
Presynaptic Release-Regulating Sphingosine 1-Phosphate 1/3 Receptors in Cortical Glutamatergic Terminals: Adaptations in EAE Mice and Impact of Therapeutic FTY720


 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. EAE Induction and Clinical Scores
2.3. Drug Treatment
2.4. Synaptosomes
2.5. Superfusion Experiments
2.6. Confocal Microscopy
2.7. Western Blot Analysis
2.8. Biotinylation Studies
2.9. Statistical Analysis
2.10. Drugs and Chemicals
3. Results
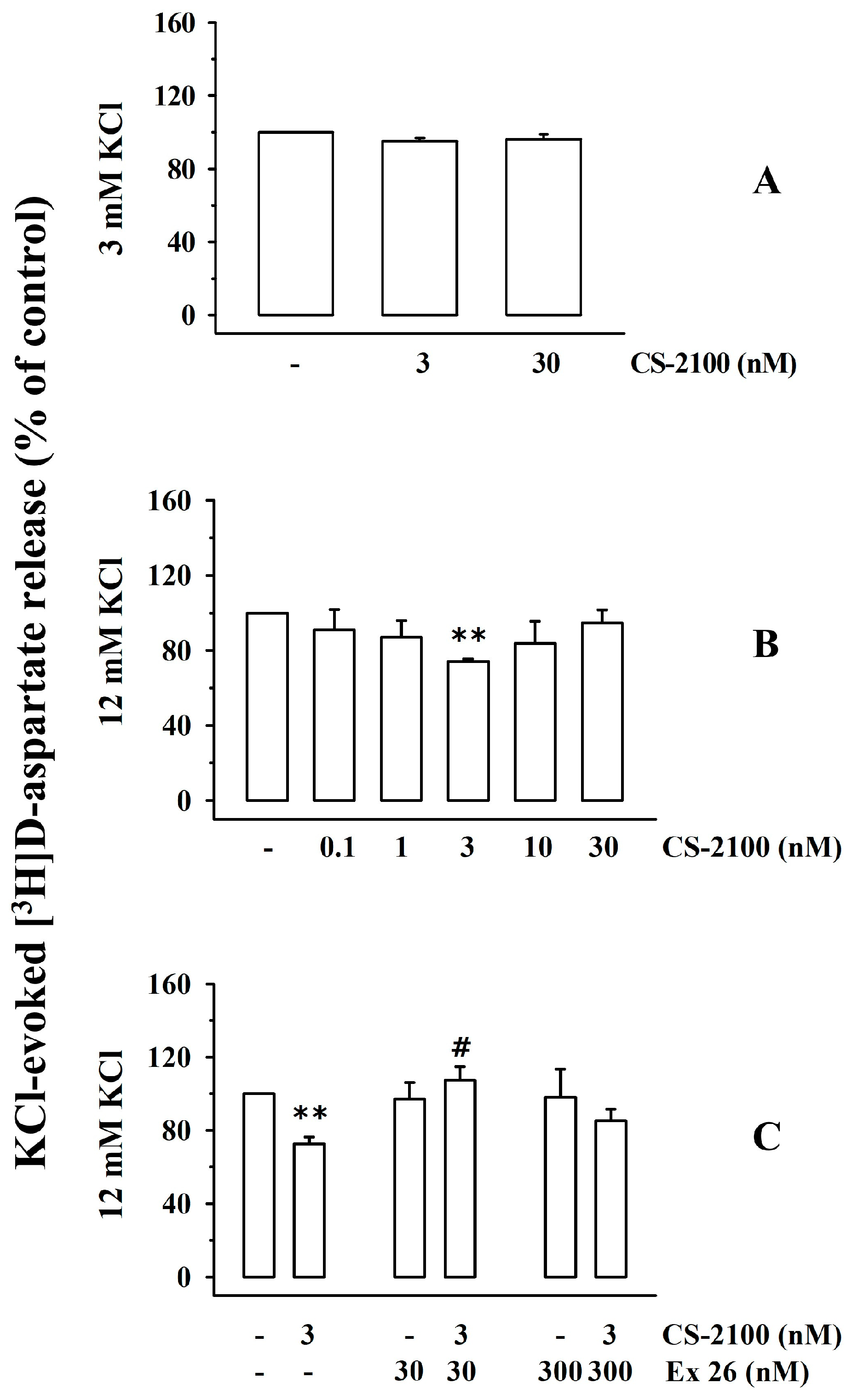
3.1. Presynaptic Release-Regulating S1P1 Receptors in Cortical Synaptosomes of Healthy Mice
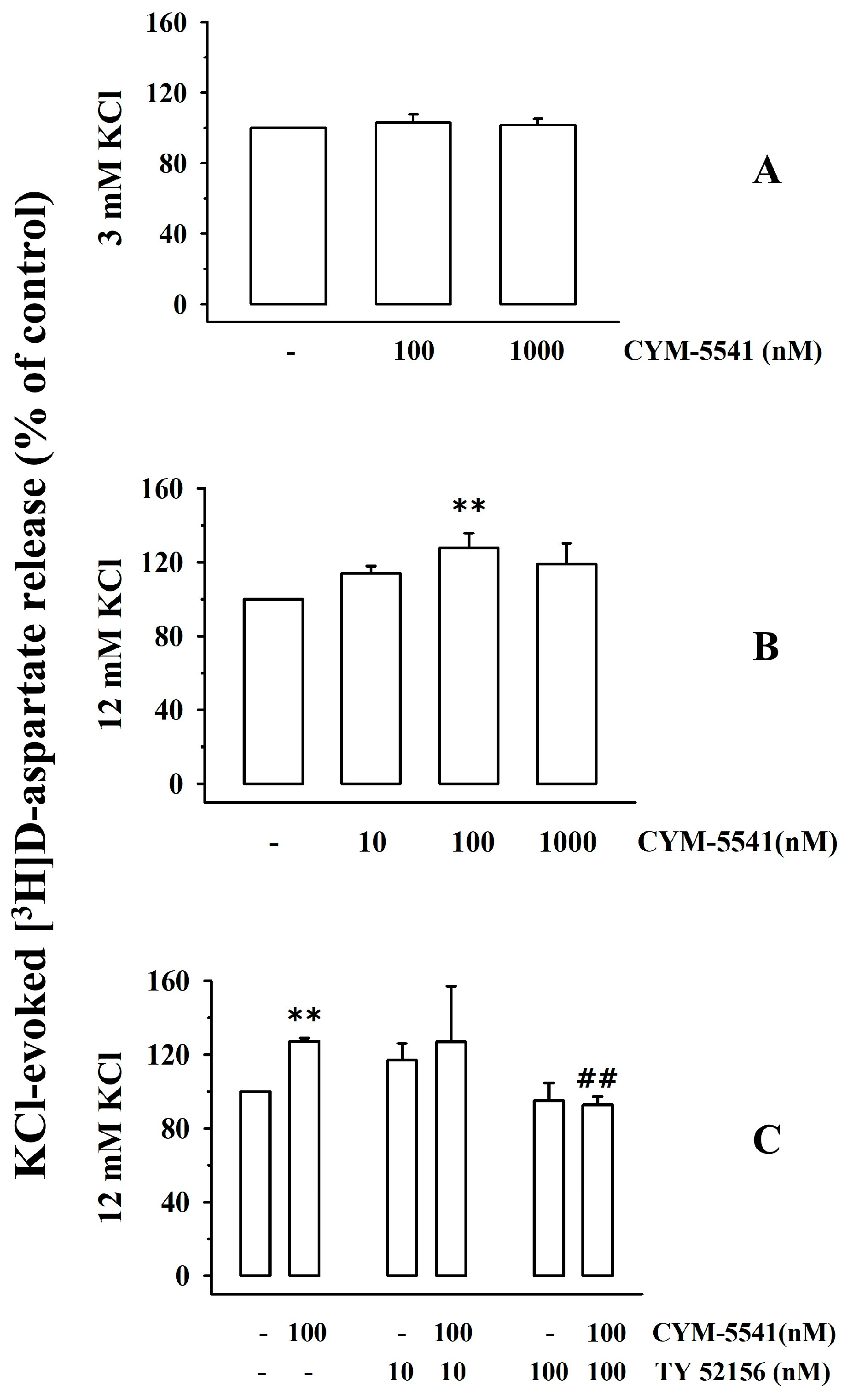
3.2. Presynaptic Release-Regulating S1P3 Receptors Exist in Cortical Synaptosomes of Healthy Mice
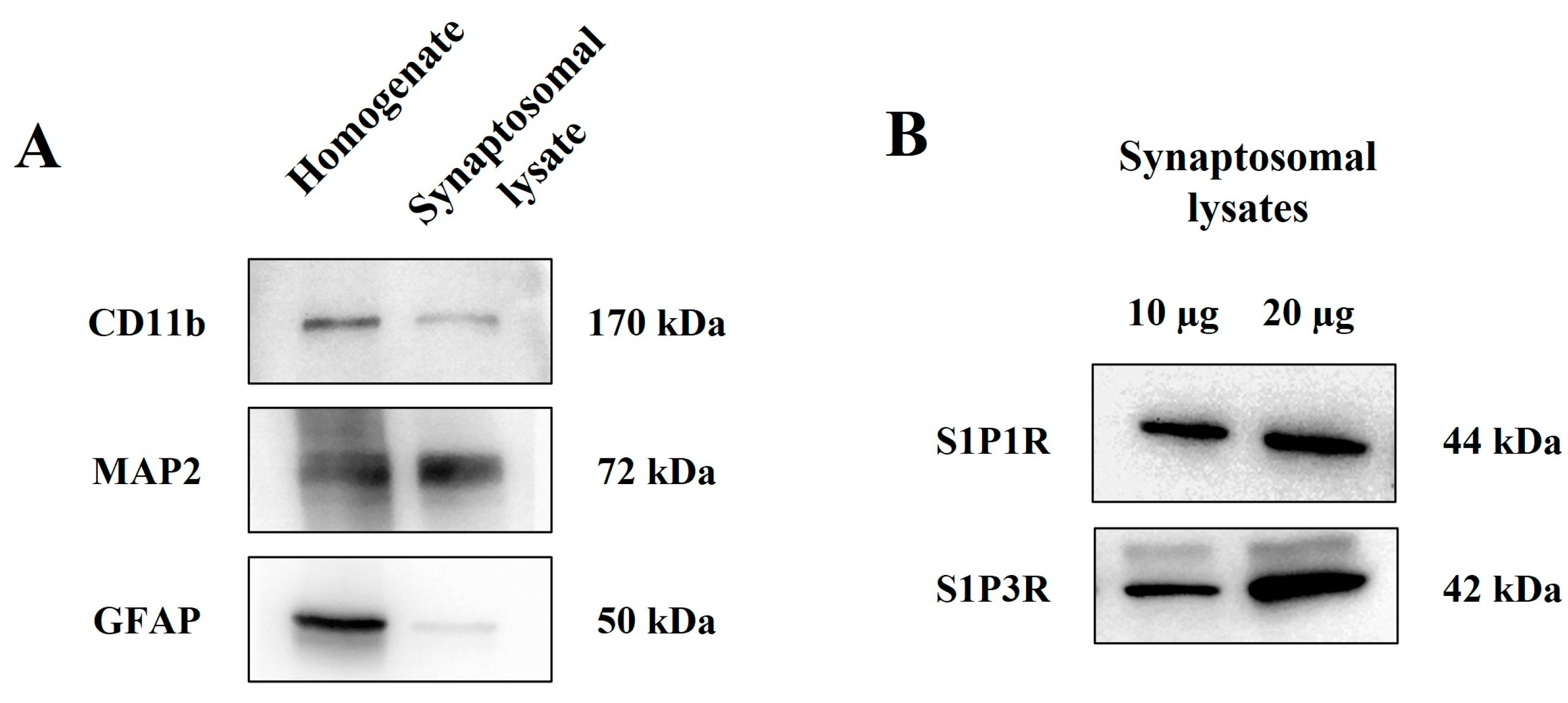
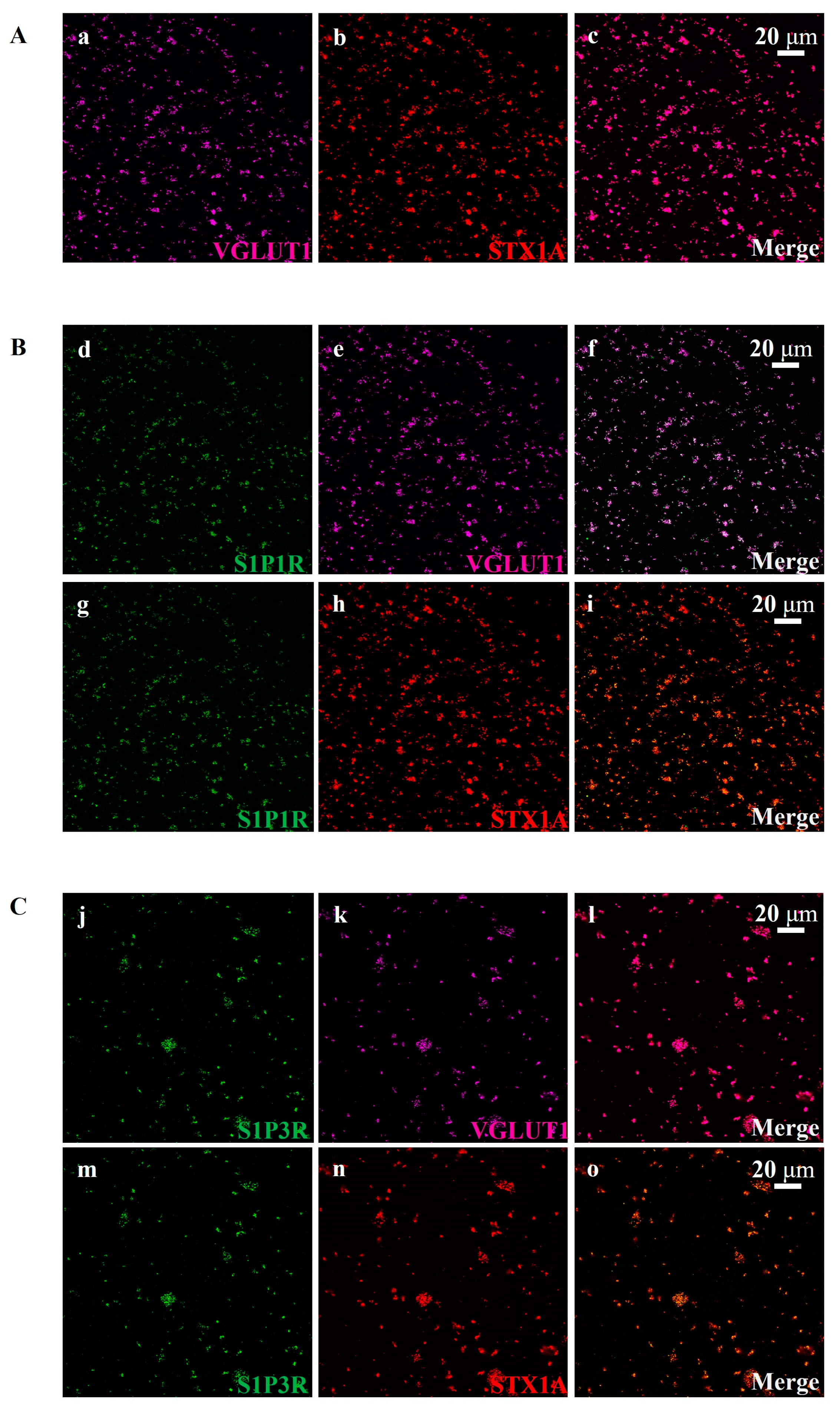
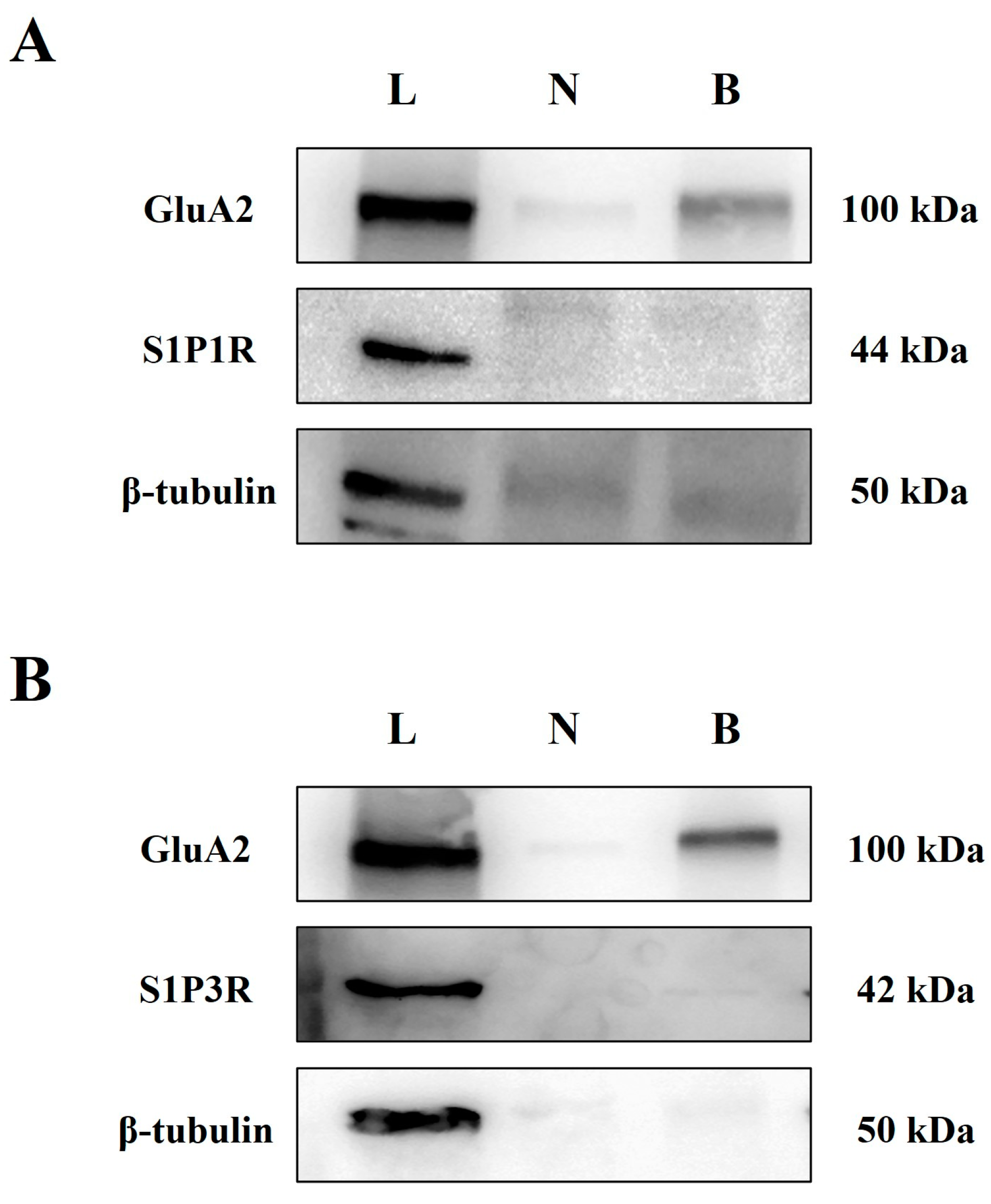
3.3. Cortical Glutamatergic Synaptosomes of Healthy Mice Are Endowed with S1P1 and S1P3 Receptor Proteins

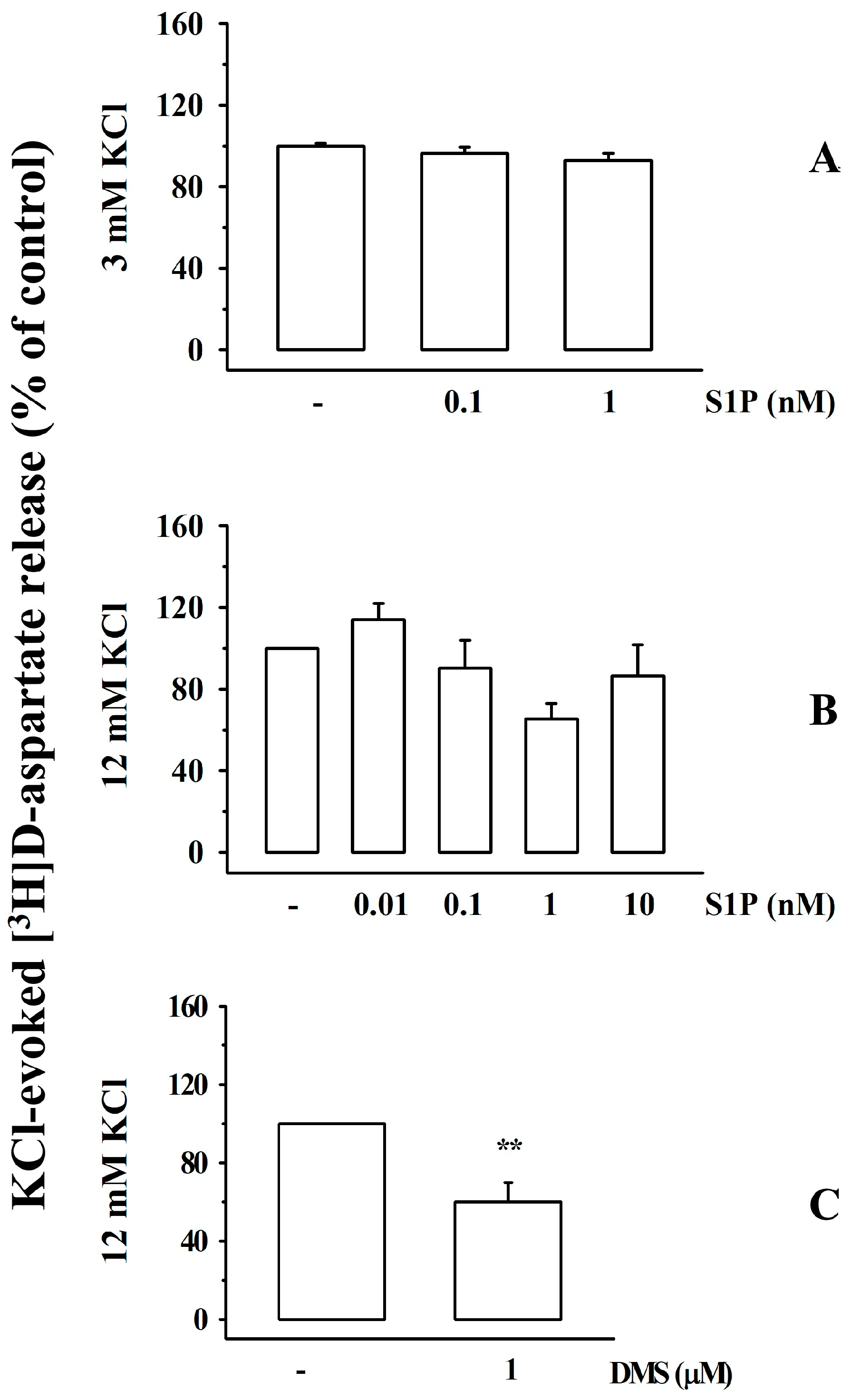
3.4. Impact of Sphingosine-1-Phosphate and N,N-Dimethylsphingosine on the Release of [3H]D-Aspartate from Cortical Synaptosomes of Healthy Mice
3.5. S1P1 and S1P3 Receptors in Cortical Synaptosomal Plasma Membranes: Biotinylation Studies
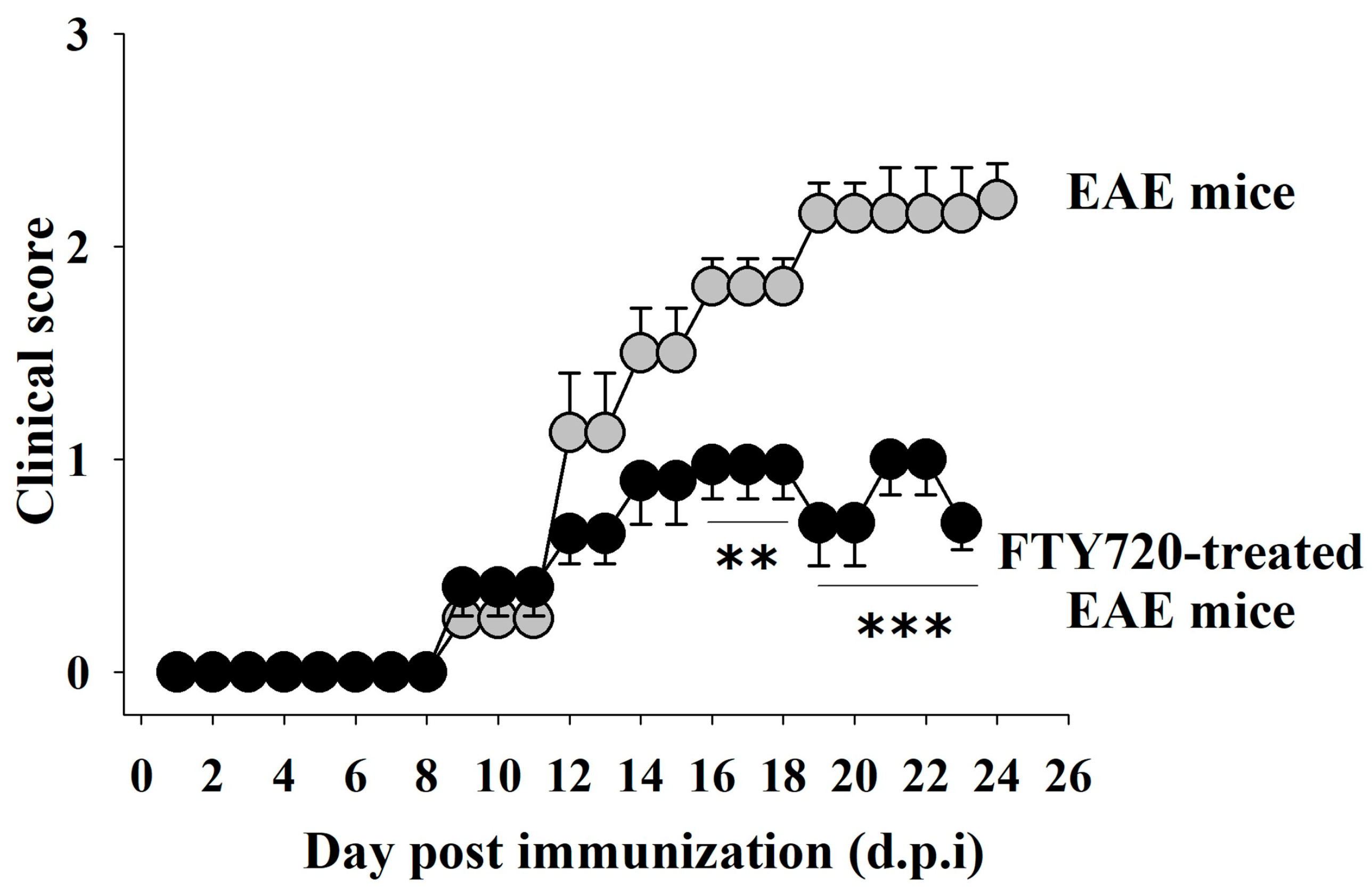
3.6. Clinical Signs in EAE Mice: Impact of FTY720
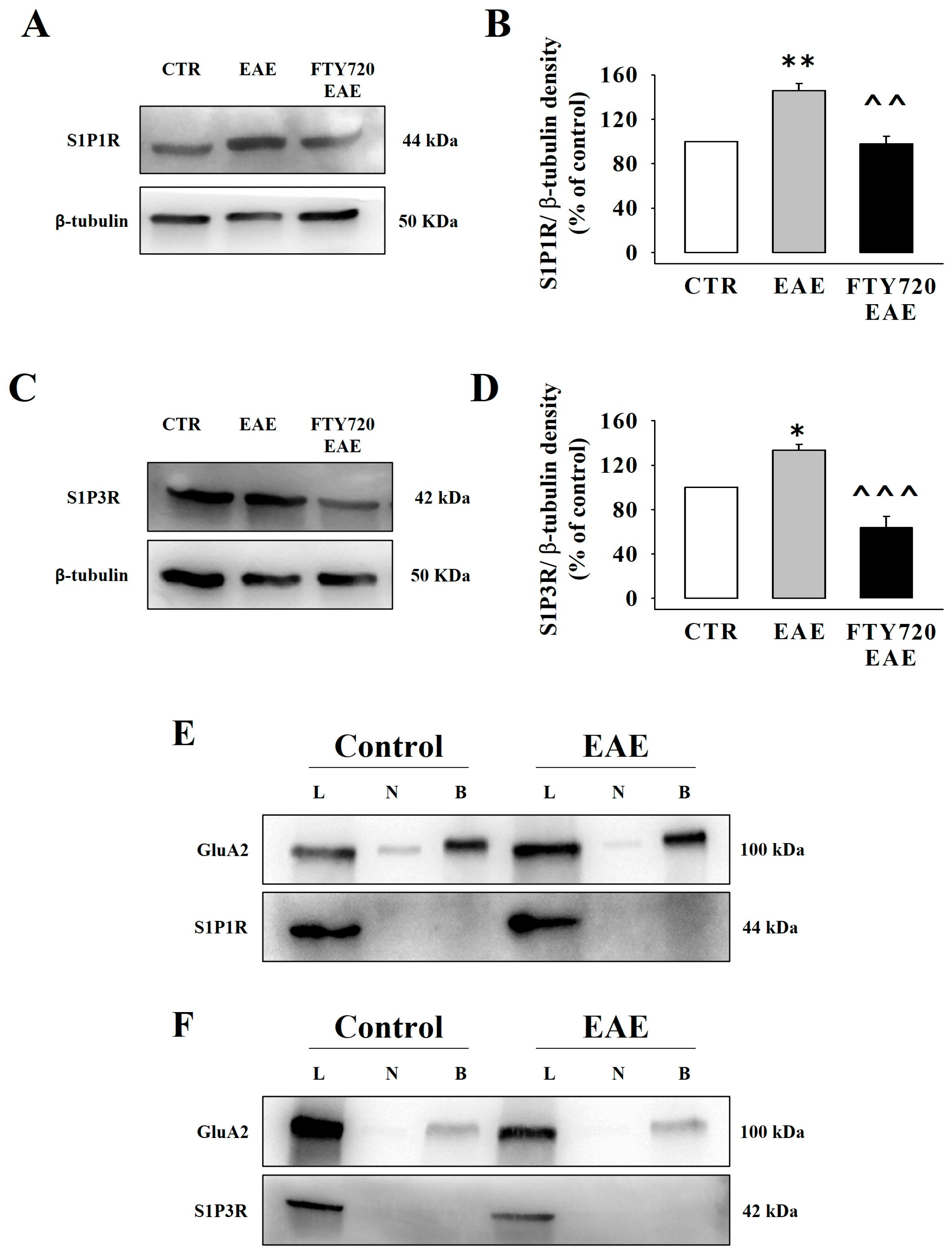
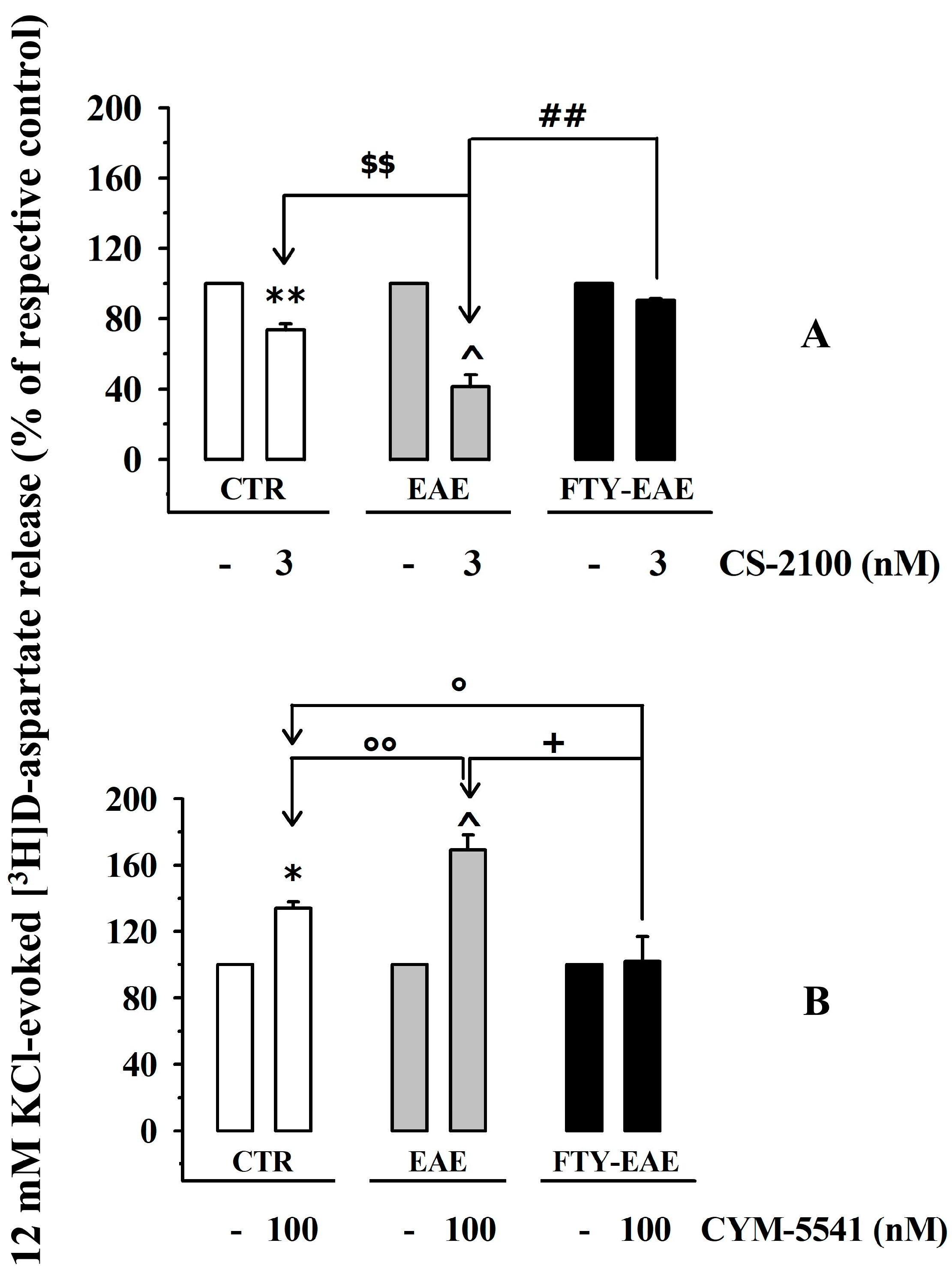
3.7. Presynaptic Release-Regulating S1P1 and S1P3 Receptors in Cortical Synaptosomes of EAE Mice Undergo Biochemical and Functional Adaptations That Are Prevented by Therapeutic Chronic FTY720
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bigaud, M.; Guerini, D.; Billich, A.; Bassilana, F.; Brinkmann, V. Second Generation S1P Pathway Modulators: Research Strategies and Clinical Developments. Biochim. Biophys. Acta 2014, 1841, 745–758. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, S.; Dev, K.K. Sphingosine-1-Phosphate Receptor Therapies: Advances in Clinical Trials for CNS-Related Diseases. Neuropharmacology 2017, 113, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Mullershausen, F.; Craveiro, L.M.; Shin, Y.; Cortes-Cros, M.; Bassilana, F.; Osinde, M.; Wishart, W.L.; Guerini, D.; Thallmair, M.; Schwab, M.E.; et al. Phosphorylated FTY720 Promotes Astrocyte Migration through Sphingosine-1-Phosphate Receptors. J. Neurochem. 2007, 102, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Gardell, S.E.; Herr, D.R.; Rivera, R.; Lee, C.-W.; Noguchi, K.; Teo, S.T.; Yung, Y.C.; Lu, M.; Kennedy, G.; et al. FTY720 (Fingolimod) Efficacy in an Animal Model of Multiple Sclerosis Requires Astrocyte Sphingosine 1-Phosphate Receptor 1 (S1P1) Modulation. Proc. Natl. Acad. Sci. USA 2011, 108, 751–756. [Google Scholar] [CrossRef]
- Anastasiadou, S.; Knöll, B. The Multiple Sclerosis Drug Fingolimod (FTY720) Stimulates Neuronal Gene Expression, Axonal Growth and Regeneration. Exp. Neurol. 2016, 279, 243–260. [Google Scholar] [CrossRef]
- Musella, A.; Gentile, A.; Guadalupi, L.; Rizzo, F.R.; De Vito, F.; Fresegna, D.; Bruno, A.; Dolcetti, E.; Vanni, V.; Vitiello, L.; et al. Central Modulation of Selective Sphingosine-1-Phosphate Receptor 1 Ameliorates Experimental Multiple Sclerosis. Cells 2020, 9, 1290. [Google Scholar] [CrossRef]
- Colombo, E.; Farina, C. Lessons from S1P Receptor Targeting in Multiple Sclerosis. Pharmacol. Ther. 2022, 230, 107971. [Google Scholar] [CrossRef]
- Mandolesi, G.; Gentile, A.; Musella, A.; Fresegna, D.; De Vito, F.; Bullitta, S.; Sepman, H.; Marfia, G.A.; Centonze, D. Synaptopathy Connects Inflammation and Neurodegeneration in Multiple Sclerosis. Nat. Rev. Neurol. 2015, 11, 711–724. [Google Scholar] [CrossRef]
- Bellingacci, L.; Mancini, A.; Gaetani, L.; Tozzi, A.; Parnetti, L.; Di Filippo, M. Synaptic Dysfunction in Multiple Sclerosis: A Red Thread from Inflammation to Network Disconnection. Int. J. Mol. Sci. 2021, 22, 9753. [Google Scholar] [CrossRef]
- Schwarz, K.; Schmitz, F. Synapse Dysfunctions in Multiple Sclerosis. Int. J. Mol. Sci. 2023, 24, 1639. [Google Scholar] [CrossRef]
- Brinkmann, V. FTY720 (Fingolimod) in Multiple Sclerosis: Therapeutic Effects in the Immune and the Central Nervous System. Br. J. Pharmacol. 2009, 158, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Gabrielli, M.; Ponzoni, L.; Pelucchi, S.; Stravalaci, M.; Beeg, M.; Mazzitelli, S.; Braida, D.; Sala, M.; Boda, E.; et al. Fingolimod Limits Acute Aβ Neurotoxicity and Promotes Synaptic Versus Extrasynaptic NMDA Receptor Functionality in Hippocampal Neurons. Sci. Rep. 2017, 7, 41734. [Google Scholar] [CrossRef] [PubMed]
- McGinley, M.P.; Cohen, J.A. Sphingosine 1-Phosphate Receptor Modulators in Multiple Sclerosis and Other Conditions. Lancet 2021, 398, 1184–1194. [Google Scholar] [CrossRef]
- Kanno, T.; Nishizaki, T.; Proia, R.L.; Kajimoto, T.; Jahangeer, S.; Okada, T.; Nakamura, S. Regulation of Synaptic Strength by Sphingosine 1-Phosphate in the Hippocampus. Neuroscience 2010, 171, 973–980. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, N.; Silva, D.G.; Barnett, M.H. Effect of Fingolimod on Brain Volume Loss in Patients with Multiple Sclerosis. CNS Drugs 2017, 31, 289–305. [Google Scholar] [CrossRef]
- Rossi, S.; Lo Giudice, T.; De Chiara, V.; Musella, A.; Studer, V.; Motta, C.; Bernardi, G.; Martino, G.; Furlan, R.; Martorana, A.; et al. Oral Fingolimod Rescues the Functional Deficits of Synapses in Experimental Autoimmune Encephalomyelitis. Br. J. Pharmacol. 2012, 165, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Bonfiglio, T.; Olivero, G.; Merega, E.; Di Prisco, S.; Padolecchia, C.; Grilli, M.; Milanese, M.; Di Cesare Mannelli, L.; Ghelardini, C.; Bonanno, G.; et al. Prophylactic versus Therapeutic Fingolimod: Restoration of Presynaptic Defects in Mice Suffering from Experimental Autoimmune Encephalomyelitis. PLoS ONE 2017, 12, e0170825. [Google Scholar] [CrossRef]
- Levite, M. Glutamate, T Cells and Multiple Sclerosis. J. Neural Transm. 2017, 124, 775–798. [Google Scholar] [CrossRef]
- Kajimoto, T.; Okada, T.; Yu, H.; Goparaju, S.K.; Jahangeer, S.; Nakamura, S. Involvement of Sphingosine-1-Phosphate in Glutamate Secretion in Hippocampal Neurons. Mol. Cell Biol. 2007, 27, 3429–3440. [Google Scholar] [CrossRef]
- Riganti, L.; Antonucci, F.; Gabrielli, M.; Prada, I.; Giussani, P.; Viani, P.; Valtorta, F.; Menna, E.; Matteoli, M.; Verderio, C. Sphingosine-1-Phosphate (S1P) Impacts Presynaptic Functions by Regulating Synapsin I Localization in the Presynaptic Compartment. J. Neurosci. 2016, 36, 4624–4634. [Google Scholar] [CrossRef]
- Wang, C.C.; Kuo, J.R.; Wang, S.J. Fingolimod Inhibits Glutamate Release through Activation of S1P1 Receptors and the G Protein Βγ Subunit-Dependent Pathway in Rat Cerebrocortical Nerve Terminals. Neuropharmacology 2021, 185, 108451. [Google Scholar] [CrossRef] [PubMed]
- Skoug, C.; Martinsson, I.; Gouras, G.K.; Meissner, A.; Duarte, J.M.N. Sphingosine 1-Phoshpate Receptors Are Located in Synapses and Control Spontaneous Activity of Mouse Neurons in Culture. Neurochem. Res. 2022, 47, 3114–3125. [Google Scholar] [CrossRef]
- Kanno, T.; Nishizaki, T. Endogenous Sphingosine 1-Phosphate Regulates Spontaneous Glutamate Release from Mossy Fiber Terminals via S1P3 Receptors. Life Sci. 2011, 89, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Vallarino, G.; Salis, A.; Lucarini, E.; Turrini, F.; Olivero, G.; Roggeri, A.; Damonte, G.; Boggia, R.; Di Cesare Mannelli, L.; Ghelardini, C.; et al. Healthy Properties of a New Formulation of Pomegranate-Peel Extract in Mice Suffering from Experimental Autoimmune Encephalomyelitis. Molecules 2022, 27, 914. [Google Scholar] [CrossRef] [PubMed]
- Dunkley, P.R.; Jarvie, P.E.; Robinson, P.J. A Rapid Percoll Gradient Procedure for Preparation of Synaptosomes. Nat. Protoc. 2008, 3, 1718–1728. [Google Scholar] [CrossRef] [PubMed]
- Grilli, M.; Raiteri, L.; Pittaluga, A. Somatostatin Inhibits Glutamate Release from Mouse Cerebrocortical Nerve Endings through Presynaptic Sst2 Receptors Linked to the Adenylyl Cyclase-Protein Kinase A Pathway. Neuropharmacology 2004, 46, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Raiteri, M.; Angelini, F.; Levi, G. A Simple Apparatus for Studying the Release of Neurotransmitters from Synaptosomes. Eur. J. Pharmacol. 1974, 25, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of Co-Localization of Objects in Dual-Colour Confocal Images. J. Microsc. 1993, 169, 375–382. [Google Scholar] [CrossRef]
- Costes, S.V.; Daelemans, D.; Cho, E.H.; Dobbin, Z.; Pavlakis, G.; Lockett, S. Automatic and Quantitative Measurement of Protein-Protein Colocalization in Live Cells. Biophys. J. 2004, 86, 3993–4003. [Google Scholar] [CrossRef]
- Nakamura, T.; Asano, M.; Sekiguchi, Y.; Mizuno, Y.; Tamaki, K.; Kimura, T.; Nara, F.; Kawase, Y.; Shimozato, T.; Doi, H.; et al. Discovery of CS-2100, a Potent, Orally Active and S1P3-Sparing S1P1 Agonist. Bioorg. Med. Chem. Lett. 2012, 22, 1788–1792. [Google Scholar] [CrossRef] [PubMed]
- Cahalan, S.M.; Gonzalez-Cabrera, P.J.; Nguyen, N.; Guerrero, M.; Cisar, E.A.G.; Leaf, N.B.; Brown, S.J.; Roberts, E.; Rosen, H. Sphingosine 1-Phosphate Receptor 1 (S1P1) Upregulation and Amelioration of Experimental Autoimmune Encephalomyelitis by an S1P1 Antagonist. Mol. Pharmacol. 2013, 83, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Murakami, A.; Takasugi, H.; Ohnuma, S.; Koide, Y.; Sakurai, A.; Takeda, S.; Hasegawa, T.; Sasamori, J.; Konno, T.; Hayashi, K.; et al. Sphingosine 1-Phosphate (S1P) Regulates Vascular Contraction via S1P3 Receptor: Investigation Based on a New S1P3 Receptor Antagonist. Mol. Pharmacol. 2010, 77, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Alloisio, S.; Cervetto, C.; Passalacqua, M.; Barbieri, R.; Maura, G.; Nobile, M.; Marcoli, M. Functional Evidence for Presynaptic P2X7 Receptors in Adult Rat Cerebrocortical Nerve Terminals. FEBS Lett. 2008, 582, 3948–3953. [Google Scholar] [CrossRef]
- Cisani, F.; Olivero, G.; Usai, C.; Van Camp, G.; Maccari, S.; Morley-Fletcher, S.; Pittaluga, A.M. Antibodies against the NH2-Terminus of the GluA Subunits Affect the AMPA-Evoked Releasing Activity: The Role of Complement. Front. Immunol. 2021, 12, 586521. [Google Scholar] [CrossRef] [PubMed]
- Gentile, A.; Musella, A.; Bullitta, S.; Fresegna, D.; De Vito, F.; Fantozzi, R.; Piras, E.; Gargano, F.; Borsellino, G.; Battistini, L.; et al. Siponimod (BAF312) Prevents Synaptic Neurodegeneration in Experimental Multiple Sclerosis. J. Neuroinflamm. 2016, 13, 207. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.F.; Bowen, J.D.; Reder, A.T. The Direct Effects of Fingolimod in the Central Nervous System: Implications for Relapsing Multiple Sclerosis. CNS Drugs 2016, 30, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, R.; Chara, J.C.; Rodríguez-Antigüedad, A.; Matute, C. FTY720 Attenuates Excitotoxicity and Neuroinflammation. J. Neuroinflamm. 2015, 12, 86. [Google Scholar] [CrossRef]
- Darios, F.D.; Jorgacevski, J.; Flašker, A.; Zorec, R.; García-Martinez, V.; Villanueva, J.; Gutiérrez, L.M.; Leese, C.; Bal, M.; Nosyreva, E.; et al. Sphingomimetic Multiple Sclerosis Drug FTY720 Activates Vesicular Synaptobrevin and Augments Neuroendocrine Secretion. Sci. Rep. 2017, 7, 5958. [Google Scholar] [CrossRef]
- Raiteri, L.; Raiteri, M. Synaptosomes Still Viable after 25 Years of Superfusion. Neurochem. Res. 2000, 25, 1265–1274. [Google Scholar] [CrossRef]
- Popoli, M.; Yan, Z.; McEwen, B.S.; Sanacora, G. The Stressed Synapse: The Impact of Stress and Glucocorticoids on Glutamate Transmission. Nat. Rev. Neurosci. 2011, 13, 22–37. [Google Scholar] [CrossRef]
- Pittaluga, A. CCL5-Glutamate Cross-Talk in Astrocyte-Neuron Communication in Multiple Sclerosis. Front. Immunol. 2017, 8, 1079. [Google Scholar] [CrossRef]
- Pittaluga, A. Acute Functional Adaptations in Isolated Presynaptic Terminals Unveil Synaptosomal Learning and Memory. Int. J. Mol. Sci. 2019, 20, 3641. [Google Scholar] [CrossRef] [PubMed]
- Bradley, M.E.; McGuinness, N.; Williams, G.; Charlton, S.J.; Dowling, M.R. The in vitro Metabolism of Sphingosine-1-Phosphate: Identification; Inhibition and Pharmacological Implications. Eur. J. Pharmacol. 2011, 672, 56–61. [Google Scholar] [CrossRef]
- Cannavo, A.; Rengo, G.; Liccardo, D.; Pagano, G.; Zincarelli, C.; De Angelis, M.C.; Puglia, R.; Di Pietro, E.; Rabinowitz, J.E.; Barone, M.V.; et al. Β1-Adrenergic Receptor and Sphingosine-1-Phosphate Receptor 1 (S1PR1) Reciprocal Downregulation Influences Cardiac Hypertrophic Response and Progression to Heart Failure: Protective Role of S1PR1 Cardiac Gene Therapy. Circulation 2013, 128, 1612–1622. [Google Scholar] [CrossRef]
- Musante, V.; Summa, M.; Cunha, R.A.; Raiteri, M.; Pittaluga, A. Pre-Synaptic Glycine GlyT1 Transporter–NMDA Receptor Interaction: Relevance to NMDA Autoreceptor Activation in the Presence of Mg2+ Ions. J. Neurochem. 2011, 117, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Mandolesi, G.; Grasselli, G.; Musumeci, G.; Centonze, D. Cognitive Deficits in Experimental Autoimmune Encephalomyelitis: Neuroinflammation and Synaptic Degeneration. Neurol. Sci. 2010, 31, S255–S259. [Google Scholar] [CrossRef] [PubMed]
- Di Prisco, S.; Merega, E.; Pittaluga, A. Functional Adaptation of Presynaptic Chemokine Receptors in EAE Mouse Central Nervous System. Synapse 2014, 68, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, F.S.; Hofereiter, J.; Rübsamen, H.; Melms, J.; Schwarz, S.; Faber, H.; Weber, P.; Pütz, B.; Loleit, V.; Weber, F.; et al. Fingolimod Induces Neuroprotective Factors in Human Astrocytes. J. Neuroinflamm. 2015, 12, 184. [Google Scholar] [CrossRef]
- Das, A.; Arifuzzaman, S.; Kim, S.H.; Lee, Y.S.; Jung, K.H.; Chai, Y.G. FTY720 (Fingolimod) Regulates Key Target Genes Essential for Inflammation in Microglial Cells as Defined by High-Resolution MRNA Sequencing. Neuropharmacology 2017, 119, 1–14. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roggeri, A.; Olivero, G.; Usai, C.; Vanmierlo, T.; Pittaluga, A. Presynaptic Release-Regulating Sphingosine 1-Phosphate 1/3 Receptors in Cortical Glutamatergic Terminals: Adaptations in EAE Mice and Impact of Therapeutic FTY720. Cells 2023, 12, 2343. https://doi.org/10.3390/cells12192343
Roggeri A, Olivero G, Usai C, Vanmierlo T, Pittaluga A. Presynaptic Release-Regulating Sphingosine 1-Phosphate 1/3 Receptors in Cortical Glutamatergic Terminals: Adaptations in EAE Mice and Impact of Therapeutic FTY720. Cells. 2023; 12(19):2343. https://doi.org/10.3390/cells12192343
Chicago/Turabian StyleRoggeri, Alessandra, Guendalina Olivero, Cesare Usai, Tim Vanmierlo, and Anna Pittaluga. 2023. "Presynaptic Release-Regulating Sphingosine 1-Phosphate 1/3 Receptors in Cortical Glutamatergic Terminals: Adaptations in EAE Mice and Impact of Therapeutic FTY720" Cells 12, no. 19: 2343. https://doi.org/10.3390/cells12192343
APA StyleRoggeri, A., Olivero, G., Usai, C., Vanmierlo, T., & Pittaluga, A. (2023). Presynaptic Release-Regulating Sphingosine 1-Phosphate 1/3 Receptors in Cortical Glutamatergic Terminals: Adaptations in EAE Mice and Impact of Therapeutic FTY720. Cells, 12(19), 2343. https://doi.org/10.3390/cells12192343