


MHC Class II Presentation in Autoimmunity

Abstract
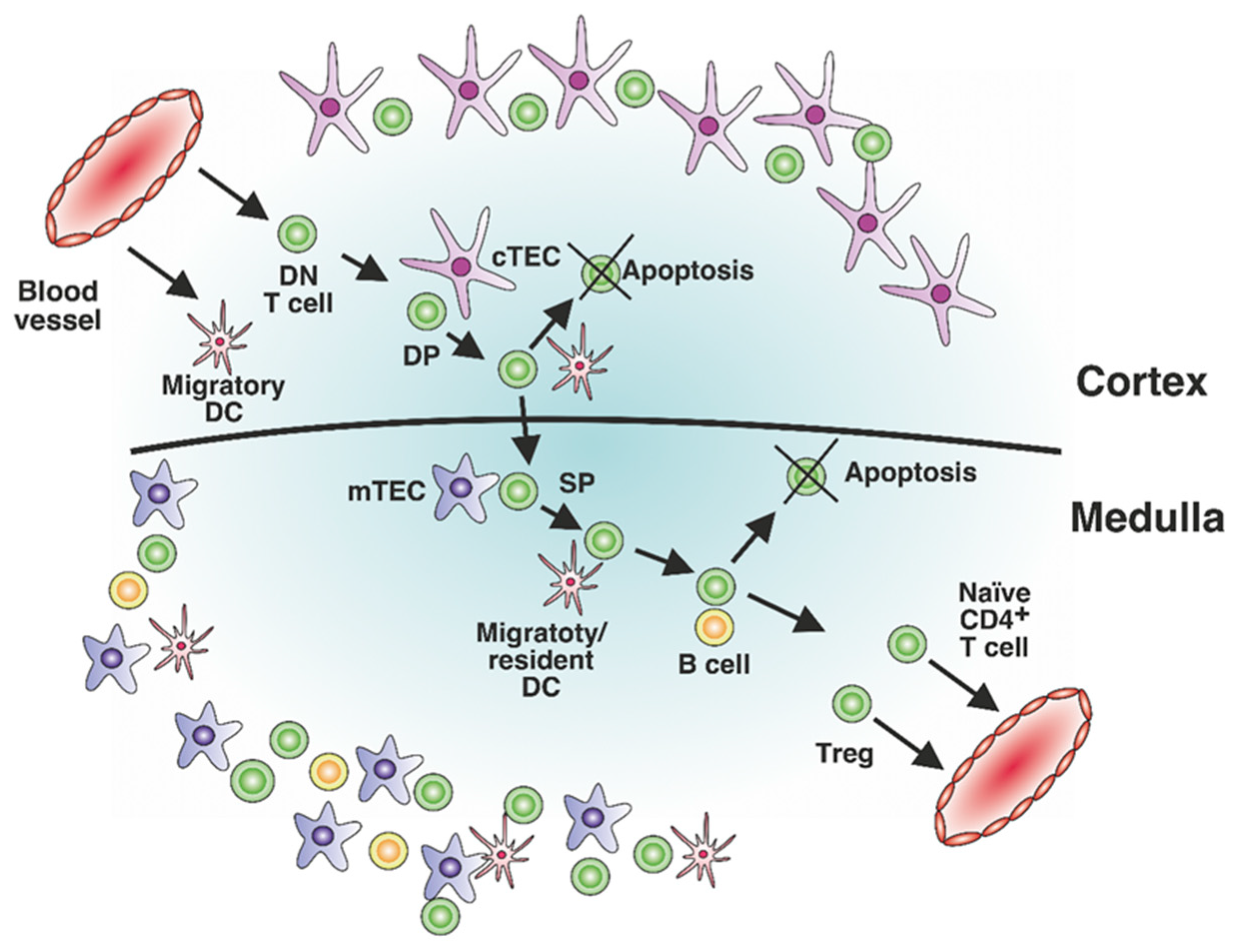
:1. Introduction
2. MHC-II Presentation of Low-Affinity Self-Antigens
3. The Peculiarities of the Interaction between TCRs and Self-pMHC Complexes: Links with Autoimmunity
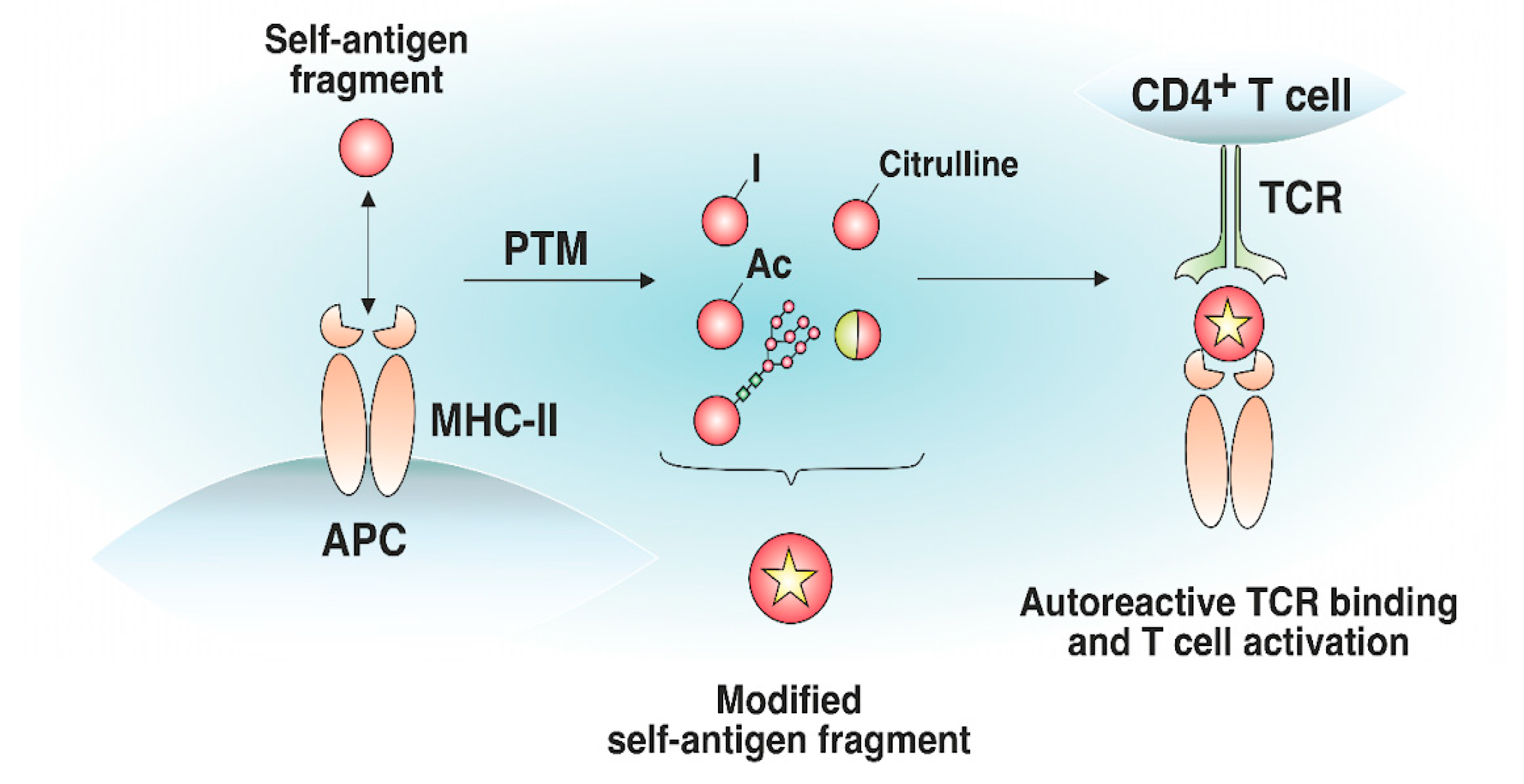
4. The Effect of MHC-II-Presented Self-Antigen Modifications on Autoreactive TCR Recognition
5. The Effect of the Proinflammatory Environment on MHC-II Antigen Presentation and Autoreactive T Cell Engagement
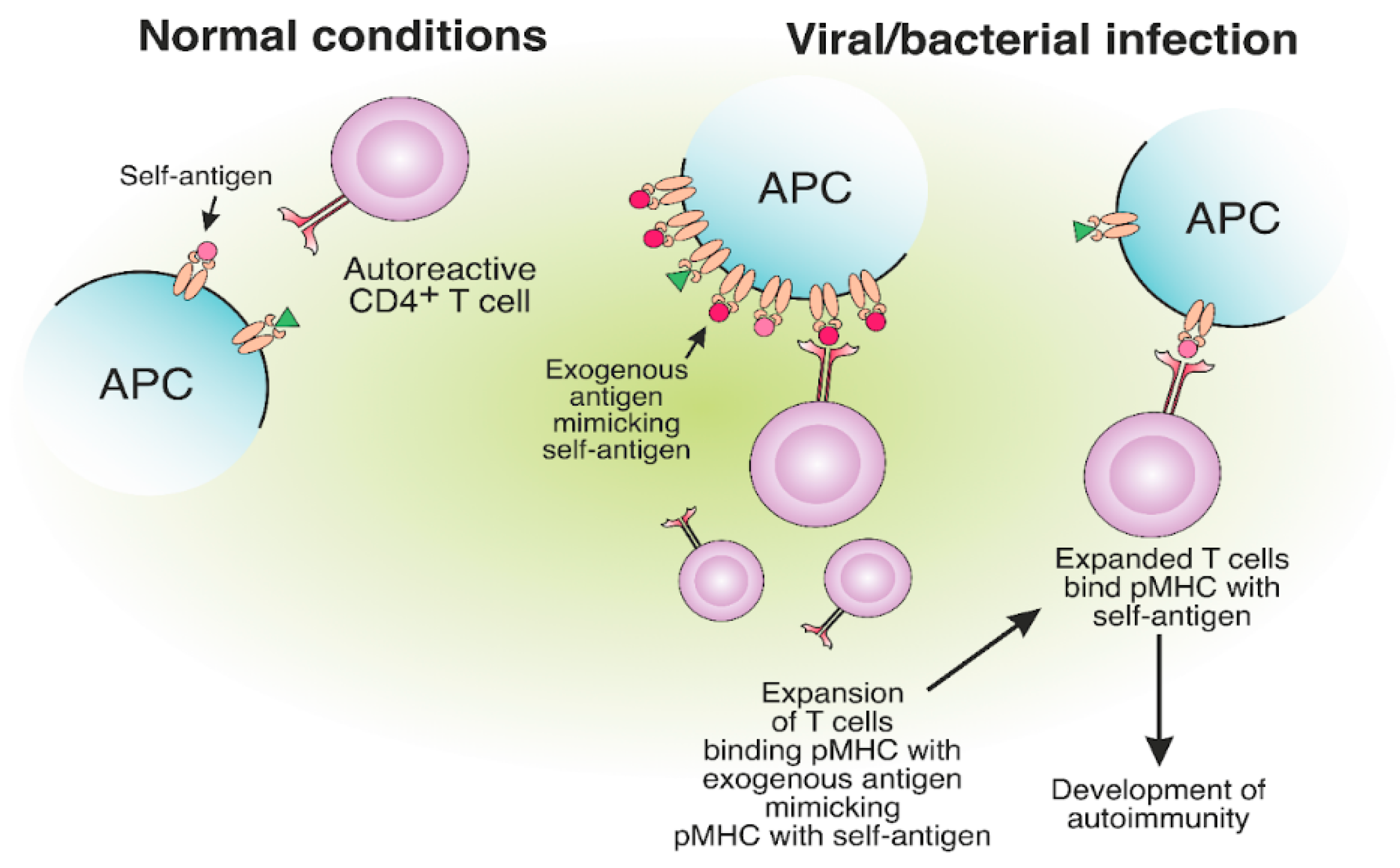
6. Molecular Mimicry between Self- and Foreign MHC-II Antigens May Lead to T Cell-Mediated Autoimmunity
7. The Role of Protective MHC-II Alleles in the Development of Autoimmune Diseases
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Debnath, M.; Banerjee, M.; Berk, M. Genetic Gateways to COVID-19 Infection: Implications for Risk, Severity, and Outcomes. FASEB J. 2020, 34, 8787–8795. [Google Scholar] [CrossRef] [PubMed]
- Colona, V.L.; Biancolella, M.; Novelli, A.; Novelli, G. Will GWAS Eventually Allow the Identification of Genomic Biomarkers for COVID-19 Severity and Mortality? J. Clin. Investig. 2021, 131, e155011. [Google Scholar] [CrossRef] [PubMed]
- Andreakos, E.; Abel, L.; Vinh, D.C.; Kaja, E.; Drolet, B.A.; Zhang, Q.; O’Farrelly, C.; Novelli, G.; Rodríguez-Gallego, C.; Haerynck, F.; et al. A Global Effort to Dissect the Human Genetic Basis of Resistance to SARS-CoV-2 Infection. Nat. Immunol. 2022, 23, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Nersisyan, S.; Zhiyanov, A.; Zakharova, M.; Ishina, I.; Kurbatskaia, I.; Mamedov, A.; Galatenko, A.; Shkurnikov, M.; Gabibov, A.; Tonevitsky, A. Alterations in SARS-CoV-2 Omicron and Delta Peptides Presentation by HLA Molecules. Peer J. 2022, 10, e13354. [Google Scholar] [CrossRef] [PubMed]
- The International HIV Controllers Study. The Major Genetic Determinants of HIV-1 Control Affect HLA Class I Peptide Presentation. Science (1979) 2010, 330, 1551–1557. [Google Scholar] [CrossRef] [Green Version]
- Košmrlj, A.; Read, E.L.; Qi, Y.; Allen, T.M.; Altfeld, M.; Deeks, S.G.; Pereyra, F.; Carrington, M.; Walker, B.D.; Chakraborty, A.K. Effects of Thymic Selection of the T-Cell Repertoire on HLA Class I-Associated Control of HIV Infection. Nature 2010, 465, 350–354. [Google Scholar] [CrossRef] [Green Version]
- Monel, B.; McKeon, A.; Lamothe-Molina, P.; Jani, P.; Boucau, J.; Pacheco, Y.; Jones, R.B.; le Gall, S.; Walker, B.D. HIV Controllers Exhibit Effective CD8+ T Cell Recognition of HIV-1-Infected Non-Activated CD4+ T Cells. Cell. Rep. 2019, 27, 142–153. [Google Scholar] [CrossRef] [Green Version]
- Ishigaki, K.; Lagattuta, K.A.; Luo, Y.; James, E.A.; Buckner, J.H.; Raychaudhuri, S. HLA Autoimmune Risk Alleles Restrict the Hypervariable Region of T Cell Receptors. Nat. Genet. 2022, 54, 393–402. [Google Scholar] [CrossRef]
- Scavuzzi, B.M.; van Drongelen, V.; Holoshitz, J. HLA-G and the MHC Cusp Theory. Front. Immunol. 2022, 13, 814967. [Google Scholar] [CrossRef]
- van Drongelen, V.; Scavuzzi, B.M.; Nogueira, S.V.; Miller, F.W.; Sawalha, A.H.; Holoshitz, J. HLA-DRB1 Allelic Epitopes That Associate with Autoimmune Disease Risk or Protection Activate Reciprocal Macrophage Polarization. Sci. Rep. 2021, 11, 2599. [Google Scholar] [CrossRef]
- Jin, H.; Kishida, K.; Arase, N.; Matsuoka, S.; Nakai, W.; Kohyama, M.; Suenaga, T.; Yamamoto, K.; Sasazuki, T.; Arase, H. Abrogation of Self-Tolerance by Misfolded Self-Antigens Complexed with MHC Class II Molecules. Sci. Adv. 2022, 8, abj9867. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-M.; Bautista, J.L.; Scott-Browne, J.; Mohan, J.F.; Hsieh, C.-S. A Broad Range of Self-Reactivity Drives Thymic Regulatory T Cell Selection to Limit Responses to Self. Immunity 2012, 37, 475–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cozzo Picca, C.; Oh, S.; Panarey, L.; Aitken, M.; Basehoar, A.; Caton, A.J. Thymocyte Deletion Can Bias Treg Formation toward Low-Abundance Self-Peptide. Eur. J. Immunol. 2009, 39, 3301–3306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagattuta, K.A.; Kang, J.B.; Nathan, A.; Pauken, K.E.; Jonsson, A.H.; Rao, D.A.; Sharpe, A.H.; Ishigaki, K.; Raychaudhuri, S. Repertoire Analyses Reveal T Cell Antigen Receptor Sequence Features That Influence T Cell Fate. Nat. Immunol. 2022, 23, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, D.; Linehan, J.L.; Dileepan, T.; Lee, Y.J.; Purtha, W.E.; Lu, J.V.; Nelson, R.W.; Fife, B.T.; Orr, H.T.; Anderson, M.S.; et al. Tolerance Is Established in Polyclonal CD4+ T Cells by Distinct Mechanisms, According to Self-Peptide Expression Patterns. Nat. Immunol. 2016, 17, 187–195. [Google Scholar] [CrossRef]
- Hahn, M.; Nicholson, M.J.; Pyrdol, J.; Wucherpfennig, K.W. Unconventional Topology of Self Peptide–Major Histocompatibility Complex Binding by a Human Autoimmune T Cell Receptor. Nat. Immunol. 2005, 6, 490–496. [Google Scholar] [CrossRef] [Green Version]
- Mariathasan, S.; Zakarian, A.; Bouchard, D.; Michie, A.M.; Zúñiga-Pflücker, J.C.; Ohashi, P.S. Duration and Strength of Extracellular Signal-Regulated Kinase Signals Are Altered During Positive Versus Negative Thymocyte Selection. J. Immunol. 2001, 167, 4966–4973. [Google Scholar] [CrossRef] [Green Version]
- Itano, A.; Salmon, P.; Kioussis, D.; Tolaini, M.; Corbella, P.; Robey, E. The Cytoplasmic Domain of CD4 Promotes the Development of CD4 Lineage T Cells. J. Exp. Med. 1996, 183, 731–741. [Google Scholar] [CrossRef]
- Fugger, L.; Svejgaard, A. Association of MHC and Rheumatoid Arthritis: HLA-DR4 and Rheumatoid Arthritis—Studies in Mice and Men. Arthritis. Res. 2000, 2, 208. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Jelcic, I.; Mühlenbruch, L.; Haunerdinger, V.; Toussaint, N.C.; Zhao, Y.; Cruciani, C.; Faigle, W.; Naghavian, R.; Foege, M.; et al. HLA-DR15 Molecules Jointly Shape an Autoreactive T Cell Repertoire in Multiple Sclerosis. Cell 2020, 183, 1264–1281.e20. [Google Scholar] [CrossRef]
- Ooi, J.D.; Petersen, J.; Tan, Y.H.; Huynh, M.; Willett, Z.J.; Ramarathinam, S.H.; Eggenhuizen, P.J.; Loh, K.L.; Watson, K.A.; Gan, P.Y.; et al. Dominant Protection from HLA-Linked Autoimmunity by Antigen-Specific Regulatory T Cells. Nature 2017, 545, 243–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamedov, A.; Vorobyeva, N.; Filimonova, I.; Zakharova, M.; Kiselev, I.; Bashinskaya, V.; Baulina, N.; Boyko, A.; Favorov, A.; Kulakova, O.; et al. Protective Allele for Multiple Sclerosis HLA-DRB1*01:01 Provides Kinetic Discrimination of Myelin and Exogenous Antigenic Peptides. Front Immunol. 2020, 10, 3088. [Google Scholar] [CrossRef] [PubMed]
- Mamedov, A.E.; Zakharova, M.Y.; Favorova, O.O.; Kulakova, O.G.; Boyko, A.N.; Knorre, V.D.; Vorobieva, N.A.; Khurs, E.N.; Kiselev, I.S.; Baulina, N.M.; et al. Loading Rate of Exogenous and Autoantigenic Determinants on Major Histocompatibility Complex Class II Mediates Resistance to Multiple Sclerosis. Dokl. Biochem. Biophys. 2019, 485, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Mamedov, A.E.; Filimonova, I.N.; Smirnov, I.V.; Belogurov, A.A. Peculiarities of the Presentation of the Encephalitogenic MBP Peptide by HLA-DR Complexes Providing Protection and Predisposition to Multiple Sclerosis. Acta Nat. 2021, 13, 127. [Google Scholar] [CrossRef] [PubMed]
- Fowler, M.J. Diabetes: Magnitude and Mechanisms. Clin. Diabetes 2010, 28, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Pociot, F.; Lernmark, Å. Genetic Risk Factors for Type 1 Diabetes. Lancet 2016, 387, 2331–2339. [Google Scholar] [CrossRef]
- Xiaomin, W.; Junbao, Y.; Eddie, J.; I-Ting, C.; Helena, R.; Kwork, W.W. Increased Islet Antigen–Specific Regulatory and Effector CD4+ T Cells in Healthy Individuals with the Type 1 Diabetes–Protective Haplotype. Sci. Immunol. 2020, 5, eaax8767. [Google Scholar] [CrossRef]
- Blum, J.S.; Wearsch, P.A.; Cresswell, P. Pathways of Antigen Processing. Annu Rev. Immunol. 2013, 31, 443–473. [Google Scholar] [CrossRef] [Green Version]
- Ishina, I.A.; Filimonova, I.N.; Zakharova, M.Y.; Ovchinnikova, L.A.; Mamedov, A.E.; Lomakin, Y.A.; Belogurov, A.A. Exhaustive Search of the Receptor Ligands by the CyCLOPS (Cytometry Cell-Labeling Operable Phage Screening) Technique. Int. J. Mol. Sci. 2020, 21, 6258. [Google Scholar] [CrossRef]
- Álvaro-Benito, M.; Freund, C. Revisiting Nonclassical HLA II Functions in Antigen Presentation: Peptide Editing and Its Modulation. HLA 2020, 96, 415–429. [Google Scholar] [CrossRef]
- Álvaro-Benito, M.; Morrison, E.; Abualrous, E.T.; Kuropka, B.; Freund, C. Quantification of HLA-DM-Dependent Major Histocompatibility Complex of Class II Immunopeptidomes by the Peptide Landscape Antigenic Epitope Alignment Utility. Front. Immunol. 2018, 9, 872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, A.B.; Kropshofer, H.; Moldenhauer, G.; Hämmerling, G.J. Kinetic Analysis of Peptide Loading onto HLA-DR Molecules Mediated by HLA-DM. Proc. Natl. Acad. Sci. USA 1996, 93, 9724–9729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvaro-Benito, M.; Morrison, E.; Ebner, F.; Abualrous, E.T.; Urbicht, M.; Wieczorek, M.; Freund, C. Distinct Editing Functions of Natural HLA-DM Allotypes Impact Antigen Presentation and CD4+ T Cell Activation. Cell. Mol. Immunol. 2020, 17, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Vargas, E.; Barker, A.P.; Zhou, Z.; He, X.; Jensen, P.E. HLA-DM Catalytically Enhances Peptide Dissociation by Sensing Peptide–MHC Class II Interactions throughout the Peptide-Binding Cleft. J. Biol. Chem. 2020, 295, 2959–2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poluektov, Y.; Kim, A.; Sadegh-Nasseri, S. HLA-DO and Its Role in MHC Class II Antigen Presentation. Front. Immunol. 2013, 4, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanaware, P.P.; Jurewicz, M.M.; Leszyk, J.D.; Shaffer, S.A.; Stern, L.J. HLA-DO Modulates the Diversity of the MHC-II Self-Peptidome. Mol. Cell. Proteom. 2019, 18, 490–503. [Google Scholar] [CrossRef] [Green Version]
- Surh, C.D.; Lee, D.-S.; Fung-Leung, W.; Karlsson, L.; Sprent, J. Thymic Selection by a Single MHC/Peptide Ligand Produces a Semidiverse Repertoire of CD4+ T Cells. Immunity 1997, 7, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Olsson, N.; Jiang, W.; Adler, L.N.; Mellins, E.D.; Elias, J.E. Tuning DO: DM Ratios Modulates MHC Class II Immunopeptidomes. Mol. Cell. Proteom. 2022, 21, 100204. [Google Scholar] [CrossRef]
- Zhou, Z.; Reyes-Vargas, E.; Escobar, H.; Chang, K.Y.; Barker, A.P.; Rockwood, A.L.; Delgado, J.C.; He, X.; Jensen, P.E. Peptidomic Analysis of Type 1 Diabetes Associated HLA-DQ Molecules and the Impact of HLA-DM on Peptide Repertoire Editing. Eur. J. Immunol. 2017, 47, 314–326. [Google Scholar] [CrossRef]
- Collado, J.A.; Alvarez, I.; Ciudad, M.T.; Espinosa, G.; Canals, F.; Pujol-Borrell, R.; Carrascal, M.; Abian, J.; Jaraquemada, D. Composition of the HLA-DR-Associated Human Thymus Peptidome. Eur. J. Immunol. 2013, 43, 2273–2282. [Google Scholar] [CrossRef]
- Nielsen, M.; Justesen, S.; Lund, O.; Lundegaard, C.; Buus, S. NetMHCIIpan-2.0—Improved Pan-Specific HLA-DR Predictions Using a Novel Concurrent Alignment and Weight Optimization Training Procedure. Immunome Res. 2010, 6, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muixí, L.; Carrascal, M.; Alvarez, I.; Daura, X.; Martí, M.; Armengol, M.P.; Pinilla, C.; Abian, J.; Pujol-Borrell, R.; Jaraquemada, D. Thyroglobulin Peptides Associate In Vivo to HLA-DR in Autoimmune Thyroid Glands. J. Immunol. 2008, 181, 795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fissolo, N.; Haag, S.; de Graaf, K.L.; Drews, O.; Stevanovic, S.; Rammensee, H.G.; Weissert, R. Naturally Presented Peptides on Major Histocompatibility Complex I and II Molecules Eluted from Central Nervous System of Multiple Sclerosis Patients. Mol. Cell. Proteom. 2009, 8, 2090–2101. [Google Scholar] [CrossRef] [Green Version]
- Martinsen, V.; Kursula, P. Multiple Sclerosis and Myelin Basic Protein: Insights into Protein Disorder and Disease. Amino. Acids 2022, 54, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Belogurov, A.A.; Kurkova, I.N.; Friboulet, A.; Thomas, D.; Misikov, V.K.; Zakharova, M.Y.; Suchkov, S.V.; Kotov, S.V.; Alehin, A.I.; Avalle, B.; et al. Recognition and Degradation of Myelin Basic Protein Peptides by Serum Autoantibodies: Novel Biomarker for Multiple Sclerosis. J. Immunol. 2008, 180, 1258–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belogurov, A.; Zakharov, K.; Lomakin, Y.; Surkov, K.; Avtushenko, S.; Kruglyakov, P.; Smirnov, I.; Makshakov, G.; Lockshin, C.; Gregoriadis, G.; et al. CD206-Targeted Liposomal Myelin Basic Protein Peptides in Patients with Multiple Sclerosis Resistant to First-Line Disease-Modifying Therapies: A First-in-Human, Proof-of-Concept Dose-Escalation Study. Neurotherapeutics 2016, 13, 895–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nokoff, N.J.; Rewers, M.; Cree Green, M. The Interplay of Autoimmunity and Insulin Resistance in Type 1 Diabetes. Discov. Med. 2012, 13, 115–122. [Google Scholar]
- Ito, Y.; Ashenberg, O.; Pyrdol, J.; Luoma, A.M.; Rozenblatt-Rosen, O.; Hofree, M.; Christian, E.; Ferrari de Andrade, L.; Tay, R.E.; Teyton, L.; et al. Rapid CLIP Dissociation from MHC II Promotes an Unusual Antigen Presentation Pathway in Autoimmunity. J. Exp. Med. 2018, 215, 2617–2635. [Google Scholar] [CrossRef]
- Busch, R.; Kollnberger, S.; Mellins, E.D. HLA Associations in Inflammatory Arthritis: Emerging Mechanisms and Clinical Implications. Nat. Rev. Rheumatol. 2019, 15, 364–381. [Google Scholar] [CrossRef] [Green Version]
- Markovic-Plese, S.; Fukaura, H.; Zhang, J.; al-Sabbagh, A.; Southwood, S.; Sette, A.; Kuchroo, V.K.; Hafler, D.A. T Cell Recognition of Immunodominant and Cryptic Proteolipid Protein Epitopes in Humans. J. Immunol. 1995, 155, 982. [Google Scholar] [CrossRef]
- Nakayama, M.; Beilke, J.N.; Jasinski, J.M.; Kobayashi, M.; Miao, D.; Li, M.; Coulombe, M.G.; Liu, E.; Elliott, J.F.; Gill, R.G.; et al. Priming and Effector Dependence on Insulin B:9-23 Peptide in NOD Islet Autoimmunity. J. Clin. Investig. 2007, 117, 1835–1843. [Google Scholar] [CrossRef] [PubMed]
- Levisetti, M.G.; Suri, A.; Petzold, S.J.; Unanue, E.R. The Insulin-Specific T Cells of Nonobese Diabetic Mice Recognize a Weak MHC-Binding Segment in More Than One Form. J. Immunol. 2007, 178, 6051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, J.F.; Petzold, S.J.; Unanue, E.R. Register Shifting of an Insulin Peptide-MHC Complex Allows Diabetogenic T Cells to Escape Thymic Deletion. J. Exp. Med. 2011, 208, 2375–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadinski, B.D.; Zhang, L.; Crawford, F.; Marrack, P.; Eisenbarth, G.S.; Kappler, J.W. Diabetogenic T Cells Recognize Insulin Bound to IAg7 in an Unexpected, Weakly Binding Register. Proc. Natl. Acad. Sci. USA 2010, 107, 10978–10983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frances, C.; Brian, S.; Niyun, J.; Aaron, M.; Maki, N.; Philip, P.; Philippa, M.; George, E.; Kappler, J.W. Specificity and Detection of Insulin-Reactive CD4+ T Cells in Type 1 Diabetes in the Nonobese Diabetic (NOD) Mouse. Proc. Natl. Acad. Sci. USA 2011, 108, 16729–16734. [Google Scholar] [CrossRef] [Green Version]
- Junbao, Y.; I-Ting, C.; Tomasz, S.; Nadia, T.-C.; Carla, J.G.; James, E.A.; Kappler, J.W.; Davidson, H.W.; Kwork, W.W. Autoreactive T Cells Specific for Insulin B:11-23 Recognize a Low-Affinity Peptide Register in Human Subjects with Autoimmune Diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 14840–14845. [Google Scholar] [CrossRef] [Green Version]
- Wan, X.; Vomund, A.N.; Peterson, O.J.; Chervonsky, A.V.; Lichti, C.F.; Unanue, E.R. The MHC-II Peptidome of Pancreatic Islets Identifies Key Features of Autoimmune Peptides. Nat. Immunol. 2020, 21, 455–463. [Google Scholar] [CrossRef]
- Loyal, L.; Braun, J.; Henze, L.; Kruse, B.; Dingeldey, M.; Reimer, U.; Kern, F.; Schwarz, T.; Mangold, M.; Unger, C.; et al. Cross-Reactive CD4+ T Cells Enhance SARS-CoV-2 Immune Responses upon Infection and Vaccination. Science (1979) 2022, 374, eabh1823. [Google Scholar] [CrossRef]
- Gao, Y.; Cai, C.; Grifoni, A.; Müller, T.R.; Niessl, J.; Olofsson, A.; Humbert, M.; Hansson, L.; Österborg, A.; Bergman, P.; et al. Ancestral SARS-CoV-2-Specific T Cells Cross-Recognize the Omicron Variant. Nat. Med. 2022, 28, 472–476. [Google Scholar] [CrossRef]
- Molodtsov, I.A.; Kegeles, E.; Mitin, A.N.; Mityaeva, O.; Musatova, O.E.; Panova, A.E.; Pashenkov, M.V.; Peshkova, I.O.; Alsalloum, A.; Asaad, W.; et al. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)–Specific T Cells and Antibodies in Coronavirus Disease 2019 (COVID-19) Protection: A Prospective Study. Clin. Infect. Dis. 2022, 75, e1–e9. [Google Scholar] [CrossRef]
- Yiyuan, Y.; Xiang, W.X.; Mariuzza, R.A.A. Crystal Structure of a Complete Ternary Complex of T-Cell Receptor, Peptide–MHC, and CD4. Proc. Natl. Acad. Sci. USA 2012, 109, 5405–5410. [Google Scholar] [CrossRef] [PubMed]
- Zareie, P.; Farenc, C.; la Gruta, N.L. MHC Restriction: Where Are We Now? Viral. Immunol. 2020, 33, 179–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mørch, A.M.; Bálint, Š.; Santos, A.M.; Davis, S.J.; Dustin, M.L. Coreceptors and TCR Signaling—The Strong and the Weak of It. Front Cell. Dev. Biol. 2020, 8, 597627. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, M.G.; Stanfield, R.L.; Wilson, I.A. How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 2006, 24, 419–466. [Google Scholar] [CrossRef] [PubMed]
- Rossjohn, J.; Gras, S.; Miles, J.J.; Turner, S.J.; Godfrey, D.I.; McCluskey, J. T Cell Antigen Receptor Recognition of Antigen-Presenting Molecules. Annu. Rev. Immunol. 2015, 33, 169–200. [Google Scholar] [CrossRef]
- Hennecke, J.; Carfi, A.; Wiley, D.C. Structure of a Covalently Stabilized Complex of a Human Aβ T-Cell Receptor, Influenza HA Peptide and MHC Class II Molecule, HLA-DR1. EMBO J. 2000, 19, 5611–5624. [Google Scholar] [CrossRef] [Green Version]
- Kato, Z.; Stern, J.N.H.; Nakamura, H.K.; Kuwata, K.; Kondo, N.; Strominger, J.L. Positioning of Autoimmune TCR-Ob.2F3 and TCR-Ob.3D1 on the MBP85–99/HLA-DR2 Complex. Proc. Natl. Acad. Sci. USA 2008, 105, 15523–15528. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Huang, Y.; Lue, J.; Quandt, J.A.; Martin, R.; Mariuzza, R.A. Structure of a Human Autoimmune TCR Bound to a Myelin Basic Protein Self-Peptide and a Multiple Sclerosis-Associated MHC Class II Molecule. EMBO J. 2005, 24, 2968–2979. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Jing, Y.; Allard, D.; Scavuzzo, M.A.; Sprouse, M.L.; Borowiak, M.; Bettini, M.L.; Bettini, M. A Dormant T-Cell Population with Autoimmune Potential Exhibits Low Self-Reactivity and Infiltrates Islets in Type 1 Diabetes. Eur. J. Immunol. 2022, 52, 1158–1170. [Google Scholar] [CrossRef]
- Sethi, D.K.; Schubert, D.A.; Anders, A.-K.; Heroux, A.; Bonsor, D.A.; Thomas, C.P.; Sundberg, E.J.; Pyrdol, J.; Wucherpfennig, K.W. A Highly Tilted Binding Mode by a Self-Reactive T Cell Receptor Results in Altered Engagement of Peptide and MHC. J. Exp. Med. 2011, 208, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.J.; Narayanan, S.; Liu, B.; Birnbaum, M.E.; Kruse, A.C.; Bowerman, N.A.; Chen, W.; Levin, A.M.; Connolly, J.M.; Zhu, C.; et al. T Cell Receptor Signaling Is Limited by Docking Geometry to Peptide-Major Histocompatibility Complex. Immunity 2011, 35, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Li, Y.; Kerzic, M.C.; Martin, R.; Mariuzza, R.A. Structure of a TCR with High Affinity for Self-Antigen Reveals Basis for Escape from Negative Selection. EMBO J. 2011, 30, 1137–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maynard, J.; Petersson, K.; Wilson, D.H.; Adams, E.J.; Blondelle, S.E.; Boulanger, M.J.; Wilson, D.B.; Garcia, K.C. Structure of an Autoimmune T Cell Receptor Complexed with Class II Peptide-MHC: Insights into MHC Bias and Antigen Specificity. Immunity 2005, 22, 81–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, D.; Bond, C.J.; Ely, L.K.; Maynard, J.; Garcia, K.C. Structural Evidence for a Germline-Encoded T Cell Receptor–Major Histocompatibility Complex Interaction “Codon”. Nat. Immunol. 2007, 8, 975–983. [Google Scholar] [CrossRef]
- Zamvil, S.S.; Mitchell, D.J.; Moore, A.C.; Kitamura, K.; Steinman, L.; Rothbard, J.B. T-Cell Epitope of the Autoantigen Myelin Basic Protein That Induces Encephalomyelitis. Nature 1986, 324, 258–260. [Google Scholar] [CrossRef]
- Raposo, B.; Merky, P.; Lundqvist, C.; Yamada, H.; Urbonaviciute, V.; Niaudet, C.; Viljanen, J.; Kihlberg, J.; Kyewski, B.; Ekwall, O.; et al. T Cells Specific for Post-Translational Modifications Escape Intrathymic Tolerance Induction. Nat. Commun. 2018, 9, 353. [Google Scholar] [CrossRef] [Green Version]
- Carayanniotis, G. Recognition of Thyroglobulin by T Cells: The Role of Iodine. Thyroid 2007, 17, 963–973. [Google Scholar] [CrossRef]
- Kwon, E.-J.; Ju, J.H. Impact of Posttranslational Modification in Pathogenesis of Rheumatoid Arthritis: Focusing on Citrullination, Carbamylation, and Acetylation. Int. J. Mol. Sci. 2021, 22, 10576. [Google Scholar] [CrossRef]
- Hill, J.A.; Southwood, S.; Sette, A.; Jevnikar, A.M.; Bell, D.A.; Cairns, E. Cutting Edge: The Conversion of Arginine to Citrulline Allows for a High-Affinity Peptide Interaction with the Rheumatoid Arthritis-Associated HLA-DRB1*0401 MHC Class II Molecule. J. Immunol. 2003, 171, 538. [Google Scholar] [CrossRef] [Green Version]
- Scherer, H.U.; Häupl, T.; Burmester, G.R. The Etiology of Rheumatoid Arthritis. J. Autoimmun. 2020, 110, 102400. [Google Scholar] [CrossRef]
- Lim, J.J.; Jones, C.M.; Loh, T.J.; Ting, Y.T.; Zareie, P.; Loh, K.L.; Rossjohn, J.; Felix, N.; Supi, A.; Baker, D.G. The Shared Susceptibility Epitope of HLA-DR4 Binds Citrullinated Self-Antigens and the TCR. Sci. Immunol. 2021, 6, eabe0896. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Schwenzer, A.; Wong, A.; Turcinov, S.; Rims, C.; Martinez, L.R.; Arribas-Layton, D.; Gerstner, C.; Muir, V.S.; Midwood, K.S. Shared Recognition of Citrullinated Tenascin-C Peptides by T and B Cells in Rheumatoid Arthritis. JCI Insight 2021, 6, e145217. [Google Scholar] [CrossRef] [PubMed]
- Ge, C.; Weisse, S.; Xu, B.; Dobritzsch, D.; Viljanen, J.; Kihlberg, J.; Do, N.-N.; Schneider, N.; Lanig, H.; Holmdahl, R.; et al. Key Interactions in the Trimolecular Complex Consisting of the Rheumatoid Arthritis—Associated DRB1 * 04:01 Molecule, the Major Glycosylated Collagen II Peptide and the T-Cell Receptor. Ann. Rheum. Dis. 2022, 81, 480. [Google Scholar] [CrossRef]
- McGinty, J.W.; Chow, I.-T.; Greenbaum, C.; Odegard, J.; Kwok, W.W.; James, E.A. Recognition of Posttranslationally Modified GAD65 Epitopes in Subjects With Type 1 Diabetes. Diabetes 2014, 63, 3033–3040. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.-L.; Horstman, S.; Gee, R.; Guyer, P.; Lam, T.T.; Kanyo, J.; Perdigoto, A.L.; Speake, C.; Greenbaum, C.J.; Callebaut, A.; et al. Citrullination of Glucokinase Is Linked to Autoimmune Diabetes. Nat. Commun. 2022, 13, 1870. [Google Scholar] [CrossRef] [PubMed]
- Mannering, S.I.; Harrison, L.C.; Williamson, N.A.; Morris, J.S.; Thearle, D.J.; Jensen, K.P.; Kay, T.W.H.; Rossjohn, J.; Falk, B.A.; Nepom, G.T.; et al. The Insulin A-Chain Epitope Recognized by Human T Cells Is Posttranslationally Modified. J. Exp. Med. 2005, 202, 1191–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Sun, D.; Whitaker, J.N. Citrullinated Myelin Basic Protein Induces Experimental Autoimmune Encephalomyelitis in Lewis Rats through a Diverse T Cell Repertoire. J. Neuroimmunol. 1998, 88, 21–29. [Google Scholar] [CrossRef]
- Carrillo-Vico, A.; Leech, M.D.; Anderton, S.M. Contribution of Myelin Autoantigen Citrullination to T Cell Autoaggression in the Central Nervous System. J. Immunol. 2010, 184, 2839. [Google Scholar] [CrossRef] [Green Version]
- Valdivia, A.O.; Agarwal, P.K.; Bhattacharya, S.K. Myelin Basic Protein Phospholipid Complexation Likely Competes with Deimination in Experimental Autoimmune Encephalomyelitis Mouse Model. ACS Omega 2020, 5, 15454–15467. [Google Scholar] [CrossRef]
- Mitchell, A.M.; Alkanani, A.A.; McDaniel, K.A.; Pyle, L.; Waugh, K.; Steck, A.K.; Nakayama, M.; Yu, L.; Gottlieb, P.A.; Rewers, M.J.; et al. T-Cell Responses to Hybrid Insulin Peptides Prior to Type 1 Diabetes Development. Proc. Natl. Acad. Sci. USA 2021, 118, e2019129118. [Google Scholar] [CrossRef]
- Aaron Wiles, T.; Powell, R.; Michel, C.R.; Scott Beard, K.; Hohenstein, A.; Bradley, B.; Reisdorph, N.; Haskins, K.; Delong, T. Identification of Hybrid Insulin Peptides (HIPs) in Mouse and Human Islets by Mass Spectrometry. J. Proteome Res. 2019, 18, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.L.; Jamison, B.L.; Wiles, T.A.; Lindsay, R.S.; Barbour, G.; Bradley, B.; Delong, T.; Friedman, R.S.; Nakayama, M.; Haskins, K. CD4 T Cells Reactive to Hybrid Insulin Peptides Are Indicators of Disease Activity in the NOD Mouse. Diabetes 2018, 67, 1836–1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, R.L.; Rihanek, M.; Hohenstein, A.C.; Nakayama, M.; Michels, A.; Gottlieb, P.A.; Haskins, K.; Delong, T. Hybrid Insulin Peptides Are Autoantigens in Type 1 Diabetes. Diabetes 2019, 68, 1830–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiles, T.A.; Hohenstein, A.; Landry, L.G.; Dang, M.; Powell, R.; Guyer, P.; James, E.A.; Nakayama, M.; Haskins, K.; Delong, T.; et al. Characterization of Human CD4 T Cells Specific for a C-Peptide/C-Peptide Hybrid Insulin Peptide. Front. Immunol. 2021, 12, 668680. [Google Scholar] [CrossRef] [PubMed]
- Delong, T.; Wiles, T.A.; Baker, R.L.; Bradley, B.; Barbour, G.; Reisdorph, R.; Haskins, K.; Elso, C.M.; Kumer, N.; Kent, S.C.; et al. Pathogenic CD4 T Cells in Type 1 Diabetes Recognize Epitopes Formed by Peptide Fusion. Science (1979) 2016, 351, 711–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arribas-Layton, D.; Guyer, P.; Delong, T.; Dang, M.; Chow, I.-T.; Speake, C.; Greenbaum, C.J.; Kwok, W.W.; Baker, R.L.; Haskins, K.; et al. Hybrid Insulin Peptides Are Recognized by Human T Cells in the Context of DRB1 * 04:01. Diabetes 2020, 69, 1492–1502. [Google Scholar] [CrossRef]
- Tran, M.T.; Faridi, P.; Lim, J.J.; Ting, Y.T.; Onwukwe, G.; Bhattacharjee, P.; Jones, C.M.; Tresoldi, E.; Cameron, F.J.; la Gruta, N.L.; et al. T Cell Receptor Recognition of Hybrid Insulin Peptides Bound to HLA-DQ8. Nat. Commun. 2021, 12, 5110. [Google Scholar] [CrossRef]
- Christen, U.; von Herrath, M.G. Infections and Autoimmunity—Good or Bad? J. Immunol. 2005, 174, 7481. [Google Scholar] [CrossRef] [Green Version]
- Markovic-Plese, S.; Hemmer, B.; Zhao, Y.; Simon, R.; Pinilla, C.; Martin, R. High Level of Cross-Reactivity in Influenza Virus Hemagglutinin-Specific CD4+ T-Cell Response: Implications for the Initiation of Autoimmune Response in Multiple Sclerosis. J. Neuroimmunol. 2005, 169, 31–38. [Google Scholar] [CrossRef]
- Hiemstra, H.S.; Schloot, N.C.; van Veelen, P.A.; Willemen, S.J.M.; Franken, K.L.M.C.; van Rood, J.J.; de Vries, R.R.P.; Chaudhuri, A.; Behan, P.O.; Drijfhout, J.W.; et al. Cytomegalovirus in Autoimmunity: T Cell Crossreactivity to Viral Antigen and Autoantigen Glutamic Acid Decarboxylase. Proc. Natl. Acad. Sci. USA 2001, 98, 3988–3991. [Google Scholar] [CrossRef] [Green Version]
- Bellucci, G.; Rinaldi, V.; Buscarinu, M.C.; Reniè, R.; Bigi, R.; Pellicciari, G.; Morena, E.; Romano, C.; Marrone, A.; Mechelli, R.; et al. Multiple Sclerosis and SARS-CoV-2: Has the Interplay Started? Front. Immunol. 2021, 12, 755333. [Google Scholar] [CrossRef] [PubMed]
- Satheesh, N.J.; Salloum-Asfar, S.; Abdulla, S.A. The Potential Role of COVID-19 in the Pathogenesis of Multiple Sclerosis—A Preliminary Report. Viruses 2021, 13, 2091. [Google Scholar] [CrossRef] [PubMed]
- Brennan, F.M.; Smith, N.M.G.; Owen, S.; Li, C.; Amjadi, P.; Green, P.; Andersson, A.; Palfreeman, A.C.; Hillyer, P.; Foey, A.; et al. Resting CD4+ effector Memory T Cells Are Precursors of Bystander-Activated Effectors: A Surrogate Model of Rheumatoid Arthritis Synovial T-Cell Function. Arthritis Res. 2008, 10, R36. [Google Scholar] [CrossRef] [Green Version]
- Martino, G.; Grohovaz, F.; Brambilla, E.; Codazzi, F.; Consiglio, A.; Clementi, E.; Filippi, M.; Comi, G.; Grimaldi, L.M.E. Proinflammatory Cytokines Regulate Antigen-Independent T-Cell Activation by Two Separate Calcium-Signaling Pathways in Multiple Sclerosis Patients. Ann. Neurol. 1998, 43, 340–349. [Google Scholar] [CrossRef]
- Steimle, V.; Siegrist, C.-A.; Mottet, A.; Lisowska-Grospierre, B.; Mach, B. Regulation of MHC Class II Expression by Interferon-γ Mediated by the Transactivator Gene CIITA. Science (1979) 1994, 265, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, B.M.; Cerase, J.; Ceracchini, C.; Levi, G.; Aloisi, F. Analysis of B7-1 and B7-2 Costimulatory Ligands in Cultured Mouse Microglia: Upregulation by Interferon-γ and Lipopolysaccharide and Downregulation by Interleukin-10, Prostaglandin E2 and Cyclic AMP-Elevating Agents. J. Neuroimmunol. 1997, 72, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Nikcevich, K.M.; Gordon, K.B.; Tan, L.; Hurst, S.D.; Kroepfl, J.F.; Gardinier, M.; Barrett, T.A.; Miller, S.D. IFN-Gamma-Activated Primary Murine Astrocytes Express B7 Costimulatory Molecules and Prime Naive Antigen-Specific T Cells. J. Immunol. 1997, 158, 614. [Google Scholar] [CrossRef]
- Russell, M.A.; Redick, S.D.; Blodgett, D.M.; Richardson, S.J.; Leete, P.; Krogvold, L.; Dahl-Jørgensen, K.; Bottino, R.; Brissova, M.; Spaeth, J.M.; et al. HLA Class II Antigen Processing and Presentation Pathway Components Demonstrated by Transcriptome and Protein Analyses of Islet β-Cells From Donors With Type 1 Diabetes. Diabetes 2019, 68, 988–1001. [Google Scholar] [CrossRef] [Green Version]
- Quesada-Masachs, E.; Zilberman, S.; Rajendran, S.; Chu, T.; McArdle, S.; Kiosses, W.B.; Lee, J.-H.M.; Yesildag, B.; Benkahla, M.A.; Pawlowska, A.; et al. Upregulation of HLA Class II in Pancreatic Beta Cells from Organ Donors with Type 1 Diabetes. Diabetologia 2022, 65, 387–401. [Google Scholar] [CrossRef]
- Zhao, Y.; Scott, N.A.; Quah, H.S.; Krishnamurthy, B.; Bond, F.; Loudovaris, T.; Mannering, S.I.; Kay, T.W.H.; Thomas, H.E. Mouse Pancreatic Beta Cells Express MHC Class II and Stimulate CD4+ T Cells to Proliferate. Eur. J. Immunol. 2015, 45, 2494–2503. [Google Scholar] [CrossRef]
- Scott, N.A.; Zhao, Y.; Krishnamurthy, B.; Mannering, S.I.; Kay, T.W.H.; Thomas, H.E. IFNγ-Induced MHC Class II Expression on Islet Endothelial Cells Is an Early Marker of Insulitis but Is Not Required for Diabetogenic CD4+ T Cell Migration. Front. Immunol. 2018, 9, 2800. [Google Scholar] [CrossRef] [PubMed]
- Gobin, S.J.P.; Montagne, L.; van Zutphen, M.; van der Valk, P.; van den Elsen, P.J.; de Groot, C.J.A. Upregulation of Transcription Factors Controlling MHC Expression in Multiple Sclerosis Lesions. Glia 2001, 36, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Falcão, A.M.; van Bruggen, D.; Marques, S.; Meijer, M.; Jäkel, S.; Agirre, E.; Samudyata; Floriddia, E.M.; Vanichkina, D.P.; ffrench-Constant, C.; et al. Disease-Specific Oligodendrocyte Lineage Cells Arise in Multiple Sclerosis. Nat. Med. 2018, 24, 1837–1844. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, S.; Ota, S.; Sekine, C.; Tada, T.; Otsuka, T.; Okamoto, T.; Sønderstrup, G.; Peterlin, B.M. Aberrant MHC Class II Expression in Mouse Joints Leads to Arthritis with Extraarticular Manifestations Similar to Rheumatoid Arthritis. Proc. Natl. Acad. Sci. USA 2006, 103, 14465–14470. [Google Scholar] [CrossRef] [Green Version]
- Fasano, R.; Malerba, E.; Prete, M.; Solimando, A.G.; Buonavoglia, A.; Silvestris, N.; Leone, P.; Racanelli, V. Impact of Antigen Presentation Mechanisms on Immune Response in Autoimmune Hepatitis. Front. Immunol. 2022, 12, 814155. [Google Scholar] [CrossRef]
- Serreze, D.V.; Wasserfall, C.; Ottendorfer, E.W.; Stalvey, M.; Pierce, M.A.; Gauntt, C.; O’Donnell, B.; Flanagan, J.B.; Campbell-Thompson, M.; Ellis, T.M.; et al. Diabetes Acceleration or Prevention by a Coxsackievirus B4 Infection: Critical Requirements for Both Interleukin-4 and Gamma Interferon. J. Virol. 2005, 79, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Nekoua, M.P.; Alidjinou, E.K.; Hober, D. Persistent Coxsackievirus B Infection and Pathogenesis of Type 1 Diabetes Mellitus. Nat. Rev. Endocrinol. 2022, 18, 503–516. [Google Scholar] [CrossRef]
- Isaacs, S.R.; Foskett, D.B.; Maxwell, A.J.; Ward, E.J.; Faulkner, C.L.; Luo, J.Y.X.; Rawlinson, W.D.; Craig, M.E.; Kim, K.W. Viruses and Type 1 Diabetes: From Enteroviruses to the Virome. Microorganisms 2021, 9, 1519. [Google Scholar] [CrossRef]
- Gabibov, A.G.; Belogurov, A.A., Jr.; Lomakin, Y.A.; Zakharova, M.Y.; Avakyan, M.E.; Dubrovskaya, V.V.; Smirnov, I.V.; Ivanov, A.S.; Molnar, A.A.; Gurtsevitch, V.E.; et al. Combinatorial Antibody Library from Multiple Sclerosis Patients Reveals Antibodies That Cross-React with Myelin Basic Protein and EBV Antigen. FASEB J. 2011, 25, 4211–4221. [Google Scholar] [CrossRef]
- Abdul-Ghaffar, A.Y.; Samaha, D.Y.; Wahba, N.S.; Hussein, A.E.A. Relation between Epstein-Barr Virus Infection and Multiple Sclerosis. QJM: Int. J. Med. 2021, 114, hcab091. [Google Scholar] [CrossRef]
- Tao, C.; Simpson-Yap, S.; Taylor, B.; Blizzard, L.; Lucas, R.; Ponsonby, A.-L.; Broadley, S.; van der Mei, I. Markers of Epstein-Barr Virus and Human Herpesvirus-6 Infection and Multiple Sclerosis Clinical Progression. Mult. Scler. Relat. Disord. 2022, 59, 103561. [Google Scholar] [CrossRef] [PubMed]
- Lanz, T.V.; Brewer, R.C.; Ho, P.P.; Moon, J.-S.; Jude, K.M.; Fernandez, D.; Fernandes, R.A.; Gomez, A.M.; Nadj, G.-S.; Bartley, C.M.; et al. Clonally Expanded B Cells in Multiple Sclerosis Bind EBV EBNA1 and GlialCAM. Nature 2022, 603, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Smatti, M.K.; Cyprian, F.S.; Nasrallah, G.K.; al Thani, A.A.; Almishal, R.O.; Yassine, H.M. Viruses and Autoimmunity: A Review on the Potential Interaction and Molecular Mechanisms. Viruses 2019, 11, 762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tengvall, K.; Huang, J.; Hellström, C.; Kammer, P.; Biström, M.; Ayoglu, B.; Bomfim, I.L.; Stridh, P.; Butt, J.; Brenner, N.; et al. Molecular Mimicry between Anoctamin 2 and Epstein-Barr Virus Nuclear Antigen 1 Associates with Multiple Sclerosis Risk. Proc. Natl. Acad. Sci. USA 2019, 116, 16955–16960. [Google Scholar] [CrossRef] [Green Version]
- Wucherpfennig, K.W.; Strominger, J.L. Molecular Mimicry in T Cell-Mediated Autoimmunity: Viral Peptides Activate Human T Cell Clones Specific for Myelin Basic Protein. Cell 1995, 80, 695–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lünemann, J.D.; Jelčić, I.; Roberts, S.; Lutterotti, A.; Tackenberg, B.; Martin, R.; Munz, C. EBNA1-Specific T Cells from Patients with Multiple Sclerosis Cross React with Myelin Antigens and Co-Produce IFN-γ and IL-2. J. Exp. Med. 2008, 205, 1763–1773. [Google Scholar] [CrossRef] [Green Version]
- Zdimerova, H.; Murer, A.; Engelmann, C.; Raykova, A.; Deng, Y.; Gujer, C.; Rühl, J.; McHugh, D.; Caduff, N.; Naghavian, R.; et al. Attenuated Immune Control of Epstein–Barr Virus in Humanized Mice Is Associated with the Multiple Sclerosis Risk Factor HLA-DR15. Eur. J. Immunol. 2021, 51, 64–75. [Google Scholar] [CrossRef]
- Latorre, D.; Kallweit, U.; Armentani, E.; Foglierini, M.; Mele, F.; Cassotta, A.; Jovic, S.; Jarrossay, D.; Mathis, J.; Zellini, F.; et al. T Cells in Patients with Narcolepsy Target Self-Antigens of Hypocretin Neurons. Nature 2018, 562, 63–68. [Google Scholar] [CrossRef]
- Luo, G.; Ambati, A.; Lin, L.; Bonvalet, M.; Partinen, M.; Ji, X.; Maecker, H.T.; Mignot, E.J.-M. Autoimmunity to Hypocretin and Molecular Mimicry to Flu in Type 1 Narcolepsy. Proc. Natl. Acad. Sci. USA 2018, 115, E12323–E12332. [Google Scholar] [CrossRef] [Green Version]
- Vuorela, A.; Freitag, T.L.; Leskinen, K.; Pessa, H.; Härkönen, T.; Stracenski, I.; Kirjavainen, T.; Olsen, P.; Saarenpää-Heikkilä, O.; Ilonen, J.; et al. Enhanced Influenza A H1N1 T Cell Epitope Recognition and Cross-Reactivity to Protein-O-Mannosyltransferase 1 in Pandemrix-Associated Narcolepsy Type 1. Nat. Commun. 2021, 12, 2283. [Google Scholar] [CrossRef]
- Ooi, J.D.; Jiang, J.-H.; Eggenhuizen, P.J.; Chua, L.L.; van Timmeren, M.; Loh, K.L.; O’Sullivan, K.M.; Gan, P.Y.; Zhong, Y.; Tsyganov, K.; et al. A Plasmid-Encoded Peptide from Staphylococcus Aureus Induces Anti-Myeloperoxidase Nephritogenic Autoimmunity. Nat. Commun. 2019, 10, 3392. [Google Scholar] [CrossRef]
- Ellis, N.M.J.; Li, Y.; Hildebrand, W.; Fischetti, V.A.; Cunningham, M.W. T Cell Mimicry and Epitope Specificity of Cross-Reactive T Cell Clones from Rheumatic Heart Disease. J. Immunol. 2005, 175, 5448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moten, D.; Teneva, I.; Apostolova, D.; Batsalova, T.; Dzhambazov, B. Molecular Mimicry of the Rheumatoid Arthritis-Related Immunodominant T-Cell Epitope within Type II Collagen (CII260-270) by the Bacterial L-Asparaginase. Int. J. Mol. Sci. 2022, 23, 9149. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lin, J.; Xiao, L.; Zhang, X.; Zhao, L.; Wang, M.; Li, L. Gut Microbiota in Systemic Lupus Erythematosus: A Fuse and a Solution. J. Autoimmun. 2022, 132, 102867. [Google Scholar] [CrossRef] [PubMed]
- Garabatos, N.; Santamaria, P. Gut Microbial Antigenic Mimicry in Autoimmunity. Front. Immunol. 2022, 13, 873607. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.T.; Roesch, L.F.W.; Ördberg, M.; Ilonen, J.; Atkinson, M.A.; Schatz, D.A.; Triplett, E.W.; Ludvigsson, J. Genetic Risk for Autoimmunity Is Associated with Distinct Changes in the Human Gut Microbiome. Nat. Commun. 2019, 10, 3621. [Google Scholar] [CrossRef] [Green Version]
- Silverman, M.; Kua, L.; Tanca, A.; Pala, M.; Palomba, A.; Tanes, C.; Bittinger, K.; Uzzau, S.; Benoist, C.; Mathis, D. Protective Major Histocompatibility Complex Allele Prevents Type 1 Diabetes by Shaping the Intestinal Microbiota Early in Ontogeny. Proc. Natl. Acad. Sci. USA 2017, 114, 9671–9676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullaney, J.A.; Stephens, J.E.; Costello, M.-E.; Fong, C.; Geeling, B.E.; Gavin, P.G.; Wright, C.M.; Spector, T.D.; Brown, M.A.; Hamilton-Williams, E.E. Type 1 Diabetes Susceptibility Alleles Are Associated with Distinct Alterations in the Gut Microbiota. Microbiome 2018, 6, 35. [Google Scholar] [CrossRef] [Green Version]
- Miyauchi, E.; Kim, S.-W.; Suda, W.; Kawasumi, M.; Onawa, S.; Taguchi-Atarashi, N.; Morita, H.; Taylor, T.D.; Hattori, M.; Ohno, H. Gut Microorganisms Act Together to Exacerbate Inflammation in Spinal Cords. Nature 2020, 585, 102–106. [Google Scholar] [CrossRef]
- Ruff, W.E.; Dehner, C.; Kim, W.J.; Pagovich, O.; Aguiar, C.L.; Yu, A.T.; Roth, A.S.; Vieira, S.M.; Kriegel, C.; Adeniyi, O.; et al. Pathogenic Autoreactive T and B Cells Cross-React with Mimotopes Expressed by a Common Human Gut Commensal to Trigger Autoimmunity. Cell Host Microbe 2019, 26, 100–113.e8. [Google Scholar] [CrossRef]
- Girdhar, K.; Huang, Q.; Chow, I.-T.; Brady, C.; Raisingani, A.; Tran, T.U.; Kwok, W.; Atkinson, M.A.; Kahn, C.R.; Altindis, E. 938-P: A Human Gut Commensal, Parabacteroides Distasonis, Enhances Type 1 Diabetes Autoimmunity via Molecular Mimicry. Diabetes 2021, 70, 938. [Google Scholar] [CrossRef]
- Girdhar, K.; Huang, Q.; Chow, I.-T.; Vatanen, T.; Brady, C.; Raisingani, A.; Autissier, P.; Atkinson, M.A.; Kwok, W.W.; Kahn, C.R.; et al. A Gut Microbial Peptide and Molecular Mimicry in the Pathogenesis of Type 1 Diabetes. Proc. Natl. Acad. Sci. USA 2022, 119, e2120028119. [Google Scholar] [CrossRef] [PubMed]
- Koning, D.; Quakkelaar, E.D.; Schellens, I.M.M.; Spierings, E.; van Baarle, D. Protective HLA Alleles Recruit Biased and Largely Similar Antigen-Specific T Cell Repertoires across Different Outcomes in HIV Infection. J. Immunol. 2022, 208, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Abualrous, E.T.; Sticht, J.; Freund, C. Major Histocompatibility Complex (MHC) Class I and Class II Proteins: Impact of Polymorphism on Antigen Presentation. Curr. Opin. Immunol. 2021, 70, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Poomarimuthu, M.; Ramasamy, T.; Govindan, R.; Andiappan, R.; Nagarajan, G.; Kadiam, S.; Mariakuttikan, J. Association of HLA-DRB1 Alleles with Rheumatic Fever and Rheumatic Heart Disease: A Meta-Analysis. Immunol. Investig. 2022, 51, 221–232. [Google Scholar] [CrossRef]
- Panhuber, A.; Lamorte, G.; Bruno, V.; Cetin, H.; Bauer, W.; Höftberger, R.; Erber, A.C.; Frommlet, F.; Koneczny, I. A Systematic Review and Meta-Analysis of HLA Class II Associations in Patients with IgG4 Autoimmunity. Sci. Rep. 2022, 12, 9229. [Google Scholar] [CrossRef]
- Zhao, L.P.; Papadopoulos, G.K.; Moustakas, A.K.; Bondinas, G.P.; Carlsson, A.; Larsson, H.E.; Ludvigsson, J.; Marcus, C.; Persson, M.; Samuelsson, U.; et al. Nine Residues in HLA-DQ Molecules Determine with Susceptibility and Resistance to Type 1 Diabetes among Young Children in Sweden. Sci. Rep. 2021, 11, 8821. [Google Scholar] [CrossRef]
- Zhao, L.P.; Papadopoulos, G.K.; Kwok, W.W.; Moustakas, A.K.; Bondinas, G.P.; Carlsson, A.; Elding Larsson, H.; Ludvigsson, J.; Marcus, C.; Samuelsson, U.; et al. Next-Generation HLA Sequence Analysis Uncovers Seven HLA-DQ Amino Acid Residues and Six Motifs Resistant to Childhood Type 1 Diabetes. Diabetes 2020, 69, 2523–2535. [Google Scholar] [CrossRef]
- Jones, E.Y.; Fugger, L.; Strominger, J.L.; Siebold, C. MHC Class II Proteins and Disease: A Structural Perspective. Nat. Rev. Immunol. 2006, 6, 271–282. [Google Scholar] [CrossRef]
- Nishimoto, H.; Kikutani, H.; Yamamura, K.; Kishimoto, T. Prevention of Autoimmune Insulitis by Expression of I–E Molecules in NOD Mice. Nature 1987, 328, 432–434. [Google Scholar] [CrossRef]
- Reich, E.-P.; Sherwin, R.S.; Kanagawa, O.; Janeway, C.A. An Explanation for the Protective Effect of the MHC Class II I–E Molecule in Murine Diabetes. Nature 1989, 341, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Verdaguer, J.; Averill, N.; Santamaria, P. A Mechanism for the Major Histocompatibility Complex–Linked Resistance to Autoimmunity. J. Exp. Med. 1997, 186, 1059–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jan, B.; Brigitte, S.; Osami, K.; Christophe, B.; Diane, M. MHC-Linked Protection from Diabetes Dissociated from Clonal Deletion of T Cells. Science (1979) 1990, 249, 293–295. [Google Scholar] [CrossRef]
- Katz, J.D.; Wang, B.; Haskins, K.; Benoist, C.; Mathis, D. Following a Diabetogenic T Cell from Genesis through Pathogenesis. Cell 1993, 74, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Wing, J.B.; Tanaka, A.; Sakaguchi, S. Human FOXP3+ Regulatory T Cell Heterogeneity and Function in Autoimmunity and Cancer. Immunity 2019, 50, 302–316. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, S.; Mikami, N.; Wing, J.B.; Tanaka, A.; Ichiyama, K.; Ohkura, N. Regulatory T Cells and Human Disease. Annu Rev. Immunol. 2020, 38, 541–566. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, A.M.; Michels, A.W. Self-Antigens Targeted by Regulatory T Cells in Type 1 Diabetes. Int. J. Mol. Sci. 2022, 23, 3155. [Google Scholar] [CrossRef]
- van Lummel, M.; Buis, D.T.P.; Ringeling, C.; de Ru, A.H.; Pool, J.; Papadopoulos, G.K.; van Veelen, P.A.; Reijonen, H.; Drijfhout, J.W.; Roep, B.O. Epitope Stealing as a Mechanism of Dominant Protection by HLA-DQ6 in Type 1 Diabetes. Diabetes 2019, 68, 787–795. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.; Serra, P.; Clemente-Casares, X.; Slattery, R.M.; Santamaria, P. Dendritic Cell–Dependent In Vivo Generation of Autoregulatory T Cells by Antidiabetogenic MHC Class II. J. Immunol. 2013, 191, 70. [Google Scholar] [CrossRef] [Green Version]
- Sue, T.; Pau, S.; Xavier, C.-C.; Jun, Y.; Shari, T.; Slattery, R.M.; Elliott, J.F.; Pere, S. Antidiabetogenic MHC Class II Promotes the Differentiation of MHC-Promiscuous Autoreactive T Cells into FOXP3+ Regulatory T Cells. Proc. Natl. Acad. Sci. USA 2013, 110, 3471–3476. [Google Scholar] [CrossRef] [Green Version]
- Beringer, D.X.; Kleijwegt, F.S.; Wiede, F.; van der Slik, A.R.; Loh, K.L.; Petersen, J.; Dudek, N.L.; Duinkerken, G.; Laban, S.; Joosten, A.; et al. T Cell Receptor Reversed Polarity Recognition of a Self-Antigen Major Histocompatibility Complex. Nat. Immunol. 2015, 16, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Stadinski, B.D.; Blevins, S.J.; Spidale, N.A.; Duke, B.R.; Huseby, P.G.; Stern, L.J.; Huseby, E.S. A Temporal Thymic Selection Switch and Ligand Binding Kinetics Constrain Neonatal Foxp3+ Treg Cell Development. Nat. Immunol. 2019, 20, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, V.V.; Khaiboullina, S.F.; Gomzikova, M.O.; Martynova, E.V.; Ferreira, A.M.; Garanina, E.E.; Sakhapov, D.I.; Lomakin, Y.A.; Khaibullin, T.I.; Granatov, E.V.; et al. Divergent Immunomodulation Capacity of Individual Myelin Peptides—Components of Liposomal Therapeutic against Multiple Sclerosis. Front. Immunol. 2017, 8, 1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belogurov, A.A., Jr.; Stepanov, A.V.; Smirnov, I.V.; Melamed, D.; Bacon, A.; Mamedov, A.E.; Boitsov, V.M.; Sashchenko, L.P.; Ponomarenko, N.A.; Sharanova, S.N.; et al. Liposome-Encapsulated Peptides Protect against Experimental Allergic Encephalitis. FASEB J. 2013, 27, 222–231. [Google Scholar] [CrossRef] [Green Version]
- Selck, C.; Dominguez-Villar, M. Antigen-Specific Regulatory T Cell Therapy in Autoimmune Diseases and Transplantation. Front. Immunol. 2021, 12, 661875. [Google Scholar] [CrossRef]
- Gupta, S.; Li, D.; Ostrov, D.A.; Nguyen, C.Q. Blocking IAg7 Class II Major Histocompatibility Complex by Drug-like Small Molecules Alleviated Sjögren’s Syndrome in NOD Mice. Life Sci. 2022, 288, 120182. [Google Scholar] [CrossRef]
- Ostrov, D.A.; Gottlieb, P.A.; Michels, A.W. Rationally Designed Small Molecules to Prevent Type 1 Diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2019, 26, 90. [Google Scholar] [CrossRef]
- Ostrov, D.A.; Alkanani, A.; McDaniel, K.A.; Case, S.; Baschal, E.E.; Pyle, L.; Ellis, S.; Pöllinger, B.; Seidl, K.J.; Shah, V.N.; et al. Methyldopa Blocks MHC Class II Binding to Disease-Specific Antigens in Autoimmune Diabetes. J. Clin. Investig. 2018, 128, 1888–1902. [Google Scholar] [CrossRef] [Green Version]
- Yi, J.; Miller, A.T.; Archambault, A.S.; Jones, A.J.; Bradstreet, T.R.; Bandla, S.; Hsu, Y.-S.; Edelson, B.T.; Zhou, Y.W.; Fremont, D.H.; et al. Antigen-Specific Depletion of CD4+ T Cells by CAR T Cells Reveals Distinct Roles of Higher- and Lower-Affinity TCRs during Autoimmunity. Sci. Immunol. 2022, 7, eabo0777. [Google Scholar] [CrossRef]
- Kobayashi, S.; Thelin, M.A.; Parrish, H.L.; Deshpande, N.R.; Lee, M.S.; Karimzadeh, A.; Niewczas, M.A.; Serwold, T.; Kuhns, M.S. A Biomimetic Five-Module Chimeric Antigen Receptor (5MCAR) Designed to Target and Eliminate Antigen-Specific T Cells. Proc. Natl. Acad. Sci. USA 2020, 117, 28950–28959. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AD | Species | MHC-II Allele | Antigenic Peptide | Proposed Molecular Mechanisms of AD Susceptibility | Reference |
|---|---|---|---|---|---|
| Low-affinity autoantigenic peptides | |||||
| T1D | Mouse | I-Ag7 | Ins. B:12–20, Ins. B:13–21 | Weak binding of CLIP by MHC-II molecule promotes loading of low-affinity peptides. | [48] |
| T1D | Mouse | I-Ag7 | Ins. B:12–20, Ins. B:14–22 | MHC-II binding of antigenic peptide in a low-affinity register leads to recognition by autoreactive TCRs. | [52,53,54,55] |
| T1D | Human | HLA-DQA1*03:01, HLA-DQB1*03:02 | Ins. B:11–23 | MHC-II binding of antigenic peptide in a low-affinity register leads to recognition by autoreactive TCRs. | [56] |
| Low-affinity autoreactive TCRs | |||||
| MS | Human | HLA-DRB1*15:01 | MBP:85–99 | Autoreactive TCRs bind pMHC complexes with low affinity and alternate topology. | [16,67] |
| MS | Human | HLA-DRB5*01:01 | MBP:84–102 | [68] | |
| High or intermediate affinity autoreactive TCRs | |||||
| MS | Human | HLA-DQA1*01:02, HLA-DQB1*05:02 | MBP:85–99 | The unusual topology of trimolecular complex impedes T cell activation by CD4 receptor. | [70] |
| MS | Human | HLA-DRB1*04:01 | MBP:111–129 | Autoreactive TCR stabilizes weak interaction between antigenic peptide and MHC-II. | [72] |
| EAE | Mouse | I-Au | MBP:1–11 (acetylated) | Autoreactive TCRs bind pMHC complexes with acetylated autoantigens in unusual binding register, where the part of peptide-binding groove is empty. | [73,74,75] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishina, I.A.; Zakharova, M.Y.; Kurbatskaia, I.N.; Mamedov, A.E.; Belogurov, A.A., Jr.; Gabibov, A.G. MHC Class II Presentation in Autoimmunity. Cells 2023, 12, 314. https://doi.org/10.3390/cells12020314
Ishina IA, Zakharova MY, Kurbatskaia IN, Mamedov AE, Belogurov AA Jr., Gabibov AG. MHC Class II Presentation in Autoimmunity. Cells. 2023; 12(2):314. https://doi.org/10.3390/cells12020314
Chicago/Turabian StyleIshina, Irina A., Maria Y. Zakharova, Inna N. Kurbatskaia, Azad E. Mamedov, Alexey A. Belogurov, Jr., and Alexander G. Gabibov. 2023. "MHC Class II Presentation in Autoimmunity" Cells 12, no. 2: 314. https://doi.org/10.3390/cells12020314
APA StyleIshina, I. A., Zakharova, M. Y., Kurbatskaia, I. N., Mamedov, A. E., Belogurov, A. A., Jr., & Gabibov, A. G. (2023). MHC Class II Presentation in Autoimmunity. Cells, 12(2), 314. https://doi.org/10.3390/cells12020314