



The Protective Pathways Activated in Kidneys of αMUPA Transgenic Mice Following Ischemia\Reperfusion-Induced Acute Kidney Injury

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Evaluation of Blood Variables and Renal Functional Parameters
2.3. Histopathology
2.4. Real-Time PCR
- Mouse primers:
- uPA: -F-AGAGTCTGAAAGTGACTATCTC,-R-CCTTCGATGTTACAGATAAGC
- uPAR: -F-TCTGGATCTTCAGAGCTTTC,-R-GCCTCTTACGGTATAACTCC
- PAI-1: -F-AGCAACAAGTTCAACTACAC,-R-CTTCCATTGTCTGATGAGTTC
- InsR: -F-AAGACCTTGGTTACCTTCTC,-R-GGATTAGTGGCATCTGTTTG
- eNOS: -F-AAAGCTGCAGGTATTTGATG,-R-AGATTGCCTCTATTTGTTGC
- ACE2: -F-CATTTGCTTGGTGATATGTG,-R-GCCTCTTGAAATATCCTTTCTG
- MasR: -F-GTTTAAGGAACTCTGGAAGATG,-R-TTAGTCAGTTAGTCAGTGGC
- Renin: -F-AGCCAAGGAGAAGAGAATAG,-R-CTCCTGTTGGGATACTGTAG
- IL-6: -F-GTCTATACCACTTCACAAGTC,-R-TGCATCATCGTTGTTCATAC
- IL-10: -F-CAGGACTTTAAGGGTTACTTG,-R-ATTTTCACAGGGGAGAAATC
- TLR4: -F-TCCCTGCATAGAGGTAGTTCC,-R-TCCAGCCACTGAAGTTCTGA
- Leptin: -F-ACATTTCACACACGCAGTCGG,-R-GGACCTGTTGATAGACTGCCA
- LC3: -F-GAACCGCAGACGCATCTCT,-R-TGATCACCGGGATCTTACTGG
- P62: -F-AATGTGATCTGTGATGGTTG,-R-GAGAGAAGCTATCAGAGAGG
- Galectine 8: -F-ATATACAAAAGCCAGGCAAG,-R-CAAATGCTTTCACATTGAGG
- TGF-β: -F-GGATACCAACTATTGCTTCAG,-R-TGTCCAGGCTCCAAATATAG
- Caspase 3: -F-CATAAGAGCACTGGAATGTC,-R-GCTCCTTTTGCTATGATCTTC
- Caspase 7: -F-CAAAACCCTGTTAGAGAAACC,-R-CCATGAGTAATAACCTGGAAC
- KIM-1: -F-CTGGAGTAATCACACTGAAG,-R-AAGTATGTACCTGGTGATAGC
- NGAL: -F-ATATGCACAGGTATCCTCAG,-R-GAAACGTTCCTTCAGTTCAG
- PGC1α: -F-TCCTCTTCAAGATCCTGTTAC,-R-CACATACAAGGGAGAATTGC
- Rpl13a: -F-AAGCAGGTACTTCTGGGCCG,-R-GGGGTTGGTATTCATCCGCT
2.5. Western Blot
2.6. Quantitative Inflammation Array
2.7. Statistical Analysis
3. Results: Impact of I/R-Induced AKI on
3.1. Body and Kidney Weight
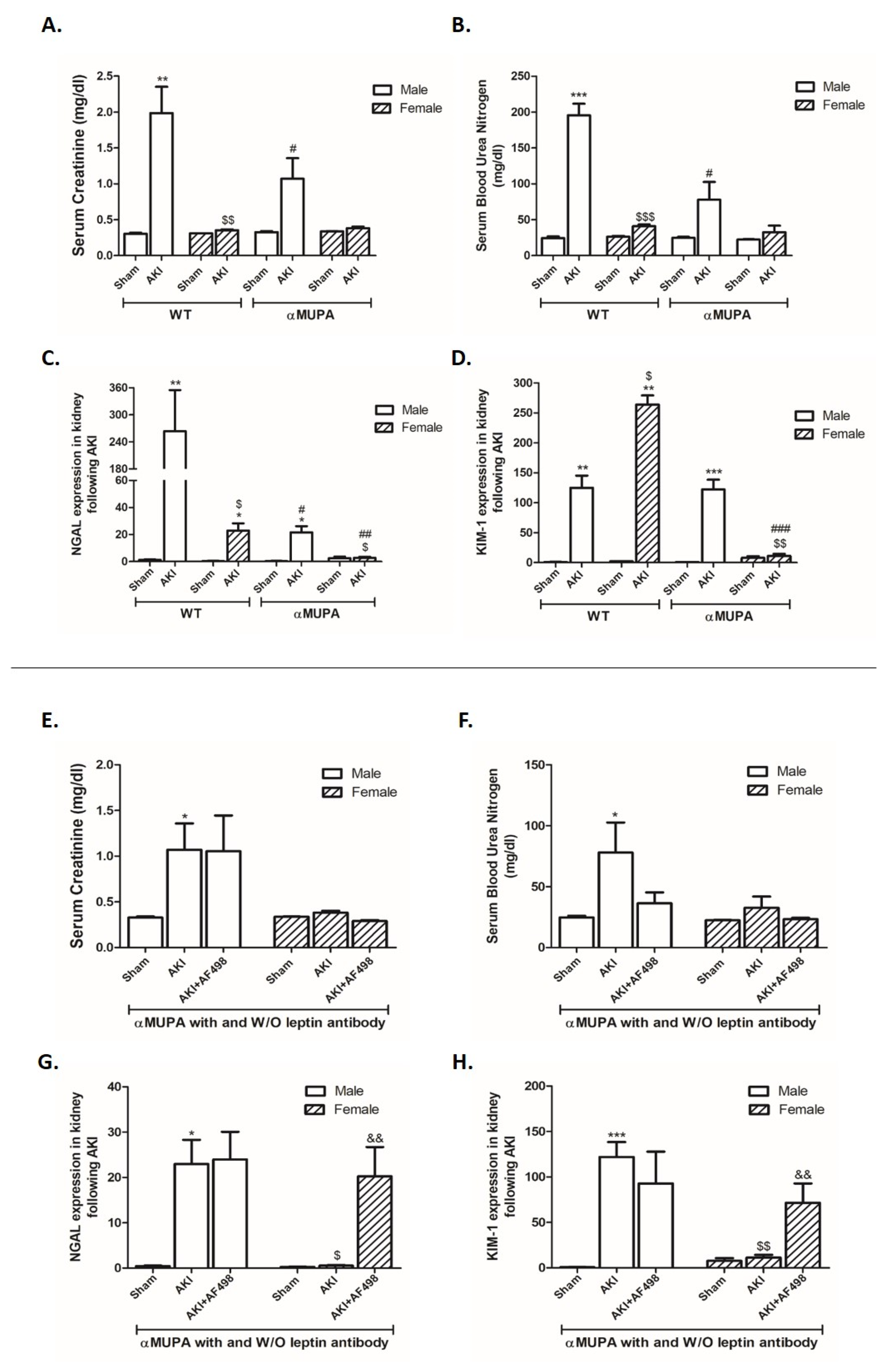
3.2. Kidney Function and Renal Injury Biomarkers
3.3. Renal Histology
3.4. Renal uPA and uPA Receptor
3.5. Renal Leptin, Insulin Receptor, and PGC1α
3.6. Renal Expression of Inflammatory and Fibrotic Markers
3.7. Renal Apoptotic and Autophagy Markers
3.8. Renal Angiotensin Converting Enzyme 2 (ACE2) and MasR
3.9. Renal Endothelial Nitric Oxide Synthase (eNOS)
3.10. Plasma Levels of Cytokines of WT and αMUPA Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bellomo, R.; Kellum, J.A.; Ronco, C. Acute kidney injury. Lancet 2012, 380, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Rewa, O.; Bagshaw, S.M. Acute kidney injury-epidemiology, outcomes and economics. Nat. Rev. Nephrol. 2014, 10, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Lameire, N.H.; Bagga, A.; Cruz, D.; De Maeseneer, J.; Endre, Z.; Kellum, J.A.; Liu, K.D.; Mehta, R.L.; Pannu, N.; Van Biesen, W.; et al. Acute kidney injury: An increasing global concern. Lancet 2013, 382, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Pakula, A.M.; Skinner, R.A. Acute Kidney Injury in the Critically Ill Patient: A Current Review of the Literature. J. Intensive Care Med. 2016, 31, 319–324. [Google Scholar] [CrossRef]
- Hoste, E.A.J.; Kellum, J.A.; Selby, N.M.; Zarbock, A.; Palevsky, P.M.; Bagshaw, S.M.; Goldstein, S.L.; Cerda, J.; Chawla, L.S. Global epidemiology and outcomes of acute kidney injury. Nat. Rev. Nephrol. 2018, 14, 607–625. [Google Scholar] [CrossRef]
- Hoste, E.A.; Bagshaw, S.M.; Bellomo, R.; Cely, C.M.; Colman, R.; Cruz, D.N.; Edipidis, K.; Forni, L.G.; Gomersall, C.D.; Govil, D.; et al. Epidemiology of acute kidney injury in critically ill patients: The multinational AKI-EPI study. Intensive Care Med. 2015, 41, 1411–1423. [Google Scholar] [CrossRef]
- Shiba, A.; Uchino, S.; Fujii, T.; Takinami, M.; Uezono, S. Association Between Intraoperative Oliguria and Acute Kidney Injury After Major Noncardiac Surgery. Anesth. Analg. 2018, 127, 1229–1235. [Google Scholar] [CrossRef]
- Legrand, M.; Rossignol, P. Cardiovascular Consequences of Acute Kidney Injury. Reply. N. Engl. J. Med. 2020, 383, 1094. [Google Scholar]
- Gonsalez, S.R.; Cortes, A.L.; Silva, R.C.D.; Lowe, J.; Prieto, M.C.; Silva Lara, L.D. Acute kidney injury overview: From basic findings to new prevention and therapy strategies. Pharmacol. Ther. 2019, 200, 1–12. [Google Scholar] [CrossRef]
- Bennett, M.R.; Devarajan, P. Proteomic analysis of acute kidney injury: Biomarkers to mechanisms. Proteom. Clin. Appl. 2011, 5, 67–77. [Google Scholar] [CrossRef]
- Mishra, J.; Ma, Q.; Prada, A.; Mitsnefes, M.; Zahedi, K.; Yang, J.; Barasch, J.; Devarajan, P. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J. Am. Soc. Nephrol. 2003, 14, 2534–2543. [Google Scholar] [CrossRef] [PubMed]
- Mishra, J.; Mori, K.; Ma, Q.; Kelly, C.; Barasch, J.; Devarajan, P. Neutrophil gelatinase-associated lipocalin: A novel early urinary biomarker for cisplatin nephrotoxicity. Am. J. Nephrol. 2004, 24, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Supavekin, S.; Zhang, W.; Kucherlapati, R.; Kaskel, F.J.; Moore, L.C.; Devarajan, P. Differential gene expression following early renal ischemia/reperfusion. Kidney Int. 2003, 63, 1714–1724. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of acute kidney injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar] [PubMed]
- Hutchens, M.P.; Fujiyoshi, T.; Komers, R.; Herson, P.S.; Anderson, S. Estrogen protects renal endothelial barrier function from ischemia-reperfusion in vitro and in vivo. Am. J. Physiol. Renal Physiol. 2012, 303, F377–F385. [Google Scholar] [CrossRef]
- Kang, K.P.; Lee, J.E.; Lee, A.S.; Jung, Y.J.; Kim, D.; Lee, S.; Hwang, H.P.; Kim, W.; Park, S.K. Effect of gender differences on the regulation of renal ischemia-reperfusion-induced inflammation in mice. Mol. Med. Rep. 2014, 9, 2061–2068. [Google Scholar] [CrossRef]
- Soranno, D.E.; Baker, P., 2nd; Kirkbride-Romeo, L.; Wennersten, S.A.; Ding, K.; Keith, B.; Cavasin, M.A.; Altmann, C.; Bagchi, R.A.; Haefner, K.R.; et al. Female and male mice have differential longterm cardiorenal outcomes following a matched degree of ischemia-reperfusion acute kidney injury. Sci. Rep. 2022, 12, 643. [Google Scholar] [CrossRef]
- Kher, A.; Meldrum, K.K.; Wang, M.; Tsai, B.M.; Pitcher, J.M.; Meldrum, D.R. Cellular and molecular mechanisms of sex differences in renal ischemia-reperfusion injury. Cardiovasc. Res. 2005, 67, 594–603. [Google Scholar] [CrossRef]
- Uchino, S.; Bellomo, R.; Morimatsu, H.; Morgera, S.; Schetz, M.; Tan, I.; Bouman, C.; Macedo, E.; Gibney, N.; Tolwani, A.; et al. Discontinuation of continuous renal replacement therapy: A post hoc analysis of a prospective multicenter observational study. Crit. Care Med. 2009, 37, 2576–2582. [Google Scholar] [CrossRef]
- Hutchens, M.P.; Dunlap, J.; Hurn, P.D.; Jarnberg, P.O. Renal ischemia: Does sex matter? Anesth. Analg. 2008, 107, 239–249. [Google Scholar] [CrossRef]
- Liu, H.B.; Meng, Q.H.; Huang, C.; Wang, J.B.; Liu, X.W. Nephroprotective Effects of Polydatin against Ischemia/Reperfusion Injury: A Role for the PI3K/Akt Signal Pathway. Oxid. Med. Cell Longev. 2015, 2015, 362158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, Q.; Zhou, Q.; Wang, R.; Xu, M.; Wang, H.; Wang, L.; Wilcox, C.S.; Liu, R.; Lai, E.Y. Protective Effect of Tempol on Acute Kidney Injury Through PI3K/Akt/Nrf2 Signaling Pathway. Kidney Blood Press. Res. 2016, 41, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Maeda, N.; Tanaka, E.; Suzuki, T.; Okumura, K.; Nomura, S.; Miyasho, T.; Haeno, S.; Yokota, H. Accurate determination of tissue steroid hormones, precursors and conjugates in adult male rat. J. Biochem. 2013, 153, 63–71. [Google Scholar] [CrossRef]
- Ikeda, M.; Swide, T.; Vayl, A.; Lahm, T.; Anderson, S.; Hutchens, M.P. Estrogen administered after cardiac arrest and cardiopulmonary resuscitation ameliorates acute kidney injury in a sex- and age-specific manner. Crit. Care 2015, 19, 332. [Google Scholar] [CrossRef] [PubMed]
- White, M.C.; Fleeman, R.; Arnold, A.C. Sex differences in the metabolic effects of the renin-angiotensin system. Biol. Sex. Differ. 2019, 10, 31. [Google Scholar] [CrossRef]
- Miller, J.A.; Cherney, D.Z.; Duncan, J.A.; Lai, V.; Burns, K.D.; Kennedy, C.R.; Zimpelmann, J.; Gao, W.; Cattran, D.C.; Scholey, J.W. Gender differences in the renal response to renin-angiotensin system blockade. J. Am. Soc. Nephrol. 2006, 17, 2554–2560. [Google Scholar] [CrossRef] [PubMed]
- Komukai, K.; Mochizuki, S.; Yoshimura, M. Gender and the renin-angiotensin-aldosterone system. Fundam. Clin. Pharmacol. 2010, 24, 687–698. [Google Scholar] [CrossRef]
- Kafami, M.; Hosseini, M.; Niazmand, S.; Farrokhi, E.; Hajzadeh, M.A.; Nazemi, S. The effects of estradiol and testosterone on renal tissues oxidative after central injection of angiotensin II in female doca—Salt treated rats. Horm. Mol. Biol. Clin. Investig. 2018, 37, 20180044. [Google Scholar] [CrossRef]
- Yanes, L.L.; Sartori-Valinotti, J.C.; Reckelhoff, J.F. Sex steroids and renal disease: Lessons from animal studies. Hypertension 2008, 51, 976–981. [Google Scholar] [CrossRef]
- Shibata, Y.; Takaoka, M.; Maekawa, D.; Kuwahara, C.; Matsumura, Y. Involvement of nitric oxide in the suppressive effect of 17beta-estradiol on endothelin-1 overproduction in ischemic acute renal failure. J. Cardiovasc. Pharmacol. 2004, 44 (Suppl. 1), S459–S461. [Google Scholar] [CrossRef]
- Rhoden, E.L.; Rhoden, C.R.; Lucas, M.L.; Pereira-Lima, L.; Zettler, C.; Bello-Klein, A. The role of nitric oxide pathway in the renal ischemia-reperfusion injury in rats. Transpl. Immunol. 2002, 10, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Chander, V.; Chopra, K. Renal protective effect of molsidomine and L-arginine in ischemia-reperfusion induced injury in rats. J. Surg. Res. 2005, 128, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Eddy, A.A. Urokinase and its receptors in chronic kidney disease. Front. Biosci. 2008, 13, 5462–5478. [Google Scholar] [CrossRef] [PubMed]
- Chapman, H.A. Plasminogen activators, integrins, and the coordinated regulation of cell adhesion and migration. Curr. Opin. Cell Biol. 1997, 9, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Ellis, V. Cellular receptors for plasminogen activators recent advances. Trends Cardiovasc. Med. 1997, 7, 227–234. [Google Scholar] [CrossRef]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGFbeta activation. J. Cell Sci. 2003, 116 Pt. 2, 217–224. [Google Scholar] [CrossRef]
- Gueler, F.; Rong, S.; Mengel, M.; Park, J.K.; Kiyan, J.; Kirsch, T.; Dumler, I.; Haller, H.; Shushakova, N. Renal urokinase-type plasminogen activator (uPA) receptor but not uPA deficiency strongly attenuates ischemia reperfusion injury and acute kidney allograft rejection. J. Immunol. 2008, 181, 1179–1189. [Google Scholar] [CrossRef]
- Tjarnstrom, J.; Holmdahl, L.; Falk, P.; Falkenberg, M.; Arnell, P.; Risberg, B. Effects of hyperbaric oxygen on expression of fibrinolytic factors of human endothelium in a simulated ischaemia/reperfusion situation. Scand. J. Clin. Lab. Investig. 2001, 61, 539–545. [Google Scholar] [CrossRef]
- Yoon, S.Y.; Lee, Y.J.; Seo, J.H.; Sung, H.J.; Park, K.H.; Choi, I.K.; Kim, S.J.; Oh, S.C.; Choi, C.W.; Kim, B.S.; et al. uPAR expression under hypoxic conditions depends on iNOS modulated ERK phosphorylation in the MDA-MB-231 breast carcinoma cell line. Cell Res. 2006, 16, 75–81. [Google Scholar] [CrossRef]
- Miskin, R.; Masos, T.; Yahav, S.; Shinder, D.; Globerson, A. AlphaMUPA mice: A transgenic model for increased life span. Neurobiol. Aging 1999, 20, 555–564. [Google Scholar] [CrossRef]
- Miskin, R.; Axelrod, J.H.; Griep, A.E.; Lee, E.; Belin, D.; Vassalli, J.D.; Westphal, H. Human and murine urokinase cDNAs linked to the murine alpha A-crystallin promoter exhibit lens and non-lens expression in transgenic mice. Eur. J. Biochem. 1990, 190, 31–38. [Google Scholar] [CrossRef]
- Booth, N. Plasminogen activators: From cloning to therapy: Edited by R. Abbate, T. Barni and A. Tsafriri; Raven Press, New York, 1991; xii+ 204 papes, $75.00. FEBS J. 1992, 310, 89. [Google Scholar] [CrossRef]
- Miskin, R.; Masos, T. Transgenic mice overexpressing urokinase-type plasminogen activator in the brain exhibit reduced food consumption, body weight and size, and increased longevity. J. Gerontol. A Biol. Sci. Med. Sci. 1997, 52, B118–B124. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.; Kornowski, R.; Gavrieli, R.; Fratty, I.; Greenberg, G.; Waldman, M.; Birk, E.; Shainberg, A.; Akirov, A.; Miskin, R.; et al. Long-Lived alphaMUPA Mice Show Attenuation of Cardiac Aging and Leptin-Dependent Cardioprotection. PLoS ONE 2015, 10, e0144593. [Google Scholar] [CrossRef] [PubMed]
- Karmazyn, M.; Purdham, D.M.; Rajapurohitam, V.; Zeidan, A. Signalling mechanisms underlying the metabolic and other effects of adipokines on the heart. Cardiovasc. Res. 2008, 79, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Coppari, R.; Bjorbaek, C. Leptin revisited: Its mechanism of action and potential for treating diabetes. Nat. Rev. Drug Discov. 2012, 11, 692–708. [Google Scholar] [CrossRef]
- Kreier, F.; Fliers, E.; Voshol, P.J.; Van Eden, C.G.; Havekes, L.M.; Kalsbeek, A.; Van Heijningen, C.L.; Sluiter, A.A.; Mettenleiter, T.C.; Romijn, J.A.; et al. Selective parasympathetic innervation of subcutaneous and intra-abdominal fat—Functional implications. J. Clin. Investig. 2002, 110, 1243–1250. [Google Scholar] [CrossRef]
- Froy, O.; Sherman, H.; Bhargava, G.; Chapnik, N.; Cohen, R.; Gutman, R.; Kronfeld-Schor, N.; Miskin, R. Spontaneous caloric restriction associated with increased leptin levels in obesity-resistant alphaMUPA mice. Int. J. Obes. 2011, 35, 226–235. [Google Scholar] [CrossRef]
- Abd Alkhaleq, H.; Kornowski, R.; Waldman, M.; Levy, E.; Zemel, R.; Nudelman, V.; Shainberg, A.; Miskin, R.; Hochhauser, E. Leptin modulates gene expression in the heart and cardiomyocytes towards mitigating ischemia-induced damage. Exp. Cell Res. 2020, 397, 112373. [Google Scholar] [CrossRef]
- Abd Alkhaleq, H.; Kornowski, R.; Waldman, M.; Zemel, R.; Lev, D.L.; Shainberg, A.; Miskin, R.; Hochhauser, E. Leptin modulates gene expression in the heart, cardiomyocytes and the adipose tissue thus mitigating LPS-induced damage. Exp. Cell Res. 2021, 404, 112647. [Google Scholar] [CrossRef]
- Pinsky, M.; Rauch, M.; Abbas, A.; Sharabi-Nov, A.; Tamir, S.; Gutman, R. Long-lived weight-reduced alphaMUPA mice show higher and longer maternal-dependent postnatal leptin surge. PLoS ONE 2017, 12, e0188658. [Google Scholar] [CrossRef] [PubMed]
- Conese, M.; Blasi, F. Urokinase/urokinase receptor system: Internalization/degradation of urokinase-serpin complexes: Mechanism and regulation. Biol. Chem. Hoppe Seyler 1995, 376, 143–155. [Google Scholar]
- Hale, L.J.; Coward, R.J. The insulin receptor and the kidney. Curr. Opin. Nephrol. Hypertens. 2013, 22, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur. Heart J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef] [PubMed]
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E., Jr.; Collins, K.J.; Dennison Himmelfarb, C.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2018, 71, e127–e248. [Google Scholar]
- Miskin, R.; Tirosh, O.; Pardo, M.; Zusman, I.; Schwartz, B.; Yahav, S.; Dubnov, G.; Kohen, R. AlphaMUPA mice: A transgenic model for longevity induced by caloric restriction. Mech. Ageing Dev. 2005, 126, 255–261. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Martinez-Moreno, J.M.; Monsalve, M.; Ramos, A.M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. The Role of PGC-1α and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules 2020, 10, 347. [Google Scholar] [CrossRef]
- Chambers, J.M.; Wingert, R.A. PGC-1alpha in Disease: Recent Renal Insights into a Versatile Metabolic Regulator. Cells 2020, 9, 2234. [Google Scholar] [CrossRef]
- Rasbach, K.A.; Schnellmann, R.G. PGC-1alpha over-expression promotes recovery from mitochondrial dysfunction and cell injury. Biochem. Biophys. Res. Commun. 2007, 355, 734–739. [Google Scholar] [CrossRef]
- Casemayou, A.; Fournel, A.; Bagattin, A.; Schanstra, J.; Belliere, J.; Decramer, S.; Marsal, D.; Gillet, M.; Chassaing, N.; Huart, A.; et al. Hepatocyte Nuclear Factor-1beta Controls Mitochondrial Respiration in Renal Tubular Cells. J. Am. Soc. Nephrol. 2017, 28, 3205–3217. [Google Scholar] [CrossRef]
- Funk, J.A.; Schnellmann, R.G. Accelerated recovery of renal mitochondrial and tubule homeostasis with SIRT1/PGC-1alpha activation following ischemia-reperfusion injury. Toxicol. Appl. Pharmacol. 2013, 273, 345–354. [Google Scholar] [CrossRef]
- Froy, O.; Chapnik, N.; Miskin, R. Long-lived alphaMUPA transgenic mice exhibit pronounced circadian rhythms. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E1017–E1024. [Google Scholar] [CrossRef] [PubMed]
- Steckler, R.; Shabtay-Yanai, A.; Pinsky, M.; Rauch, M.; Tamir, S.; Gutman, R. Long-Lived alphaMUPA Mice Show Reduced Sexual Dimorphism in Lifespan, and in Energy and Circadian Homeostasis-Related Parameters. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 451–460. [Google Scholar] [CrossRef] [PubMed]
- DiBona, G.F.; Kopp, U.C. Neural control of renal function. Physiol. Rev. 1997, 77, 75–197. [Google Scholar] [CrossRef] [PubMed]
- Denton, K.M.; Luff, S.E.; Shweta, A.; Anderson, W.P. Differential neural control of glomerular ultrafiltration. Clin. Exp. Pharmacol. Physiol. 2004, 31, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, M.; Liu, Y.; Shin, E.J.; Sweeney, G. Leptin protects H9c2 rat cardiomyocytes from H2O2-induced apoptosis. FEBS J. 2008, 275, 3136–3144. [Google Scholar] [CrossRef]
- Zheng, J.; Fang, J.; Yin, Y.J.; Wang, X.C.; Ren, A.J.; Bai, J.; Sun, X.J.; Yuan, W.J.; Lin, L. Leptin protects cardiomyocytes from serum-deprivation-induced apoptosis by increasing anti-oxidant defence. Clin. Exp. Pharmacol. Physiol. 2010, 37, 955–962. [Google Scholar] [CrossRef]
- Yu, L.; Zhao, Y.; Xu, S.; Jin, C.; Wang, M.; Fu, G. Leptin confers protection against TNF-alpha-induced apoptosis in rat cardiomyocytes. Biochem. Biophys. Res. Commun. 2014, 455, 126–132. [Google Scholar] [CrossRef]
- Heyman, S.N.; Evans, R.G.; Rosen, S.; Rosenberger, C. Cellular adaptive changes in AKI: Mitigating renal hypoxic injury. Nephrol. Dial. Transplant. 2012, 27, 1721–1728. [Google Scholar] [CrossRef]
- Mazzieri, R.; Masiero, L.; Zanetta, L.; Monea, S.; Onisto, M.; Garbisa, S.; Mignatti, P. Control of type IV collagenase activity by components of the urokinase-plasmin system: A regulatory mechanism with cell-bound reactants. EMBO J. 1997, 16, 2319–2332. [Google Scholar] [CrossRef]
- Roelofs, J.J.; Rowshani, A.T.; van den Berg, J.G.; Claessen, N.; Aten, J.; ten Berge, I.J.; Weening, J.J.; Florquin, S. Expression of urokinase plasminogen activator and its receptor during acute renal allograft rejection. Kidney Int. 2003, 64, 1845–1853. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Friess, H.; di Mola, F.F.; Schilling, M.; Maurer, C.; Graber, H.U.; Dervenis, C.; Zimmermann, A.; Buchler, M.W. Activation of the serine proteinase system in chronic kidney rejection. Transplantation 1998, 65, 1628–1634. [Google Scholar] [CrossRef] [PubMed]
- Hahm, E.; Wei, C.; Fernandez, I.; Li, J.; Tardi, N.J.; Tracy, M.; Wadhwani, S.; Cao, Y.; Peev, V.; Zloza, A.; et al. Bone marrow-derived immature myeloid cells are a main source of circulating suPAR contributing to proteinuric kidney disease. Nat. Med. 2017, 23, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Hayek, S.S.; Koh, K.H.; Grams, M.E.; Wei, C.; Ko, Y.A.; Li, J.; Samelko, B.; Lee, H.; Dande, R.R.; Lee, H.W.; et al. A tripartite complex of suPAR, APOL1 risk variants and α(v)β(3) integrin on podocytes mediates chronic kidney disease. Nat. Med. 2017, 23, 945–953. [Google Scholar] [CrossRef]
- Hayek, S.S.; Sever, S.; Ko, Y.A.; Trachtman, H.; Awad, M.; Wadhwani, S.; Altintas, M.M.; Wei, C.; Hotton, A.L.; French, A.L.; et al. Soluble Urokinase Receptor and Chronic Kidney Disease. N. Engl. J. Med. 2015, 373, 1916–1925. [Google Scholar] [CrossRef]
- Leth, J.M.; Leth-Espensen, K.Z.; Kristensen, K.K.; Kumari, A.; Lund Winther, A.M.; Young, S.G.; Ploug, M. Evolution and Medical Significance of LU Domain-Containing Proteins. Int. J. Mol. Sci. 2019, 20, 2760. [Google Scholar] [CrossRef]
- Wei, C.; Li, J.; Adair, B.D.; Zhu, K.; Cai, J.; Merchant, M.; Samelko, B.; Liao, Z.; Koh, K.H.; Tardi, N.J.; et al. uPAR isoform 2 forms a dimer and induces severe kidney disease in mice. J. Clin. Investig. 2019, 129, 1946–1959. [Google Scholar] [CrossRef]
- Huai, Q.; Mazar, A.P.; Kuo, A.; Parry, G.C.; Shaw, D.E.; Callahan, J.; Li, Y.; Yuan, C.; Bian, C.; Chen, L.; et al. Structure of human urokinase plasminogen activator in complex with its receptor. Science 2006, 311, 656–659. [Google Scholar] [CrossRef]
- Thunø, M.; Macho, B.; Eugen-Olsen, J. suPAR: The molecular crystal ball. Dis. Markers 2009, 27, 157–172. [Google Scholar] [CrossRef]
- Wei, C.; Moller, C.C.; Altintas, M.M.; Li, J.; Schwarz, K.; Zacchigna, S.; Xie, L.; Henger, A.; Schmid, H.; Rastaldi, M.P.; et al. Modification of kidney barrier function by the urokinase receptor. Nat. Med. 2008, 14, 55–63. [Google Scholar] [CrossRef]
- Hayek, S.S.; Ko, Y.A.; Awad, M.; Ahmed, H.; Gray, B.; Hosny, K.M.; Aida, H.; Tracy, M.J.; Wei, C.; Sever, S.; et al. Cardiovascular Disease Biomarkers and suPAR in Predicting Decline in Renal Function: A Prospective Cohort Study. Kidney Int. Rep. 2017, 2, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Hayek, S.S.; Landsittel, D.P.; Wei, C.; Zeier, M.; Yu, A.S.L.; Torres, V.E.; Roth, S.; Pao, C.S.; Reiser, J. Soluble Urokinase Plasminogen Activator Receptor and Decline in Kidney Function in Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2019, 30, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Coresh, J.; Tin, A.; Rebholz, C.M.; Chen, T.K.; Hayek, S.S.; Tracy, M.; Lipkowitz, M.S.; Appel, L.J.; Levey, A.S.; et al. Soluble Urokinase-Type Plasminogen Activator Receptor in Black Americans with CKD. Clin. J. Am. Soc. Nephrol. 2018, 13, 1013–1021. [Google Scholar] [CrossRef]
- Maile, L.A.; Busby, W.H.; Gollahon, K.A.; Flowers, W.; Garbacik, N.; Garbacik, S.; Stewart, K.; Nichols, T.; Bellinger, D.; Patel, A.; et al. Blocking ligand occupancy of the alphaVbeta3 integrin inhibits the development of nephropathy in diabetic pigs. Endocrinology 2014, 155, 4665–4675. [Google Scholar] [CrossRef]
- Hayek, S.S.; Leaf, D.E.; Samman Tahhan, A.; Raad, M.; Sharma, S.; Waikar, S.S.; Sever, S.; Camacho, A.; Wang, X.; Dande, R.R.; et al. Soluble Urokinase Receptor and Acute Kidney Injury. N. Engl. J. Med. 2020, 382, 416–426. [Google Scholar] [CrossRef]
- Mondino, A.; Blasi, F. uPA and uPAR in fibrinolysis, immunity and pathology. Trends Immunol. 2004, 25, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Woodhead, V.E.; Stonehouse, T.J.; Binks, M.H.; Speidel, K.; Fox, D.A.; Gaya, A.; Hardie, D.; Henniker, A.J.; Horejsi, V.; Sagawa, K.; et al. Novel molecular mechanisms of dendritic cell-induced T cell activation. Int. Immunol. 2000, 12, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Nykjaer, A.; Møller, B.; Todd, R.F., 3rd; Christensen, T.; Andreasen, P.A.; Gliemann, J.; Petersen, C.M. Urokinase receptor. An activation antigen in human T lymphocytes. J. Immunol. 1994, 152, 505–516. [Google Scholar] [CrossRef]
- Sitrin, R.G.; Shollenberger, S.B.; Strieter, R.M.; Gyetko, M.R. Endogenously produced urokinase amplifies tumor necrosis factor-alpha secretion by THP-1 mononuclear phagocytes. J. Leukoc. Biol. 1996, 59, 302–311. [Google Scholar] [CrossRef]
- Abraham, E.; Gyetko, M.R.; Kuhn, K.; Arcaroli, J.; Strassheim, D.; Park, J.S.; Shetty, S.; Idell, S. Urokinase-type plasminogen activator potentiates lipopolysaccharide-induced neutrophil activation. J. Immunol. 2003, 170, 5644–5651. [Google Scholar] [CrossRef]
- Goldfarb, R.H.; Timonen, T.; Herberman, R.B. Production of plasminogen activator by human natural killer cells. Large granular lymphocytes. J. Exp. Med. 1984, 159, 935–951. [Google Scholar] [CrossRef] [PubMed]
- Gyetko, M.R.; Chen, G.H.; McDonald, R.A.; Goodman, R.; Huffnagle, G.B.; Wilkinson, C.C.; Fuller, J.A.; Toews, G.B. Urokinase is required for the pulmonary inflammatory response to Cryptococcus neoformans. A murine transgenic model. J. Clin. Investig. 1996, 97, 1818–1826. [Google Scholar] [CrossRef] [PubMed]
- Gyetko, M.R.; Sud, S.; Kendall, T.; Fuller, J.A.; Newstead, M.W.; Standiford, T.J. Urokinase receptor-deficient mice have impaired neutrophil recruitment in response to pulmonary Pseudomonas aeruginosa infection. J. Immunol. 2000, 165, 1513–1519. [Google Scholar] [CrossRef]
- May, A.E.; Kanse, S.M.; Lund, L.R.; Gisler, R.H.; Imhof, B.A.; Preissner, K.T. Urokinase receptor (CD87) regulates leukocyte recruitment via beta 2 integrins in vivo. J. Exp. Med. 1998, 188, 1029–1037. [Google Scholar] [CrossRef]
- Shushakova, N.; Tkachuk, N.; Dangers, M.; Tkachuk, S.; Park, J.K.; Hashimoto, K.; Haller, H.; Dumler, I. Urokinase-induced activation of the gp130/Tyk2/Stat3 pathway mediates a pro-inflammatory effect in human mesangial cells via expression of the anaphylatoxin C5a receptor. J. Cell Sci. 2005, 118 Pt 12, 2743–2753. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Body Weight before Surgery (g) | Body Weight after Surgery (g) | % Change in BW | Kidney Weight (KW) (g) | Kidney/Body Weight Ratio (%) |
|---|---|---|---|---|---|
| WT male—sham | 28.64 ± 0.77 | 27.57 ± 0.72 | −3.7% | 0.197 ± 0.007 | 0.71 ± 0.02 |
| WT male—AKI | 30.37 ± 0.82 | 26.94 ± 0.95 @ | −11.3% | 0.265 ± 0.01 | 0.99 ± 0.05 *** |
| WT female—sham | 23.63 ± 0.3 | 22.95 ± 1.1 | −2.8% | 0.13 ± 0.007 | 0.59 ± 0.0005 $ |
| WT female—AKI | 22.81 ± 1.04 | 20.96 ± 0.63 | −8% | 0.17 ± 0.008 | 0.81 ± 0.02 **,$ |
| αMUPA male—sham | 22.84 ± 0.75 | 23.63 ± 1.4 | +3.4% | 0.16 ± 0.007 | 0.71 ± 0.01 |
| αMUPA male—AKI | 23.49 ± 0.42 | 21.91 ± 0.28 @@ | −6.7% | 0.174 ± 0.005 | 0.79 ± 0.02 *,# |
| αMUPA male—AKI + AF498 | 28.73 ± 0.58 | 26.26 ± 0.69 | −8.6% | 0.254 ± 0.02 | 0.96 ± 0.05 |
| αMUPA female—sham | 20.42 ± 1 | 21.56 ± 0.13 | +5.5% | 0.113 ± 0.003 | 0.52 ± 0.02 $$ |
| αMUPA female—AKI | 20.75 ± 0.65 | 19.21 ± 0.47 | −7.4% | 0.114 ± 0.003 | 0.59 ± 0.02 $$$,### |
| αMUPA male—AKI + AF498 | 20.08 ± 1.03 $$ | 17.74 ± 0.67 $$ | −11.25% | 0.147 ± 0.02 | 0.82 ± 0.09 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkhaleq, H.A.; Karram, T.; Fokra, A.; Hamoud, S.; Kabala, A.; Abassi, Z. The Protective Pathways Activated in Kidneys of αMUPA Transgenic Mice Following Ischemia\Reperfusion-Induced Acute Kidney Injury. Cells 2023, 12, 2497. https://doi.org/10.3390/cells12202497
Alkhaleq HA, Karram T, Fokra A, Hamoud S, Kabala A, Abassi Z. The Protective Pathways Activated in Kidneys of αMUPA Transgenic Mice Following Ischemia\Reperfusion-Induced Acute Kidney Injury. Cells. 2023; 12(20):2497. https://doi.org/10.3390/cells12202497
Chicago/Turabian StyleAlkhaleq, Heba Abd, Tony Karram, Ahmad Fokra, Shadi Hamoud, Aviva Kabala, and Zaid Abassi. 2023. "The Protective Pathways Activated in Kidneys of αMUPA Transgenic Mice Following Ischemia\Reperfusion-Induced Acute Kidney Injury" Cells 12, no. 20: 2497. https://doi.org/10.3390/cells12202497
APA StyleAlkhaleq, H. A., Karram, T., Fokra, A., Hamoud, S., Kabala, A., & Abassi, Z. (2023). The Protective Pathways Activated in Kidneys of αMUPA Transgenic Mice Following Ischemia\Reperfusion-Induced Acute Kidney Injury. Cells, 12(20), 2497. https://doi.org/10.3390/cells12202497