dev. Ther. 2017, 11, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, I.; Lovegren, M.; Wirtz, V.; Larouche, R.; Tanguay, M.; Anderson, M.S.; Hartmann, S.; Coric, V.; Berman, R.M. A Pharmacokinetic Bioequivalence Study Comparing Sublingual Riluzole (BHV-0223) and Oral Tablet Formulation of Riluzole in Healthy Volunteers. Clin. Pharm. Drug Dev. 2020, 9, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Wymer, J.; Apple, S.; Harrison, A.; Hill, B.A. Pharmacokinetics, Bioavailability, and Swallowing Safety with Riluzole Oral Film. Clin. Pharm. Drug Dev. 2023, 12, 57–64. [Google Scholar] [CrossRef]
- Watanabe, K.; Tanaka, M.; Yuki, S.; Hirai, M.; Yamamoto, Y. How is edaravone effective against acute ischemic stroke and amyotrophic lateral sclerosis? J. Clin. Biochem. Nutr. 2018, 62, 20–38. [Google Scholar] [CrossRef]
- Ikeda, K.; Iwasaki, Y.; Kaji, R. Neuroprotective effect of ultra-high dose methylcobalamin in wobbler mouse model of amyotrophic lateral sclerosis. J. Neurol. Sci. 2015, 354, 70–74. [Google Scholar] [CrossRef]
- Watanabe, T.; Yuki, S.; Egawa, M.; Nishi, H. Protective effects of MCI-186 on cerebral ischemia: Possible involvement of free radical scavenging and antioxidant actions. J. Pharm. Exp. 1994, 268, 1597–1604. [Google Scholar]
- Yamamoto, T.; Yuki, S.; Watanabe, T.; Mitsuka, M.; Saito, K.I.; Kogure, K. Delayed neuronal death prevented by inhibition of increased hydroxyl radical formation in a transient cerebral ischemia. Brain Res. 1997, 762, 240–242. [Google Scholar] [CrossRef] [PubMed]
- The Writing Group; Edaravone (MCI-186) ALS 19 Study Group. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef] [PubMed]
- EDARAVONE (MCI-186) ALS 16 STUDY GROUP. A post-hoc subgroup analysis of outcomes in the first phase III clinical study of edaravone (MCI-186) in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Witzel, S.; Maier, A.; Steinbach, R.; Grosskreutz, J.; Koch, J.C.; Sarikidi, A.; Petri, S.; Günther, R.; Wolf, J.; Hermann, A.; et al. Safety and Effectiveness of Long-term Intravenous Administration of Edaravone for Treatment of Patients with Amyotrophic Lateral Sclerosis. JAMA Neurol. 2022, 79, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Lunetta, C.; Moglia, C.; Lizio, A.; Caponnetto, C.; Dubbioso, R.; Giannini, F.; Matà, S.; Mazzini, L.; Sabatelli, M.; Siciliano, G.; et al. The Italian multicenter experience with edaravone in amyotrophic lateral sclerosis. J. Neurol. 2020, 267, 3258–3267. [Google Scholar] [CrossRef]
- Cruz, M.P. Edaravone (Radicava): A Novel Neuroprotective Agent for the Treatment of Amyotrophic Lateral Sclerosis. Pharm. Ther. 2018, 43, 25–28. [Google Scholar]
- Yoshino, H.; Kimura, A. Investigation of the therapeutic effects of edaravone, a free radical scavenger, on amyotrophic lateral sclerosis (Phase II study). Amyotroph. Lateral Scler. 2006, 7, 241–245. [Google Scholar] [CrossRef]
- Peña-Quintana, L.; Llarena, M.; Reyes-Suárez, D.; Aldámiz-Echevarria, L. Profile of sodium phenylbutyrate granules for the treatment of urea-cycle disorders: Patient perspectives. Patient Prefer. Adherence 2017, 11, 1489–1496. [Google Scholar] [CrossRef]
- Hofmann, A.F. The continuing importance of bile acids in liver and intestinal disease. Arch. Intern. Med. 1999, 159, 2647–2658. [Google Scholar] [CrossRef]
- Del Signore, S.J.; Amante, D.J.; Kim, J.; Stack, E.C.; Goodrich, S.; Cormier, K.; Smith, K.; Cudkowicz, M.E.; Ferrante, R.J. Combined riluzole and sodium phenylbutyrate therapy in transgenic amyotrophic lateral sclerosis mice. Amyotroph. Lateral Scler. 2009, 10, 85–94. [Google Scholar] [CrossRef]
- Ryu, H.; Smith, K.; Camelo, S.I.; Carreras, I.; Lee, J.; Iglesias, A.H.; Dangond, F.; Cormier, K.A.; Cudkowicz, M.E.; Brown, R.H.; et al. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J. Neurochem. 2005, 93, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Kusaczuk, M. Tauroursodeoxycholate-Bile Acid with Chaperoning Activity: Molecular and Cellular Effects and Therapeutic Perspectives. Cells 2019, 8, 1471. [Google Scholar] [CrossRef] [PubMed]
- Dionísio, P.A.; Amaral, J.D.; Ribeiro, M.F.; Lo, A.C.; D’Hooge, R.; Rodrigues, C.M.P. Amyloid-β pathology is attenuated by tauroursodeoxycholic acid treatment in APP/PS1 mice after disease onset. Neurobiol. Aging 2015, 36, 228–240. [Google Scholar] [CrossRef]
- Paganoni, S.; Hendrix, S.; Dickson, S.P.; Knowlton, N.; Macklin, E.A.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; et al. Long-term survival of participants in the CENTAUR trial of sodium phenylbutyrate-taurursodiol in amyotrophic lateral sclerosis. Muscle Nerve 2021, 63, 31–39. [Google Scholar] [CrossRef]
- Paganoni, S.; Hendrix, S.; Dickson, S.P.; Knowlton, N.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; et al. Effect of sodium phenylbutyrate/taurursodiol on tracheostomy/ventilation-free survival and hospitalisation in amyotrophic lateral sclerosis: Long-term results from the CENTAUR trial. J. Neurol. Neurosurg. Psychiatry 2022, 93, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.; Jozsa, F.; Al-Chalabi, A. Nonmotor Symptoms in Amyotrophic Lateral Sclerosis: A Systematic Review. Int. Rev. Neurobiol. 2017, 134, 1409–1441. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.; Pioro, E.; Myers, K.; Sirdofsky, M.; Goslin, K.; Meekins, G.; Yu, H.; Wymer, J.; Cudkowicz, M.; Macklin, E.A.; et al. Enhanced Bulbar Function in Amyotrophic Lateral Sclerosis: The Nuedexta Treatment Trial. Neurotherapeutics 2017, 14, 762–772. [Google Scholar] [CrossRef]
- Chiò, A.; Bottacchi, E.; Buffa, C.; Mutani, R.; Mora, G.; The PARALS. Positive effects of tertiary centres for amyotrophic lateral sclerosis on outcome and use of hospital facilities. J. Neurol. Neurosurg. Psychiatry 2006, 77, 948–950. [Google Scholar] [CrossRef]
- Paipa, A.J.; Povedano, M.; Barcelo, A.; Domínguez, R.; Saez, M.; Turon, J.; Prats, E.; Farrero, E.; Virgili, N.; Martínez, J.A.; et al. Survival benefit of multidisciplinary care in amyotrophic lateral sclerosis in Spain: Association with noninvasive mechanical ventilation. J. Multidiscip. Healthc. 2019, 12, 465–470. [Google Scholar] [CrossRef]
- Hogden, A.; Foley, G.; Henderson, R.D.; James, N.; Aoun, S.M. Amyotrophic lateral sclerosis: Improving care with a multidisciplinary approach. J. Multidiscip. Healthc. 2017, 10, 205–215. [Google Scholar] [CrossRef]
- Albert, S.M.; Murphy, P.L.; Del Bene, M.L.; Rowland, L.P. Prospective study of palliative care in ALS: Choice, timing, outcomes. J. Neurol. Sci. 1999, 169, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Munroe, C.A.; Sirdofsky, M.D.; Kuru, T.; Anderson, E.D. End-of-life decision making in 42 patients with amyotrophic lateral sclerosis. Respir. Care 2007, 52, 996–999. [Google Scholar] [PubMed]
- Gould, R.L.; Coulson, M.C.; Brown, R.G.; Goldstein, L.H.; Al-Chalabi, A.; Howard, R.J. Psychotherapy and pharmacotherapy interventions to reduce distress or improve well-being in people with amyotrophic lateral sclerosis: A systematic review. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 293–302. [Google Scholar] [CrossRef]
- Fulton, J.J.; Newins, A.R.; Porter, L.S.; Ramos, K. Psychotherapy Targeting Depression and Anxiety for Use in Palliative Care: A Meta-Analysis. J. Palliat. Med. 2018, 21, 1024–1037. [Google Scholar] [CrossRef] [PubMed]
- Lunetta, C.; Lizio, A.; Sansone, V.A.; Cellotto, N.M.; Maestri, E.; Bettinelli, M.; Gatti, V.; Melazzini, M.G.; Meola, G.; Corbo, M. Strictly monitored exercise programs reduce motor deterioration in ALS: Preliminary results of a randomized controlled trial. J. Neurol. 2016, 263, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Clawson, L.L.; Cudkowicz, M.; Krivickas, L.; Brooks, B.R.; Sanjak, M.; Allred, P.; Atassi, N.; Swartz, A.; Steinhorn, G.; Uchil, A.; et al. A randomized controlled trial of resistance and endurance exercise in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 250–258. [Google Scholar] [CrossRef]
- Zhu, Y.; Xu, Y.; Xuan, R.; Huang, J.; István, B.; Fekete, G.; Gu, Y. Mixed Comparison of Different Exercise Interventions for Function, Respiratory, Fatigue, and Quality of Life in Adults with Amyotrophic Lateral Sclerosis: Systematic Review and Network Meta-Analysis. Front. Aging Neurosci. 2022, 14, 919059. [Google Scholar] [CrossRef]
- Maier, A.; Eicher, C.; Kiselev, J.; Klebbe, R.; Greuèl, M.; Kettemann, D.; Gaudlitz, M.; Walter, B.; Oleimeulen, U.; Münch, C.; et al. Acceptance of Enhanced Robotic Assistance Systems in People with Amyotrophic Lateral Sclerosis-Associated Motor Impairment: Observational Online Study. JMIR Rehabil. Assist. Technol. 2021, 8, e18972. [Google Scholar] [CrossRef]
- Driessen, M.J.; Dekker, J.; Lankhorst, G.; van der Zee, J. Occupational therapy for patients with chronic diseases: CVA, rheumatoid arthritis and progressive diseases of the central nervous system. Disabil. Rehabil. 1997, 19, 198–204. [Google Scholar] [CrossRef]
- Janiszewski, D.W.; Caroscio, J.T.; Wisham, L.H. Amyotrophic lateral sclerosis: A comprehensive rehabilitation approach. Arch. Phys. Med. Rehabil. 1983, 64, 304–307. [Google Scholar]
- Braile, L.E. Support for the drooping head. Am. J. Occup. 1981, 35, 661–662. [Google Scholar] [CrossRef] [PubMed]
- Raglio, A.; Giovanazzi, E.; Pain, D.; Baiardi, P.; Imbriani, C.; Imbriani, M.; Mora, G. Active music therapy approach in amyotrophic lateral sclerosis: A randomized-controlled trial. Int. J. Rehabil. Res. 2016, 39, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.R. Aquatic therapy for an ALS patient. Am. J. Occup. 1988, 42, 115–120. [Google Scholar] [CrossRef]
- Klebbe, R.; Scherzinger, S.; Eicher, C. Assistive Robots for Patients with Amyotrophic Lateral Sclerosis: Exploratory Task-Based Evaluation Study with an Early-Stage Demonstrator. JMIR Rehabil. Assist. Technol. 2022, 9, e35304. [Google Scholar] [CrossRef]
- Maheu, V.; Frappier, J.; Archambault, P.S.; Routhier, F. Evaluation of the JACO robotic arm: Clinico-economic study for powered wheelchair users with upper-extremity disabilities. IEEE Int. Conf. Rehabil. Robot. 2011, 2011, 5975397. [Google Scholar] [CrossRef]
- Proietti, T.; O’Neill, C.; Gerez, L.; Cole, T.; Mendelowitz, S.; Nuckols, K.; Hohimer, C.; Lin, D.; Paganoni, S.; Walsh, C. Restoring arm function with a soft robotic wearable for individuals with amyotrophic lateral sclerosis. Sci. Transl. Med. 2023, 15, eadd1504. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Koyama, S.; Sakurai, H.; Teranishi, T.; Kanada, Y.; Tanabe, S. Wearable robotic exoskeleton for gait reconstruction in patients with spinal cord injury: A literature review. J. Orthop. Transl. 2021, 28, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Olar, M.-L.; Leba, M.; Risteiu, M. Exoskeleton—Wearable devices. Literature review. MATEC Web Conf. 2021, 342, 05005. [Google Scholar] [CrossRef]
- Ward, A.L.; Sanjak, M.; Duffy, K.; Bravver, E.; Williams, N.; Nichols, M.; Brooks, B.R. Power wheelchair prescription, utilization, satisfaction, and cost for patients with amyotrophic lateral sclerosis: Preliminary data for evidence-based guidelines. Arch. Phys. Med. Rehabil. 2010, 91, 268–272. [Google Scholar] [CrossRef]
- Ward, A.L.; Hammond, S.; Holsten, S.; Bravver, E.; Brooks, B.R. Power Wheelchair Use in Persons with Amyotrophic Lateral Sclerosis: Changes Over Time. Assist. Technol. 2015, 27, 238–245. [Google Scholar] [CrossRef]
- Elliott, M.A.; Malvar, H.; Maassel, L.L.; Campbell, J.; Kulkarni, H.; Spiridonova, I.; Sophy, N.; Beavers, J.; Paradiso, A.; Needham, C.; et al. Eye-controlled, power wheelchair performs well for ALS patients. Muscle Nerve 2019, 60, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Manero, A.C.; McLinden, S.L.; Sparkman, J.; Oskarsson, B. Evaluating surface EMG control of motorized wheelchairs for amyotrophic lateral sclerosis patients. J. Neuroeng. Rehabil. 2022, 19, 88. [Google Scholar] [CrossRef] [PubMed]
- Grewal, H.; Matthews, A.; Tea, R.; George, K. LIDAR-based autonomous wheelchair. In Proceedings of the 2017 IEEE Sensors Applications Symposium (SAS), Glassboro, NJ, USA, 13–15 March 2017; pp. 1–6. [Google Scholar] [CrossRef]
- Mistry, K.S.; Pelayo, P.; Anil, D.G.; George, K. An SSVEP based brain computer interface system to control electric wheelchairs. In Proceedings of the 2018 IEEE International Instrumentation and Measurement Technology Conference (I2MTC), Houston, TX, USA, 4–17 May 2018; pp. 1–6. [Google Scholar] [CrossRef]
- Wanluk, N.; Visitsattapongse, S.; Juhong, A.; Pintavirooj, C. Smart wheelchair based on eye tracking. In Proceedings of the 2016 9th Biomedical Engineering International Conference (BMEiCON), Laung Prabang, Laos, 7–9 December 2016; pp. 1–4. [Google Scholar] [CrossRef]
- Dahmani, M.; Chowdhury, M.E.H.; Khandakar, A.; Rahman, T.; Al-Jayyousi, K.; Hefny, A.; Kiranyaz, S. An Intelligent and Low-Cost Eye-Tracking System for Motorized Wheelchair Control. Sensors 2020, 20, 3936. [Google Scholar] [CrossRef]
- Wang, H.; Li, Y.; Long, J.; Yu, T.; Gu, Z. An asynchronous wheelchair control by hybrid EEG-EOG brain-computer interface. Cogn. Neurodyn. 2014, 8, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Bona, S.; Donvito, G.; Cozza, F.; Malberti, I.; Vaccari, P.; Lizio, A.; Greco, L.; Carraro, E.; Sansone, V.A.; Lunetta, C. The development of an augmented reality device for the autonomous management of the electric bed and the electric wheelchair for patients with amyotrophic lateral sclerosis: A pilot study. Disabil. Rehabil. Assist. Technol. 2021, 16, 513–519. [Google Scholar] [CrossRef]
- Londral, A.; Pinto, A.; Pinto, S.; Azevedo, L.; De Carvalho, M. Quality of life in amyotrophic lateral sclerosis patients and caregivers: Impact of assistive communication from early stages. Muscle Nerve 2015, 52, 933–941. [Google Scholar] [CrossRef]
- Vansteensel, M.J.; Klein, E.; van Thiel, G.; Gaytant, M.; Simmons, Z.; Wolpaw, J.R.; Vaughan, T.M. Towards clinical application of implantable brain-computer interfaces for people with late-stage ALS: Medical and ethical considerations. J. Neurol. 2023, 270, 1323–1336. [Google Scholar] [CrossRef]
- Willett, F.; Kunz, E.; Fan, C.; Avansino, D.; Wilson, G.; Choi, E.Y.; Kamdar, F.; Hochberg, L.R.; Druckmann, S.; Shenoy, K.V.; et al. A high-performance speech neuroprosthesis. bioRxiv 2023. [Google Scholar] [CrossRef]
- Sellers, E.W.; Vaughan, T.M.; Wolpaw, J.R. A brain-computer interface for long-term independent home use. Amyotroph. Lateral Scler. 2010, 11, 449–455. [Google Scholar] [CrossRef]
- Vansteensel, M.J.; Pels, E.G.M.; Bleichner, M.G.; Branco, M.P.; Denison, T.; Freudenburg, Z.V.; Gosselaar, P.; Leinders, S.; Ottens, T.H.; Van Den Boom, M.A.; et al. Fully Implanted Brain-Computer Interface in a Locked-In Patient with ALS. N. Engl. J. Med. 2016, 375, 2060–2066. [Google Scholar] [CrossRef]
- Wolpaw, J.R.; Bedlack, R.S.; Reda, D.J.; Ringer, R.J.; Banks, P.G.; Vaughan, T.M.; Heckman, S.M.; McCane, L.M.; Carmack, C.S.; Winden, S.; et al. Independent home use of a brain-computer interface by people with amyotrophic lateral sclerosis. Neurology 2018, 91, e258–e267. [Google Scholar] [CrossRef] [PubMed]
- Kühnlein, P.; Gdynia, H.-J.; Sperfeld, A.-D.; Lindner-Pfleghar, B.; Ludolph, A.C.; Prosiegel, M.; Riecker, A. Diagnosis and treatment of bulbar symptoms in amyotrophic lateral sclerosis. Nat. Clin. Pract. Neurol. 2008, 4, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.M.; Elackattu, A.; Noordzij, J.P.; Walsh, M.J.; Langmore, S.E. Palliative treatment of dysphonia and dysarthria. Otolaryngol. Clin. N. Am. 2009, 42, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Pattee, G.L.; Plowman, E.K.; Focht Garand, K.L.; Costello, J.; Brooks, B.R.; Berry, J.D.; Smith, R.A.; Atassi, N.; Chapin, J.L.; Yunusova, Y.; et al. Provisional best practices guidelines for the evaluation of bulbar dysfunction in amyotrophic lateral sclerosis. Muscle Nerve 2019, 59, 531–536. [Google Scholar] [CrossRef]
- Collis, J.; Bloch, S. Survey of UK speech and language therapists’ assessment and treatment practices for people with progressive dysarthria. Int. J. Lang. Commun. Disord. 2012, 47, 725–737. [Google Scholar] [CrossRef]
- Körner, S.; Sieniawski, M.; Kollewe, K.; Rath, K.J.; Krampfl, K.; Zapf, A.; Dengler, R.; Petri, S. Speech therapy and communication device: Impact on quality of life and mood in patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 20–25. [Google Scholar] [CrossRef]
- D’Antona, S.; Caramenti, M.; Porro, D.; Castiglioni, I.; Cava, C. Amyotrophic Lateral Sclerosis: A Diet Review. Foods 2021, 10, 3128. [Google Scholar] [CrossRef]
- Shimizu, T.; Nakayama, Y.; Matsuda, C.; Haraguchi, M.; Bokuda, K.; Ishikawa-Takata, K.; Kawata, A.; Isozaki, E. Prognostic significance of body weight variation after diagnosis in ALS: A single-centre prospective cohort study. J. Neurol. 2019, 266, 1412–1420. [Google Scholar] [CrossRef]
- Mariosa, D.; Beard, J.D.; Umbach, D.M.; Bellocco, R.; Keller, J.; Peters, T.L.; Allen, K.D.; Ye, W.; Sandler, D.P.; Schmidt, S.; et al. Body Mass Index and Amyotrophic Lateral Sclerosis: A Study of US Military Veterans. Am. J. Epidemiol. 2017, 185, 362–371. [Google Scholar] [CrossRef]
- Moglia, C.; Calvo, A.; Grassano, M.; Canosa, A.; Manera, U.; D’Ovidio, F.; Bombaci, A.; Bersano, E.; Mazzini, L.; Mora, G.; et al. Early weight loss in amyotrophic lateral sclerosis: Outcome relevance and clinical correlates in a population-based cohort. J. Neurol. Neurosurg. Psychiatry 2019, 90, 666–673. [Google Scholar] [CrossRef]
- Mandrioli, J.; Malerba, S.A.; Beghi, E.; Fini, N.; Fasano, A.; Zucchi, E.; De Pasqua, S.; Guidi, C.; Terlizzi, E.; Sette, E.; et al. Riluzole and other prognostic factors in ALS: A population-based registry study in Italy. J. Neurol. 2018, 265, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Steyn, F.J.; Ioannides, Z.A.; van Eijk, R.P.A.; Heggie, S.; Thorpe, K.A.; Ceslis, A.; Heshmat, S.; Henders, A.K.; Wray, N.R.; van den Berg, L.H.; et al. Hypermetabolism in ALS is associated with greater functional decline and shorter survival. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Holm, T.; Maier, A.; Wicks, P.; Lang, D.; Linke, P.; Münch, C.; Steinfurth, L.; Meyer, R.; Meyer, T. Severe loss of appetite in amyotrophic lateral sclerosis patients: Online self-assessment study. Interact. J. Med. Res. 2013, 2, e8. [Google Scholar] [CrossRef] [PubMed]
- Burgos, R.; Bretón, I.; Cereda, E.; Desport, J.C.; Dziewas, R.; Genton, L.; Gomes, F.; Jésus, P.; Leischker, A.; Muscaritoli, M.; et al. ESPEN guideline clinical nutrition in neurology. Clin. Nutr. 2018, 37, 354–396. [Google Scholar] [CrossRef]
- Padilla, G.V.; Grant, M.; Wong, H.; Hansen, B.W.; Hanson, R.L.; Bergstrom, N.; Kubo, W.R. Subjective distresses of nasogastric tube feeding. J. Parenter. Enter. Nutr. 1979, 3, 53–57. [Google Scholar] [CrossRef]
- Gomes, C.A.R.; Andriolo, R.B.; Bennett, C.; Lustosa, S.A.S.; Matos, D.; Waisberg, D.R.; Waisberg, J. Percutaneous endoscopic gastrostomy versus nasogastric tube feeding for adults with swallowing disturbances. Cochrane Database Syst. Rev. 2015, 2015, CD008096. [Google Scholar] [CrossRef]
- Bond, L.; Ganguly, P.; Khamankar, N.; Mallet, N.; Bowen, G.; Green, B.; Mitchell, C.S. A Comprehensive Examination of Percutaneous Endoscopic Gastrostomy and Its Association with Amyotrophic Lateral Sclerosis Patient Outcomes. Brain Sci. 2019, 9, 223. [Google Scholar] [CrossRef]
- Burkhardt, C.; Neuwirth, C.; Sommacal, A.; Andersen, P.M.; Weber, M. Is survival improved by the use of NIV and PEG in amyotrophic lateral sclerosis (ALS)? A post-mortem study of 80 ALS patients. PLoS ONE 2017, 12, e0177555. [Google Scholar] [CrossRef]
- Fasano, A.; Fini, N.; Ferraro, D.; Ferri, L.; Vinceti, M.; Errals; Mandrioli, J. Percutaneous endoscopic gastrostomy, body weight loss and survival in amyotrophic lateral sclerosis: A population-based registry study. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 233–242. [Google Scholar] [CrossRef]
- Dorst, J.; Ludolph, A.C. Non-invasive ventilation in amyotrophic lateral sclerosis. Adv. Neurol. Disord. 2019, 12, 1756286419857040. [Google Scholar] [CrossRef]
- Boentert, M. Sleep disturbances in patients with amyotrophic lateral sclerosis: Current perspectives. Nat. Sci. Sleep 2019, 11, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Kleopa, K.A.; Sherman, M.; Neal, B.; Romano, G.J.; Heiman-Patterson, T. Bipap improves survival and rate of pulmonary function decline in patients with ALS. J. Neurol. Sci. 1999, 164, 82–88. [Google Scholar] [CrossRef]
- Pinto, A.C.; Evangelista, T.; Carvalho, M.; Alves, M.A.; Sales Luís, M.L. Respiratory assistance with a non-invasive ventilator (Bipap) in MND/ALS patients: Survival rates in a controlled trial. J. Neurol. Sci. 1995, 129, 19–26. [Google Scholar] [CrossRef]
- Ackrivo, J.; Hsu, J.Y.; Hansen-Flaschen, J.; Elman, L.; Kawut, S.M. Noninvasive Ventilation Use Is Associated with Better Survival in Amyotrophic Lateral Sclerosis. Ann. Am. Thorac. Soc. 2021, 18, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Bourke, S.C.; Bullock, R.E.; Williams, T.L.; Shaw, P.J.; Gibson, G.J. Noninvasive ventilation in ALS: Indications and effect on quality of life. Neurology 2003, 61, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Vandoorne, E.; Vrijsen, B.; Belge, C.; Testelmans, D.; Buyse, B. Noninvasive ventilation in amyotrophic lateral sclerosis: Effects on sleep quality and quality of life. Acta Clin. Belg. 2016, 71, 389–394. [Google Scholar] [CrossRef]
- Hayashi, N.; Atsuta, N.; Yokoi, D.; Nakamura, R.; Nakatochi, M.; Katsuno, M.; Izumi, Y.; Kanai, K.; Hattori, N.; Taniguchi, A.; et al. Prognosis of amyotrophic lateral sclerosis patients undergoing tracheostomy invasive ventilation therapy in Japan. J. Neurol. Neurosurg. Psychiatry 2020, 91, 285–290. [Google Scholar] [CrossRef]
- Spittel, S.; Maier, A.; Kettemann, D.; Walter, B.; Koch, B.; Krause, K.; Norden, J.; Münch, C.; Meyer, T. Non-invasive and tracheostomy invasive ventilation in amyotrophic lateral sclerosis: Utilization and survival rates in a cohort study over 12 years in Germany. Eur. J. Neurol. 2021, 28, 1160–1171. [Google Scholar] [CrossRef]
- Turner, M.R.; Faull, C.; McDermott, C.J.; Nickol, A.H.; Palmer, J.; Talbot, K. Tracheostomy in motor neurone disease. Pract. Neurol. 2019, 19, 467–475. [Google Scholar] [CrossRef]
- Kaub-Wittemer, D.; von Steinbüchel, N.; Wasner, M.; Laier-Groeneveld, G.; Borasio, G.D. Quality of life and psychosocial issues in ventilated patients with amyotrophic lateral sclerosis and their caregivers. J. Pain Symptom Manag. 2003, 26, 890–896. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, Y.; Deng, M. New developments and opportunities in drugs being trialed for amyotrophic lateral sclerosis from 2020 to 2022. Front. Pharm. 2022, 13, 1054006. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.; Funke, A.; Münch, C.; Kettemann, D.; Maier, A.; Walter, B.; Thomas, A.; Spittel, S. Real world experience of patients with amyotrophic lateral sclerosis (ALS) in the treatment of spasticity using tetrahydrocannabinol:cannabidiol (THC:CBD). BMC Neurol. 2019, 19, 222. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Bátkai, S.; Kunos, G. The Endocannabinoid System as an Emerging Target of Pharmacotherapy. Pharm. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef] [PubMed]
- Urbi, B.; Broadley, S.; Bedlack, R.; Russo, E.; Sabet, A. Study protocol for a randomised, double-blind, placebo-controlled study evaluating the Efficacy of cannabis-based Medicine Extract in slowing the disease pRogression of Amyotrophic Lateral sclerosis or motor neurone Disease: The EMERALD trial. BMJ Open 2019, 9, e029449. [Google Scholar] [CrossRef]
- Wong, C.; Dakin, R.S.; Williamson, J.; Newton, J.; Steven, M.; Colville, S.; Stavrou, M.; Gregory, J.M.; Elliott, E.; Mehta, A.R.; et al. Motor Neuron Disease Systematic Multi-Arm Adaptive Randomised Trial (MND-SMART): A multi-arm, multi-stage, adaptive, platform, phase III randomised, double-blind, placebo-controlled trial of repurposed drugs in motor neuron disease. BMJ Open 2022, 12, e064173. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Raphael, A.R.; LaDow, E.S.; McGurk, L.; Weber, R.; Trojanowski, J.Q.; Lee, V.M.-Y.; Finkbeiner, S.; Gitler, A.D.; Bonini, N.M. Therapeutic modulation of eIF2α-phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat. Genet. 2014, 46, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Westergard, T.; McAvoy, K.; Russell, K.; Wen, X.; Pang, Y.; Morris, B.; Pasinelli, P.; Trotti, D.; Haeusler, A. Repeat-associated non-AUG translation in C9orf72-ALS/FTD is driven by neuronal excitation and stress. EMBO Mol. Med. 2019, 11, e9423. [Google Scholar] [CrossRef]
- Ionescu, A.; Gradus, T.; Altman, T.; Maimon, R.; Saraf Avraham, N.; Geva, M.; Hayden, M.; Perlson, E. Targeting the Sigma-1 Receptor via Pridopidine Ameliorates Central Features of ALS Pathology in a SOD1G93A Model. Cell Death Dis. 2019, 10, 210. [Google Scholar] [CrossRef] [PubMed]
- Moreau, C.; Danel, V.; Devedjian, J.C.; Grolez, G.; Timmerman, K.; Laloux, C.; Petrault, M.; Gouel, F.; Jonneaux, A.; Dutheil, M.; et al. Could Conservative Iron Chelation Lead to Neuroprotection in Amyotrophic Lateral Sclerosis? Antioxid. Redox Signal. 2018, 29, 742–748. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, Y.; Zhang, J.; Shao, Y.; Li, S.; Wang, Y.; Cai, H.; Feng, Y.; Le, W. Fingerprint analysis of Huolingshengji Formula and its neuroprotective effects in SOD1G93A mouse model of amyotrophic lateral sclerosis. Sci. Rep. 2018, 8, 1668. [Google Scholar] [CrossRef]
- Yang, E.J. A Novel Supplement Attenuates Oxidative Stress-Induced TDP-43-Related Pathogenesis in TDP-43-Expressed Cells. Evid.-Based Complement. Altern. Med. 2021, 2021, e6773260. [Google Scholar] [CrossRef] [PubMed]
- Gold, J.; Rowe, D.B.; Kiernan, M.C.; Vucic, S.; Mathers, S.; van Eijk, R.P.A.; Nath, A.; Garcia Montojo, M.; Norato, G.; Santamaria, U.A.; et al. Safety and tolerability of Triumeq in amyotrophic lateral sclerosis: The Lighthouse trial. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, B.; Maragakis, N.; Bedlack, R.S.; Goyal, N.; Meyer, J.A.; Genge, A.; Bodkin, C.; Maiser, S.; Staff, N.; Zinman, L.; et al. MN-166 (ibudilast) in amyotrophic lateral sclerosis in a Phase IIb/III study: COMBAT-ALS study design. Neurodegener. Dis. Manag. 2021, 11, 431–443. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, H.; Ying, Z.; Gao, Q. Ibudilast enhances the clearance of SOD1 and TDP-43 aggregates through TFEB-mediated autophagy and lysosomal biogenesis: The new molecular mechanism of ibudilast and its implication for neuroprotective therapy. Biochem. Biophys. Res. Commun. 2020, 526, 231–238. [Google Scholar] [CrossRef]
- Mora, J.S.; Genge, A.; Chio, A.; Estol, C.J.; Chaverri, D.; Hernández, M.; Marín, S.; Mascias, J.; Rodriguez, G.E.; Povedano, M.; et al. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: A randomized clinical trial. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.S.; Bradley, W.G.; Chaverri, D.; Hernández-Barral, M.; Mascias, J.; Gamez, J.; Gargiulo-Monachelli, G.M.; Moussy, A.; Mansfield, C.D.; Hermine, O.; et al. Long-term survival analysis of masitinib in amyotrophic lateral sclerosis. Adv. Neurol. Disord. 2021, 14, 17562864211030364. [Google Scholar] [CrossRef]
- Stefanova, N.; Georgievska, B.; Eriksson, H.; Poewe, W.; Wenning, G.K. Myeloperoxidase inhibition ameliorates multiple system atrophy-like degeneration in a transgenic mouse model. Neurotox. Res. 2012, 21, 393–404. [Google Scholar] [CrossRef]
- Xiong, L.; McCoy, M.; Komuro, H.; West, X.Z.; Yakubenko, V.; Gao, D.; Dudiki, T.; Milo, A.; Chen, J.; Podrez, E.A.; et al. Inflammation-dependent oxidative stress metabolites as a hallmark of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2022, 178, 125–133. [Google Scholar] [CrossRef]
- Castillo, K.; Nassif, M.; Valenzuela, V.; Rojas, F.; Matus, S.; Mercado, G.; Court, F.A.; van Zundert, B.; Hetz, C. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 2013, 9, 1308–1320. [Google Scholar] [CrossRef]
- Li, Y.; Guo, Y.; Wang, X.; Yu, X.; Duan, W.; Hong, K.; Wang, J.; Han, H.; Li, C. Trehalose decreases mutant SOD1 expression and alleviates motor deficiency in early but not end-stage amyotrophic lateral sclerosis in a SOD1-G93A mouse model. Neuroscience 2015, 298, 12–25. [Google Scholar] [CrossRef]
- Shefner, J.M.; Andrews, J.A.; Genge, A.; Jackson, C.; Lechtzin, N.; Miller, T.M.; Cockroft, B.M.; Meng, L.; Wei, J.; Wolff, A.A.; et al. A Phase 2, Double-Blind, Randomized, Dose-Ranging Trial of Reldesemtiv in Patients with ALS. Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Oki, R.; Izumi, Y.; Fujita, K.; Miyamoto, R.; Nodera, H.; Sato, Y.; Sakaguchi, S.; Nokihara, H.; Kanai, K.; Tsunemi, T.; et al. Efficacy and Safety of Ultrahigh-Dose Methylcobalamin in Early-Stage Amyotrophic Lateral Sclerosis: A Randomized Clinical Trial. JAMA Neurol. 2022, 79, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolny, G.; Giacinti, C.; Pelosi, L.; Nicoletti, C.; Winn, N.; Barberi, L.; Molinaro, M.; Rosenthal, N.; Musarò, A. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J. Cell Biol. 2005, 168, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Lepore, A.C.; Haenggeli, C.; Gasmi, M.; Bishop, K.M.; Bartus, R.T.; Maragakis, N.J.; Rothstein, J.D. Intraparenchymal spinal cord delivery of adeno-associated virus IGF-1 is protective in the SOD1G93A model of ALS. Brain Res. 2007, 1185, 256–265. [Google Scholar] [CrossRef]
- Dodge, J.C.; Treleaven, C.M.; Fidler, J.A.; Hester, M.; Haidet, A.; Handy, C.; Rao, M.; Eagle, A.; Matthews, J.C.; Taksir, T.V.; et al. AAV4-mediated Expression of IGF-1 and VEGF within Cellular Components of the Ventricular System Improves Survival Outcome in Familial ALS Mice. Mol. Ther. 2010, 18, 2075–2084. [Google Scholar] [CrossRef]
- Lin, H.; Hu, H.; Duan, W.; Liu, Y.; Tan, G.; Li, Z.; Liu, Y.; Deng, B.; Song, X.; Wang, W.; et al. Intramuscular Delivery of scAAV9-hIGF1 Prolongs Survival in the hSOD1G93A ALS Mouse Model via Upregulation of D-Amino Acid Oxidase. Mol. Neurobiol. 2018, 55, 682–695. [Google Scholar] [CrossRef]
- Sorenson, E.J.; Windbank, A.J.; Mandrekar, J.N.; Bamlet, W.R.; Appel, S.H.; Armon, C.; Barkhaus, P.E.; Bosch, P.; Boylan, K.; David, W.S.; et al. Subcutaneous IGF-1 is not beneficial in 2-year ALS trial. Neurology 2008, 71, 1770–1775. [Google Scholar] [CrossRef]
- Robinson, A.P.; Zhang, J.Z.; Titus, H.E.; Karl, M.; Merzliakov, M.; Dorfman, A.R.; Karlik, S.; Stewart, M.G.; Watt, R.K.; Facer, B.D.; et al. Nanocatalytic activity of clean-surfaced, faceted nanocrystalline gold enhances remyelination in animal models of multiple sclerosis. Sci. Rep. 2020, 10, 1936. [Google Scholar] [CrossRef]
- Aartsma-Rus, A. FDA Approval of Nusinersen for Spinal Muscular Atrophy Makes 2016 the Year of Splice Modulating Oligonucleotides. Nucleic Acid. 2017, 27, 67–69. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 2017, 27, 1–3. [Google Scholar] [CrossRef]
- Boros, B.D.; Schoch, K.M.; Kreple, C.J.; Miller, T.M. Antisense Oligonucleotides for the Study and Treatment of ALS. Neurotherapeutics 2022, 19, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.; Cudkowicz, M.; Shaw, P.J.; Andersen, P.M.; Atassi, N.; Bucelli, R.C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A.L.; et al. Phase 1-2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2020, 383, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.M.; Cudkowicz, M.E.; Genge, A.; Shaw, P.J.; Sobue, G.; Bucelli, R.C.; Chiò, A.; Van Damme, P.; Ludolph, A.C.; Glass, J.D.; et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2022, 387, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Korobeynikov, V.A.; Lyashchenko, A.K.; Blanco-Redondo, B.; Jafar-Nejad, P.; Shneider, N.A. Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat. Med. 2022, 28, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lin, S.; Staats, K.A.; Li, Y.; Chang, W.-H.; Hung, S.-T.; Hendricks, E.; Linares, G.R.; Wang, Y.; Son, E.Y.; et al. Haploinsufficiency Leads to Neurodegeneration in C9ORF72 ALS/FTD Human Induced Motor Neurons. Nat. Med. 2018, 24, 313–325. [Google Scholar] [CrossRef]
- McEachin, Z.T.; Parameswaren, J.; Raj, N.; Bassell, G.J.; Jiang, J. RNA-mediated toxicity in C9orf72 ALS and FTD. Neurobiol. Dis. 2020, 145, 105055. [Google Scholar] [CrossRef]
- Velázquez-Pérez, L.C.; Rodríguez-Labrada, R.; Fernandez-Ruiz, J. Spinocerebellar Ataxia Type 2: Clinicogenetic Aspects, Mechanistic Insights, and Management Approaches. Front. Neurol. 2017, 8, 472. [Google Scholar] [CrossRef]
- Elden, A.C.; Kim, H.-J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef]
- Becker, L.A.; Huang, B.; Bieri, G.; Ma, R.; Knowles, D.A.; Jafar-Nejad, P.; Messing, J.; Kim, H.J.; Soriano, A.; Auburger, G.; et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 2017, 544, 367–371. [Google Scholar] [CrossRef]
- Amado, D.A.; Davidson, B.L. Gene therapy for ALS: A review. Mol. Ther. 2021, 29, 3345–3358. [Google Scholar] [CrossRef]
- Mueller, C.; Berry, J.D.; McKenna-Yasek, D.M.; Gernoux, G.; Owegi, M.A.; Pothier, L.M.; Douthwright, C.L.; Gelevski, D.; Luppino, S.D.; Blackwood, M.; et al. SOD1 Suppression with Adeno-Associated Virus and MicroRNA in Familial ALS. N. Engl. J. Med. 2020, 383, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Paré, B.; Lehmann, M.; Beaudin, M.; Nordström, U.; Saikali, S.; Julien, J.-P.; Gilthorpe, J.D.; Marklund, S.L.; Cashman, N.R.; Andersen, P.M.; et al. Misfolded SOD1 pathology in sporadic Amyotrophic Lateral Sclerosis. Sci. Rep. 2018, 8, 14223. [Google Scholar] [CrossRef] [PubMed]
- Zancanella, V.; van Rooijen, K.; van Vliet, R.; Pereira, I.; de Ruiter, A.; Shtefaniuk, V.; Kieper, S.; Wartel, M.; Dobrynin, G.; Kovačević, J.; et al. AAV-miQURE®-mediated targeting of hexanucleotide repeat expansion-containing transcripts in ALS C9orf72 mouse models. Hum. Gene Ther. 2022, 33, A72–A73. [Google Scholar]
- Redman, M.; King, A.; Watson, C.; King, D. What is CRISPR/Cas9? Arch. Dis. Child. Educ. Pract. 2016, 101, 213–215. [Google Scholar] [CrossRef]
- Meijboom, K.E.; Abdallah, A.; Fordham, N.P.; Nagase, H.; Rodriguez, T.; Kraus, C.; Gendron, T.F.; Krishnan, G.; Esanov, R.; Andrade, N.S.; et al. CRISPR/Cas9-mediated excision of ALS/FTD-causing hexanucleotide repeat expansion in C9ORF72 rescues major disease mechanisms in vivo and in vitro. Nat. Commun. 2022, 13, 6286. [Google Scholar] [CrossRef]
- Deng, H.-X.; Zhai, H.; Shi, Y.; Liu, G.; Lowry, J.; Liu, B.; Ryan, É.B.; Yan, J.; Yang, Y.; Zhang, N.; et al. Efficacy and long-term safety of CRISPR/Cas9 genome editing in the SOD1-linked mouse models of ALS. Commun. Biol. 2021, 4, 396. [Google Scholar] [CrossRef]
- Deneault, E.; Chaineau, M.; Nicouleau, M.; Castellanos Montiel, M.J.; Franco Flores, A.K.; Haghi, G.; Chen, C.X.-Q.; Abdian, N.; Shlaifer, I.; Beitel, L.K.; et al. A streamlined CRISPR workflow to introduce mutations and generate isogenic iPSCs for modeling amyotrophic lateral sclerosis. Methods 2022, 203, 297–310. [Google Scholar] [CrossRef]
- Beel, S.; Herdewyn, S.; Fazal, R.; De Decker, M.; Moisse, M.; Robberecht, W.; Van Den Bosch, L.; Van Damme, P. Progranulin reduces insoluble TDP-43 levels, slows down axonal degeneration and prolongs survival in mutant TDP-43 mice. Mol. Neurodegener. 2018, 13, 55. [Google Scholar] [CrossRef]
- Schymick, J.C.; Yang, Y.; Andersen, P.M.; Vonsattel, J.P.; Greenway, M.; Momeni, P.; Elder, J.; Chiò, A.; Restagno, G.; Robberecht, W.; et al. Progranulin mutations and amyotrophic lateral sclerosis or amyotrophic lateral sclerosis–frontotemporal dementia phenotypes. J. Neurol. Neurosurg. Psychiatry 2007, 78, 754–756. [Google Scholar] [CrossRef]
- Cannon, A.; Fujioka, S.; Rutherford, N.J.; Ferman, T.J.; Broderick, D.F.; Boylan, K.B.; Graff-Radford, N.R.; Uitti, R.J.; Rademakers, R.; Wszolek, Z.K.; et al. Clinicopathologic variability of the GRN A9D mutation, including amyotrophic lateral sclerosis. Neurology 2013, 80, 1771–1777. [Google Scholar] [CrossRef]
- Maier, M.; Welt, T.; Wirth, F.; Montrasio, F.; Preisig, D.; McAfoose, J.; Vieira, F.G.; Kulic, L.; Späni, C.; Stehle, T.; et al. A human-derived antibody targets misfolded SOD1 and ameliorates motor symptoms in mouse models of amyotrophic lateral sclerosis. Sci. Transl. Med. 2018, 10, eaah3924. [Google Scholar] [CrossRef] [PubMed]
- Sironi, F.; De Marchi, F.; Mazzini, L.; Bendotti, C. Cell therapy in ALS: An update on preclinical and clinical studies. Brain Res. Bull. 2023, 194, 64–81. [Google Scholar] [CrossRef] [PubMed]
- Cudkowicz, M.E.; Lindborg, S.R.; Goyal, N.A.; Miller, R.G.; Burford, M.J.; Berry, J.D.; Nicholson, K.A.; Mozaffar, T.; Katz, J.S.; Jenkins, L.J.; et al. A randomized placebo-controlled phase 3 study of mesenchymal stem cells induced to secrete high levels of neurotrophic factors in amyotrophic lateral sclerosis. Muscle Nerve 2022, 65, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.-W.; Noh, M.-Y.; Kwon, M.-S.; Kim, H.Y.; Oh, S.; Park, J.; Kim, H.-J.; Ki, C.-S.; Kim, S.H. Repeated Intrathecal Mesenchymal Stem Cells for Amyotrophic Lateral Sclerosis. Ann. Neurol. 2018, 84, 361–373. [Google Scholar] [CrossRef]
- Arenas, A.; Chen, J.; Kuang, L.; Barnett, K.R.; Kasarskis, E.J.; Gal, J.; Zhu, H. Lysine acetylation regulates the RNA binding, subcellular localization and inclusion formation of FUS. Hum. Mol. Genet. 2020, 29, 2684–2697. [Google Scholar] [CrossRef]
- Schaffert, L.-N.; Carter, W.G. Do Post-Translational Modifications Influence Protein Aggregation in Neurodegenerative Diseases: A Systematic Review. Brain Sci. 2020, 10, 232. [Google Scholar] [CrossRef]
- Rhoads, S.N.; Monahan, Z.T.; Yee, D.S.; Shewmaker, F.P. The Role of Post-Translational Modifications on Prion-Like Aggregation and Liquid-Phase Separation of FUS. Int. J. Mol. Sci. 2018, 19, 886. [Google Scholar] [CrossRef]
- Sanna, S.; Esposito, S.; Masala, A.; Sini, P.; Nieddu, G.; Galioto, M.; Fais, M.; Iaccarino, C.; Cestra, G.; Crosio, C. HDAC1 inhibition ameliorates TDP-43-induced cell death in vitro and in vivo. Cell Death Dis. 2020, 11, 369. [Google Scholar] [CrossRef]
- Buratti, E. TDP-43 post-translational modifications in health and disease. Expert Opin. Ther. Targets 2018, 22, 279–293. [Google Scholar] [CrossRef]
- Banks, C.J.; Andersen, J.L. Mechanisms of SOD1 regulation by post-translational modifications. Redox Biol. 2019, 26, 101270. [Google Scholar] [CrossRef]
- Trist, B.G.; Genoud, S.; Roudeau, S.; Rookyard, A.; Abdeen, A.; Cottam, V.; Hare, D.J.; White, M.; Altvater, J.; Fifita, J.A.; et al. Altered SOD1 maturation and post-translational modification in amyotrophic lateral sclerosis spinal cord. Brain 2022, 145, 3108–3130. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef]
- Teng, C.-S.; Wu, B.-H.; Yen, M.-R.; Chen, P.-Y. MethGET: Web-based bioinformatics software for correlating genome-wide DNA methylation and gene expression. BMC Genom. 2020, 21, 375. [Google Scholar] [CrossRef]
- Hunt, C.R.; Ramnarain, D.; Horikoshi, N.; Iyengar, P.; Pandita, R.K.; Shay, J.W.; Pandita, T.K. Histone Modifications and DNA Double-Strand Break Repair after Exposure to Ionizing Radiations. Radiat. Res. 2013, 179, 383–392. [Google Scholar] [CrossRef]
- Bennett, S.A.; Tanaz, R.; Cobos, S.N.; Torrente, M.P. Epigenetics in amyotrophic lateral sclerosis: A role for histone post-translational modifications in neurodegenerative disease. Transl. Res. 2019, 204, 19–30. [Google Scholar] [CrossRef]
- Cobos, S.N.; Bennett, S.A.; Torrente, M.P. The impact of histone post-translational modifications in neurodegenerative diseases. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 1982–1991. [Google Scholar] [CrossRef]
- Cobos, S.N.; Torrente, M.P. Epidrugs in Amyotrophic Lateral Sclerosis/Frontotemporal Dementia: Contextualizing a Role for Histone Kinase Inhibition in Neurodegenerative Disease. ACS Pharmacol. Transl. Sci. 2022, 5, 134–137. [Google Scholar] [CrossRef]
- Klingl, Y.E.; Pakravan, D.; Bosch, L.V.D. Opportunities for histone deacetylase inhibition in amyotrophic lateral sclerosis. Br. J. Pharmacol. 2021, 178, 1353–1372. [Google Scholar] [CrossRef]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovás, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 2017, 8, 861. [Google Scholar] [CrossRef] [PubMed]
- Burg, T.; Rossaert, E.; Moisse, M.; Van Damme, P.; Van Den Bosch, L. Histone Deacetylase Inhibition Regulates Lipid Homeostasis in a Mouse Model of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 11224. [Google Scholar] [CrossRef] [PubMed]
- Tejido, C.; Pakravan, D.; Bosch, L.V.D. Potential Therapeutic Role of HDAC Inhibitors in FUS-ALS. Front. Mol. Neurosci. 2021, 14, 154. [Google Scholar] [CrossRef]
- Fazal, R.; Boeynaems, S.; Swijsen, A.; De Decker, M.; Fumagalli, L.; Moisse, M.; Vanneste, J.; Guo, W.; Boon, R.; Vercruysse, T.; et al. HDAC6 inhibition restores TDP-43 pathology and axonal transport defects in human motor neurons with TARDBP mutations. EMBO J. 2021, 40, e106177. [Google Scholar] [CrossRef]
- Boutillier, A.-L.; Tzeplaeff, L.; Dupuis, L. The dark side of HDAC inhibition in ALS. EBioMedicine 2019, 41, 38–39. [Google Scholar] [CrossRef] [PubMed]
- Sahana, T.G.; Zhang, K. Mitogen-Activated Protein Kinase Pathway in Amyotrophic Lateral Sclerosis. Biomedicines 2021, 9, 969. [Google Scholar] [CrossRef]
- Buratti, E. Targeting TDP-43 proteinopathy with drugs and drug-like small molecules. Br. J. Pharmacol. 2021, 178, 1298–1315. [Google Scholar] [CrossRef]
- Lingor, P.; Weber, M.; Camu, W.; Friede, T.; Hilgers, R.; Leha, A.; Neuwirth, C.; Günther, R.; Benatar, M.; Kuzma-Kozakiewicz, M.; et al. ROCK-ALS: Protocol for a Randomized, Placebo-Controlled, Double-Blind Phase IIa Trial of Safety, Tolerability and Efficacy of the Rho Kinase (ROCK) Inhibitor Fasudil in Amyotrophic Lateral Sclerosis. Front. Neurol. 2019, 10, 293. [Google Scholar] [CrossRef]
- Koch, J.C.; Tatenhorst, L.; Roser, A.-E.; Saal, K.-A.; Tönges, L.; Lingor, P. ROCK inhibition in models of neurodegeneration and its potential for clinical translation. Pharm. 2018, 189, 1–21. [Google Scholar] [CrossRef]
- Hop, P.J.; Zwamborn, R.A.J.; Hannon, E.; Shireby, G.L.; Nabais, M.F.; Walker, E.M.; van Rheenen, W.; van Vugt, J.J.F.A.; Dekker, A.M.; Westeneng, H.-J.; et al. Genome-wide study of DNA methylation shows alterations in metabolic, inflammatory, and cholesterol pathways in ALS. Sci. Transl. Med. 2022, 14, eabj0264. [Google Scholar] [CrossRef]
- Chestnut, B.A.; Chang, Q.; Price, A.; Lesuisse, C.; Wong, M.; Martin, L.J. Epigenetic regulation of motor neuron cell death through DNA methylation. J. Neurosci. 2011, 31, 16619–16636. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Wong, M. Aberrant regulation of DNA methylation in amyotrophic lateral sclerosis: A new target of disease mechanisms. Neurotherapeutics 2013, 10, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Fumagalli, L.; Bosch, L.V.D.; Guo, W.; Fumagalli, L.; Bosch, L.V.D. Targeting Axonal Transport: A New Therapeutic Avenue for ALS; IntechOpen: London, UK, 2020. [Google Scholar] [CrossRef]
- Vanneste, J.; Van Den Bosch, L. The Role of Nucleocytoplasmic Transport Defects in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 12175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9ORF72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef]
- Chou, C.-C.; Zhang, Y.; Umoh, M.E.; Vaughan, S.W.; Lorenzini, I.; Liu, F.; Sayegh, M.; Donlin-Asp, P.G.; Chen, Y.H.; Duong, D.M.; et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci. 2018, 21, 228–239. [Google Scholar] [CrossRef]
- Jovičić, A.; Mertens, J.; Boeynaems, S.; Bogaert, E.; Chai, N.; Yamada, S.B.; Paul, J.W.; Sun, S.; Herdy, J.R.; Bieri, G.; et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci. 2015, 18, 1226–1229. [Google Scholar] [CrossRef]
- Guo, L.; Fare, C.M.; Shorter, J. Therapeutic dissolution of aberrant phases by nuclear-import receptors. Trends Cell Biol. 2019, 29, 308–322. [Google Scholar] [CrossRef]
- Konopka, A.; Atkin, J.D. DNA Damage, Defective DNA Repair, and Neurodegeneration in Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2022, 14, 786420. [Google Scholar] [CrossRef]
- McGurk, L.; Mojsilovic-Petrovic, J.; Van Deerlin, V.M.; Shorter, J.; Kalb, R.G.; Lee, V.M.; Trojanowski, J.Q.; Lee, E.B.; Bonini, N.M. Nuclear poly(ADP-ribose) activity is a therapeutic target in amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2018, 6, 84. [Google Scholar] [CrossRef]
- Brown, D.G.; Shorter, J.; Wobst, H.J. Emerging small-molecule therapeutic approaches for amyotrophic lateral sclerosis and frontotemporal dementia. Bioorganic Med. Chem. Lett. 2020, 30, 126942. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, Y.; Fang, Q.; Zhang, N.; Kumar, G.; Zhang, J.; Song, L.-J.; Yu, J.; Zhao, L.; Zhang, H.-T.; et al. The Rho kinase inhibitor fasudil attenuates Aβ1–42-induced apoptosis via the ASK1/JNK signal pathway in primary cultures of hippocampal neurons. Metab. Brain Dis. 2019, 34, 1787–1801. [Google Scholar] [CrossRef] [PubMed]
- Gugliandolo, A.; Pollastro, F.; Bramanti, P.; Mazzon, E. Cannabidiol exerts protective effects in an in vitro model of Parkinson’s disease activating AKT/mTOR pathway. Fitoterapia 2020, 143, 104553. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Lopes, D.H.J.; Du, Z.; Pang, E.S.; Shanmugam, A.; Lomakin, A.; Talbiersky, P.; Tennstaedt, A.; McDaniel, K.; Bakshi, R.; et al. Lysine-specific molecular tweezers are broad-spectrum inhibitors of assembly and toxicity of amyloid proteins. J. Am. Chem. Soc. 2011, 133, 16958–16969. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.; Meng, H.; Wongkongkathep, P.; Corrales, C.I.; Sepanj, N.; Atlasi, R.S.; Klärner, F.-G.; Schrader, T.; Spencer, M.J.; Loo, J.A.; et al. The molecular tweezer CLR01 inhibits aberrant superoxide dismutase 1 (SOD1) self-assembly in vitro and in the G93A-SOD1 mouse model of ALS. J. Biol. Chem. 2019, 294, 3501–3513. [Google Scholar] [CrossRef]
- Samanta, N.; Ruiz-Blanco, Y.B.; Fetahaj, Z.; Gnutt, D.; Lantz, C.; Loo, J.A.; Sanchez-Garcia, E.; Ebbinghaus, S. Superoxide Dismutase 1 Folding Stability as a Target for Molecular Tweezers in SOD1-Related Amyotrophic Lateral Sclerosis. ChemBioChem 2022, 23, e202200396. [Google Scholar] [CrossRef]
- Di, J.; Siddique, I.; Li, Z.; Malki, G.; Hornung, S.; Dutta, S.; Hurst, I.; Ishaaya, E.; Wang, A.; Tu, S.; et al. The molecular tweezer CLR01 improves behavioral deficits and reduces tau pathology in P301S-tau transgenic mice. Alzheimer’s Res. Ther. 2021, 13, 6. [Google Scholar] [CrossRef]
- Goyal, N.A.; Berry, J.D.; Windebank, A.; Staff, N.P.; Maragakis, N.J.; van den Berg, L.H.; Genge, A.; Miller, R.; Baloh, R.H.; Kern, R.; et al. Addressing heterogeneity in amyotrophic lateral sclerosis CLINICAL TRIALS. Muscle Nerve 2020, 62, 156–166. [Google Scholar] [CrossRef]
- Goutman, S.A.; Feldman, E.L. Clinical Trials of Therapies for Amyotrophic Lateral Sclerosis: One Size Does Not Fit All. JAMA Neurol. 2015, 72, 743–744. [Google Scholar] [CrossRef]
- Olney, N.T.; Spina, S.; Miller, B.L. Frontotemporal Dementia. Neurol. Clin. 2017, 35, 339–374. [Google Scholar] [CrossRef]
- Ishaque, A.; Mah, D.; Seres, P.; Luk, C.; Eurich, D.; Johnston, W.; Yang, Y.-H.; Kalra, S. Evaluating the cerebral correlates of survival in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2018, 5, 1350–1361. [Google Scholar] [CrossRef]
- Chiò, A.; Calvo, A.; Moglia, C.; Mazzini, L.; Mora, G.; PARALS Study Group. Phenotypic heterogeneity of amyotrophic lateral sclerosis: A population based study. J. Neurol. Neurosurg. Psychiatry 2011, 82, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.-Q.; Chen, Y.; Chen, X.; Cao, B.; Ou, R.; Zhang, L.; Hou, Y.; Shang, H. Clinical and prognostic features of ALS/MND in different phenotypes-data from a hospital-based registry. Brain Res. Bull. 2018, 142, 403–408. [Google Scholar] [CrossRef]
- Westeneng, H.-J.; Debray, T.P.A.; Visser, A.E.; van Eijk, R.P.A.; Rooney, J.P.K.; Calvo, A.; Martin, S.; McDermott, C.J.; Thompson, A.G.; Pinto, S.; et al. Prognosis for patients with amyotrophic lateral sclerosis: Development and validation of a personalised prediction model. Lancet Neurol. 2018, 17, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Ackrivo, J.; Hansen-Flaschen, J.; Wileyto, E.P.; Schwab, R.J.; Elman, L.; Kawut, S.M. Development of a prognostic model of respiratory insufficiency or death in amyotrophic lateral sclerosis. Eur. Respir. J. 2019, 53, 1802237. [Google Scholar] [CrossRef] [PubMed]
- Salmon, K.; Anoja, N.; Breiner, A.; Chum, M.; Dionne, A.; Dupré, N.; Fiander, A.; Fok, D.; Ghavanini, A.; Gosselin, S.; et al. Genetic testing for amyotrophic lateral sclerosis in Canada—An assessment of current practices. Amyotroph. Lateral Scler. Front. Degener. 2022, 23, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.; Oh, K.-W.; Nordin, A.; Panthi, S.; Kim, S.H.; Nordin, F.; Freischmidt, A.; Ludolph, A.C.; Ki, C.S.; Forsberg, K.; et al. De novo mutations in SOD1 are a cause of ALS. J. Neurol. Neurosurg. Psychiatry 2022, 93, 201–206. [Google Scholar] [CrossRef]
- Grassano, M.; Calvo, A.; Moglia, C.; Sbaiz, L.; Brunetti, M.; Barberis, M.; Casale, F.; Manera, U.; Vasta, R.; Canosa, A.; et al. Systematic evaluation of genetic mutations in ALS: A population-based study. J. Neurol. Neurosurg. Psychiatry 2022, 93, 1190–1193. [Google Scholar] [CrossRef]
- Benatar, M.; Wuu, J.; Andersen, P.M.; Bucelli, R.C.; Andrews, J.A.; Otto, M.; Farahany, N.A.; Harrington, E.A.; Chen, W.; Mitchell, A.A.; et al. Design of a Randomized, Placebo-Controlled, Phase 3 Trial of Tofersen Initiated in Clinically Presymptomatic SOD1 Variant Carriers: The ATLAS Study. Neurotherapeutics 2022, 19, 1248–1258. [Google Scholar] [CrossRef]
- Benatar, M.; Wuu, J.; McHutchison, C.; Postuma, R.B.; Boeve, B.F.; Petersen, R.; Ross, C.A.; Rosen, H.; Arias, J.J.; Fradette, S.; et al. Preventing amyotrophic lateral sclerosis: Insights from pre-symptomatic neurodegenerative diseases. Brain 2022, 145, 27–44. [Google Scholar] [CrossRef]
- Vidovic, M.; Müschen, L.H.; Brakemeier, S.; Machetanz, G.; Naumann, M.; Castro-Gomez, S. Current State and Future Directions in the Diagnosis of Amyotrophic Lateral Sclerosis. Cells 2023, 12, 736. [Google Scholar] [CrossRef]
- Sturmey, E.; Malaspina, A. Blood biomarkers in ALS: Challenges, applications and novel frontiers. Acta Neurol. Scand. 2022, 146, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Gaetani, L.; Blennow, K.; Calabresi, P.; Di Filippo, M.; Parnetti, L.; Zetterberg, H. Neurofilament light chain as a biomarker in neurological disorders. J. Neurol. Neurosurg. Psychiatry 2019, 90, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Gaiottino, J.; Norgren, N.; Dobson, R.; Topping, J.; Nissim, A.; Malaspina, A.; Bestwick, J.P.; Monsch, A.U.; Regeniter, A.; Lindberg, R.L.; et al. Increased neurofilament light chain blood levels in neurodegenerative neurological diseases. PLoS ONE 2013, 8, e75091. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.-H.; Macdonald-Wallis, C.; Gray, E.; Pearce, N.; Petzold, A.; Norgren, N.; Giovannoni, G.; Fratta, P.; Sidle, K.; Fish, M.; et al. Neurofilament light chain: A prognostic biomarker in amyotrophic lateral sclerosis. Neurology 2015, 84, 2247–2257. [Google Scholar] [CrossRef]
- Thompson, A.G.; Gray, E.; Verber, N.; Bobeva, Y.; Lombardi, V.; Shepheard, S.R.; Yildiz, O.; Feneberg, E.; Farrimond, L.; Dharmadasa, T.; et al. Multicentre appraisal of amyotrophic lateral sclerosis biofluid biomarkers shows primacy of blood neurofilament light chain. Brain Commun. 2022, 4, fcac029. [Google Scholar] [CrossRef]
- Benatar, M.; Wuu, J.; Lombardi, V.; Jeromin, A.; Bowser, R.; Andersen, P.M.; Malaspina, A. Neurofilaments in pre-symptomatic ALS and the impact of genotype. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 538–548. [Google Scholar] [CrossRef]
- Vacchiano, V.; Mastrangelo, A.; Zenesini, C.; Masullo, M.; Quadalti, C.; Avoni, P.; Polischi, B.; Cherici, A.; Capellari, S.; Salvi, F.; et al. Plasma and CSF Neurofilament Light Chain in Amyotrophic Lateral Sclerosis: A Cross-Sectional and Longitudinal Study. Front. Aging Neurosci. 2021, 13, 753242. [Google Scholar] [CrossRef]
- Benatar, M.; Wuu, J.; Andersen, P.M.; Lombardi, V.; Malaspina, A. Neurofilament light: A candidate biomarker of presymptomatic amyotrophic lateral sclerosis and phenoconversion. Ann. Neurol. 2018, 84, 130–139. [Google Scholar] [CrossRef]
- Magen, I.; Yacovzada, N.S.; Yanowski, E.; Coenen-Stass, A.; Grosskreutz, J.; Lu, C.-H.; Greensmith, L.; Malaspina, A.; Fratta, P.; Hornstein, E. Circulating miR-181 is a prognostic biomarker for amyotrophic lateral sclerosis. Nat. Neurosci. 2021, 24, 1534–1541. [Google Scholar] [CrossRef]
- Ren, Y.; Li, S.; Chen, S.; Sun, X.; Yang, F.; Wang, H.; Li, M.; Cui, F.; Huang, X. TDP-43 and Phosphorylated TDP-43 Levels in Paired Plasma and CSF Samples in Amyotrophic Lateral Sclerosis. Front. Neurol. 2021, 12, 663637. [Google Scholar] [CrossRef]
- Henkel, J.S.; Beers, D.R.; Wen, S.; Rivera, A.L.; Toennis, K.M.; Appel, J.E.; Zhao, W.; Moore, D.H.; Powell, S.Z.; Appel, S.H. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol. Med. 2013, 5, 64–79. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Henkel, J.S.; Zhao, W.; Wang, J.; Huang, A.; Wen, S.; Liao, B.; Appel, S.H. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain 2011, 134, 1293–1314. [Google Scholar] [CrossRef] [PubMed]
- Sheean, R.K.; McKay, F.C.; Cretney, E.; Bye, C.R.; Perera, N.D.; Tomas, D.; Weston, R.A.; Scheller, K.J.; Djouma, E.; Menon, P.; et al. Association of Regulatory T-Cell Expansion with Progression of Amyotrophic Lateral Sclerosis: A Study of Humans and a Transgenic Mouse Model. JAMA Neurol. 2018, 75, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Shepheard, S.R.; Karnaros, V.; Benyamin, B.; Schultz, D.W.; Dubowsky, M.; Wuu, J.; Chataway, T.; Malaspina, A.; Benatar, M.; Rogers, M.-L. Urinary neopterin: A novel biomarker of disease progression in amyotrophic lateral sclerosis. Eur. J. Neurol. 2022, 29, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Shepheard, S.R.; Wuu, J.; Cardoso, M.; Wiklendt, L.; Dinning, P.G.; Chataway, T.; Schultz, D.; Benatar, M.; Rogers, M.-L. Urinary p75ECD: A prognostic, disease progression, and pharmacodynamic biomarker in ALS. Neurology 2017, 88, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Ami, D.; Duse, A.; Mereghetti, P.; Cozza, F.; Ambrosio, F.; Ponzini, E.; Grandori, R.; Lunetta, C.; Tavazzi, S.; Pezzoli, F.; et al. Tear-Based Vibrational Spectroscopy Applied to Amyotrophic Lateral Sclerosis. Anal. Chem. 2021, 93, 16995–17002. [Google Scholar] [CrossRef]
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chiò, A.; Traynor, B.J. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun. 2016, 7, 12408. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tzeplaeff, L.; Wilfling, S.; Requardt, M.V.; Herdick, M. Current State and Future Directions in the Therapy of ALS. Cells 2023, 12, 1523. https://doi.org/10.3390/cells12111523
Tzeplaeff L, Wilfling S, Requardt MV, Herdick M. Current State and Future Directions in the Therapy of ALS. Cells. 2023; 12(11):1523. https://doi.org/10.3390/cells12111523
Chicago/Turabian StyleTzeplaeff, Laura, Sibylle Wilfling, Maria Viktoria Requardt, and Meret Herdick. 2023. "Current State and Future Directions in the Therapy of ALS" Cells 12, no. 11: 1523. https://doi.org/10.3390/cells12111523
APA StyleTzeplaeff, L., Wilfling, S., Requardt, M. V., & Herdick, M. (2023). Current State and Future Directions in the Therapy of ALS. Cells, 12(11), 1523. https://doi.org/10.3390/cells12111523