
Mineralocorticoid Receptor Antagonism Prevents Type 2 Familial Partial Lipodystrophy Brown Adipocyte Dysfunction
 , , , ,
, , , ,  ,
,  ,
,  , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
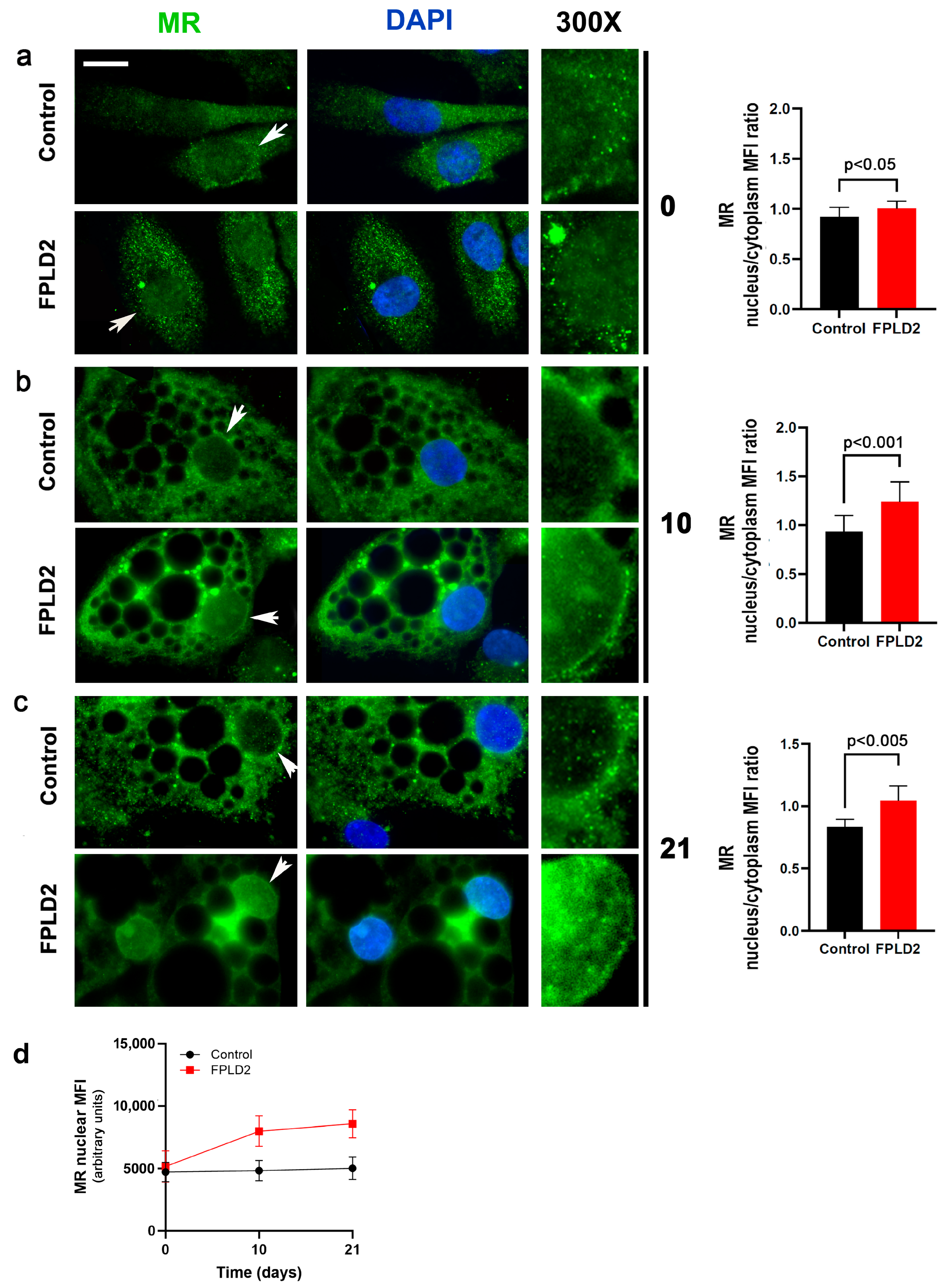
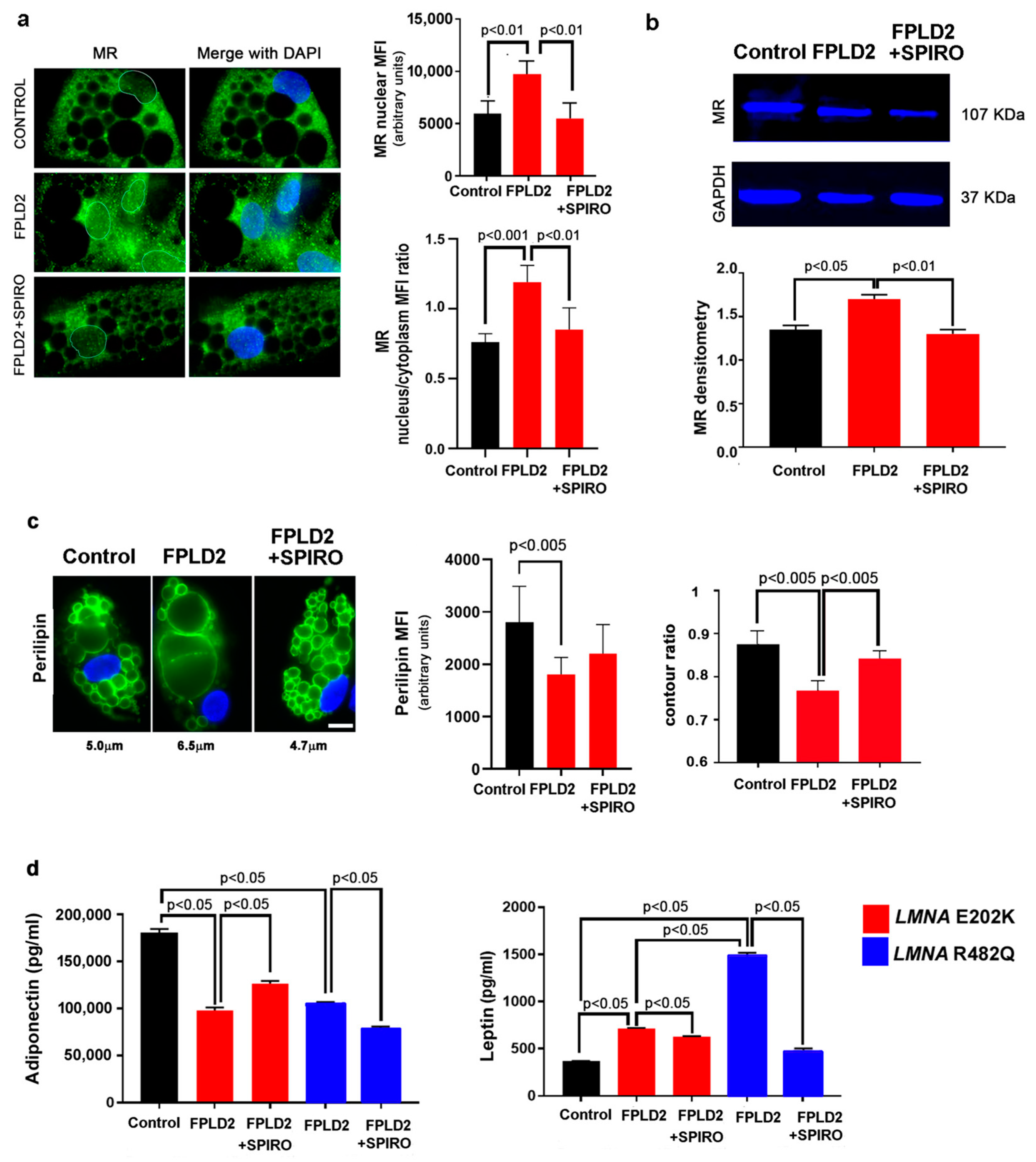
3.1. MR Localization
3.1.1. MR Localization in FPLD2 Brown Preadipocytes
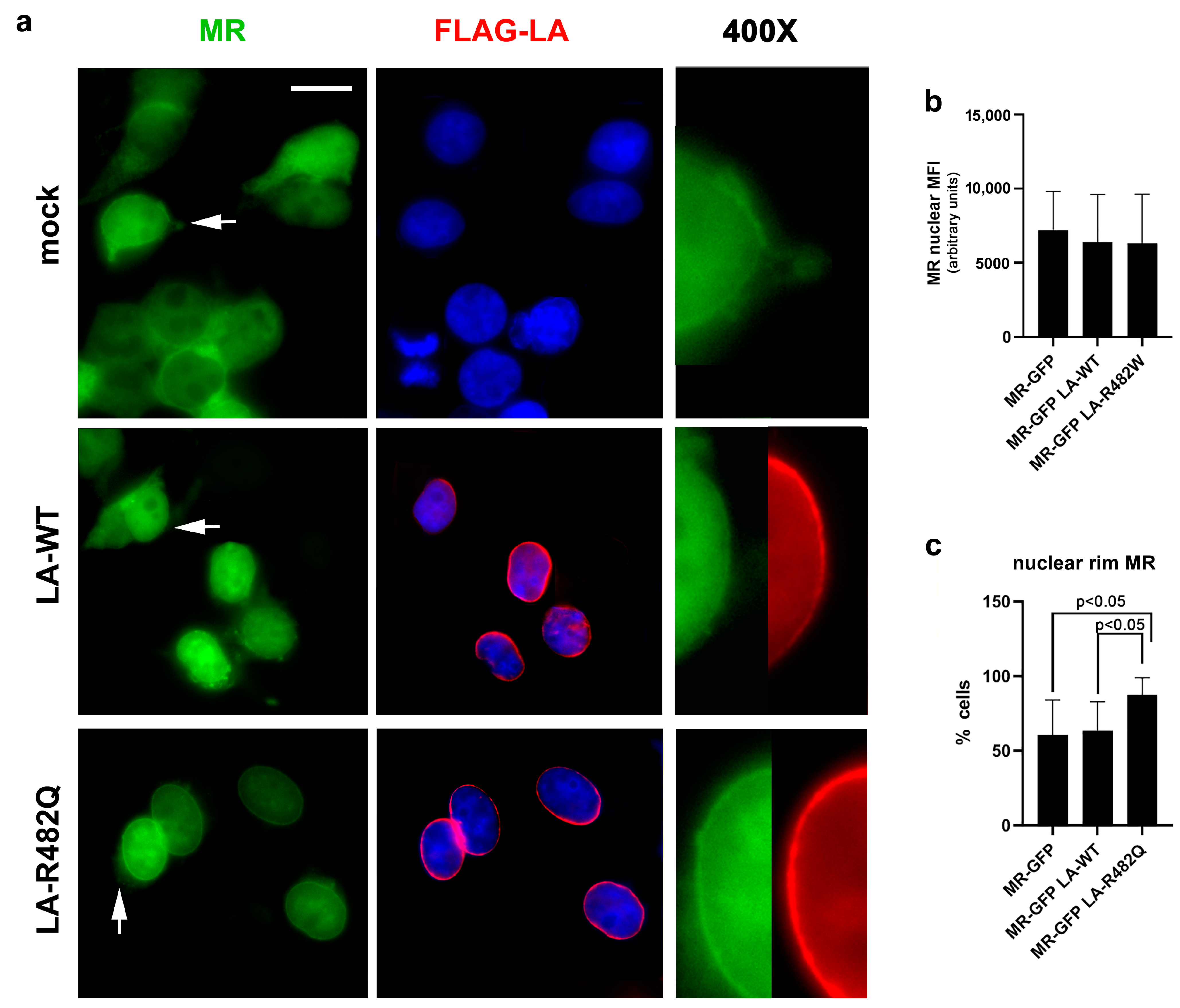
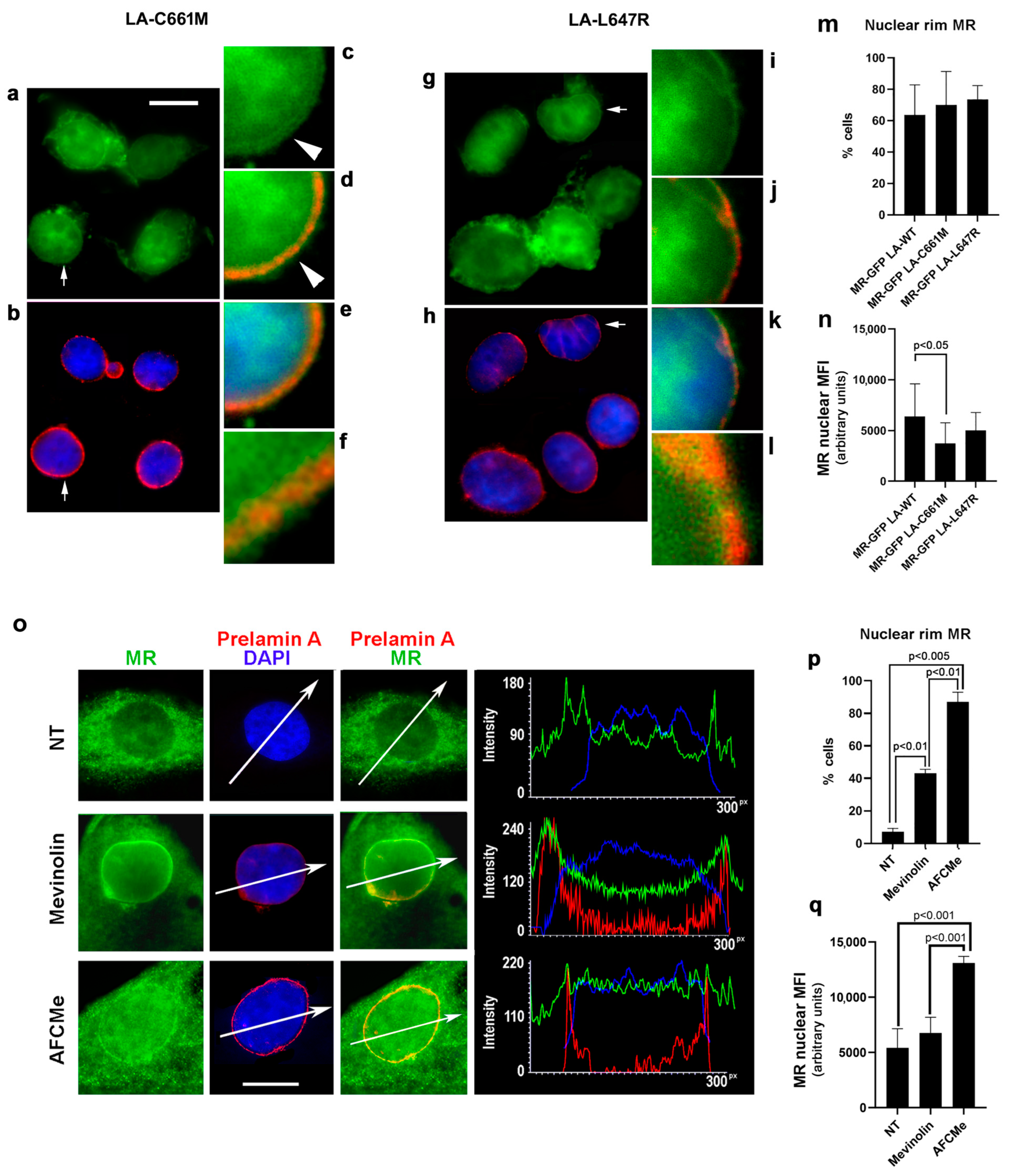
3.1.2. MR Localization in Cells Overexpressing Prelamin A
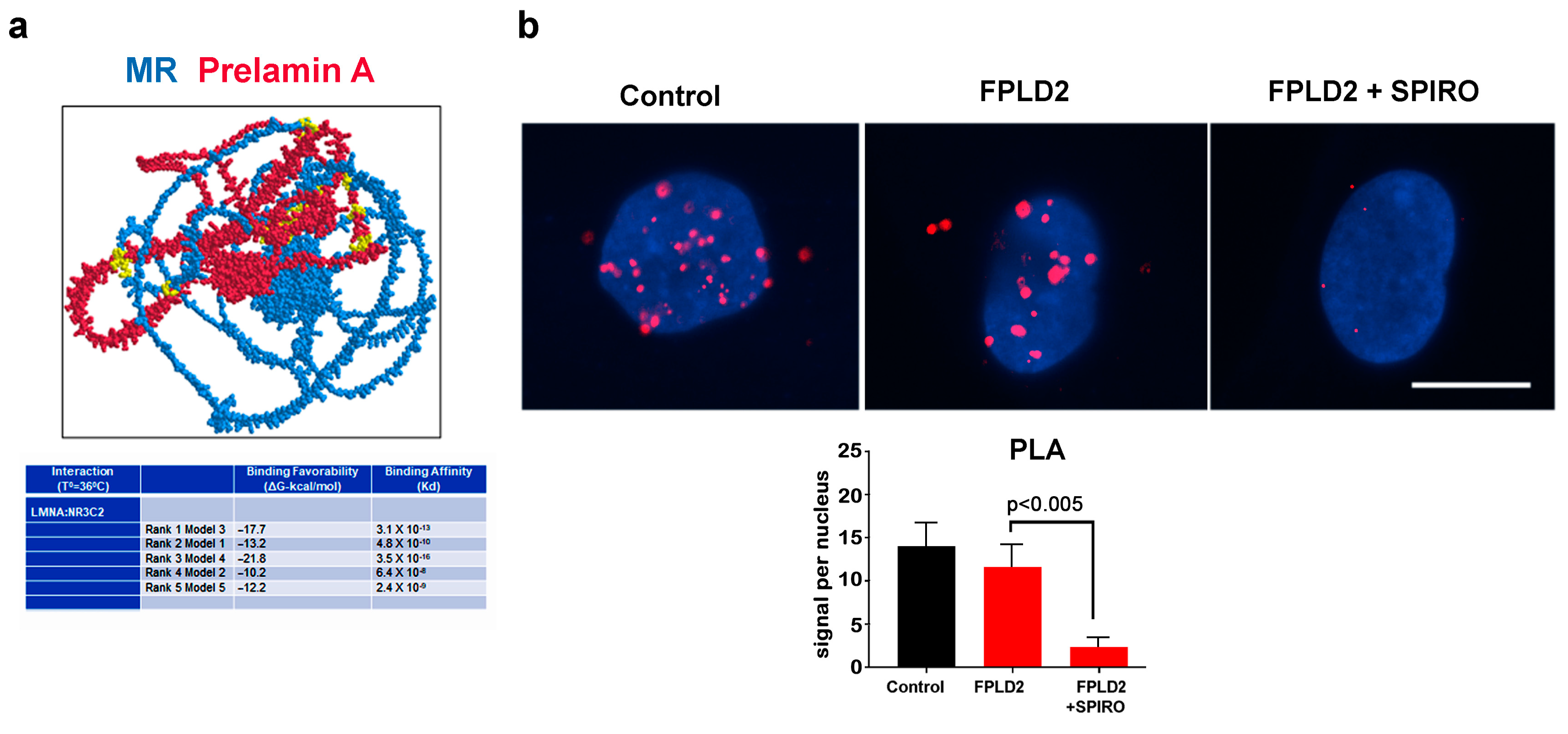
3.2. MR Binding to Prelamin A
3.3. MR Antagonism Redirects FPLD2 Brown Preadipocyte Differentiation
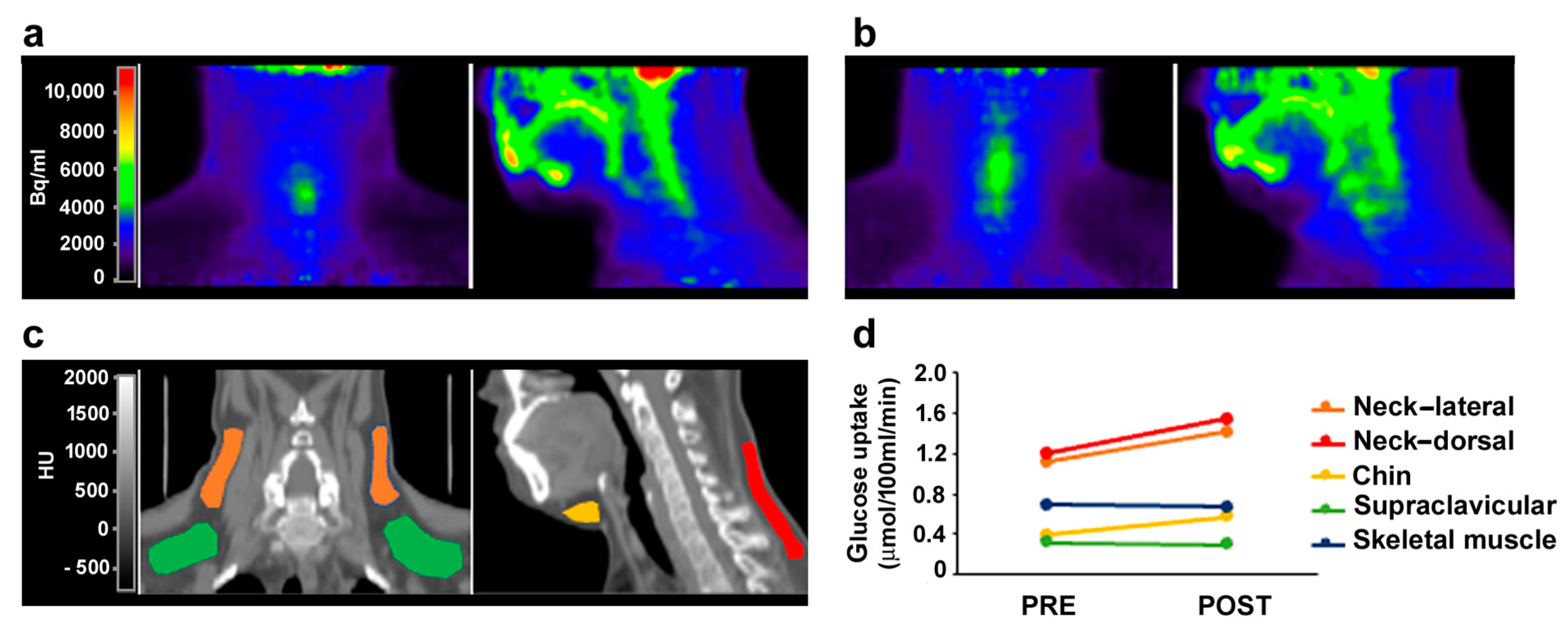
3.4. MR Antagonism in FPLD2
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pellegrini, C.; Columbaro, M.; Schena, E.; Prencipe, S.; Andrenacci, D.; Iozzo, P.; Angela Guzzardi, M.; Capanni, C.; Mattioli, E.; Loi, M.; et al. Altered adipocyte differentiation and unbalanced autophagy in type 2 Familial Partial Lipodystrophy: An in vitro and in vivo study of adipose tissue browning. Exp. Mol. Med. 2019, 51, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Shackleton, S.; Lloyd, D.J.; Jackson, S.N.; Evans, R.; Niermeijer, M.F.; Singh, B.M.; Schmidt, H.; Brabant, G.; Kumar, S.; Durrington, P.N.; et al. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat. Genet. 2000, 24, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Pombo, A.; Diaz-Lopez, E.J.; Castro, A.I.; Sanchez-Iglesias, S.; Cobelo-Gomez, S.; Prado-Morana, T.; Araujo-Vilar, D. Clinical Spectrum of LMNA-Associated Type 2 Familial Partial Lipodystrophy: A Systematic Review. Cells 2023, 12, 725. [Google Scholar] [CrossRef] [PubMed]
- von Schnurbein, J.; Adams, C.; Akinci, B.; Ceccarini, G.; D’Apice, M.R.; Gambineri, A.; Hennekam, R.C.M.; Jeru, I.; Lattanzi, G.; Miehle, K.; et al. European lipodystrophy registry: Background and structure. Orphanet J. Rare Dis. 2020, 15, 17. [Google Scholar] [CrossRef]
- Araujo-Vilar, D.; Fernandez-Pombo, A.; Victoria, B.; Mosquera-Orgueira, A.; Cobelo-Gomez, S.; Castro-Pais, A.; Hermida-Ameijeiras, A.; Loidi, L.; Sanchez-Iglesias, S. Variable Expressivity and Allelic Heterogeneity in Type 2 Familial Partial Lipodystrophy: The p.(Thr528Met) LMNA Variant. J. Clin. Med. 2021, 10, 1497. [Google Scholar] [CrossRef]
- Araujo-Vilar, D.; Lado-Abeal, J.; Palos-Paz, F.; Lattanzi, G.; Bandin, M.A.; Bellido, D.; Dominguez-Gerpe, L.; Calvo, C.; Perez, O.; Ramazanova, A.; et al. A novel phenotypic expression associated with a new mutation in LMNA gene, characterized by partial lipodystrophy, insulin resistance, aortic stenosis and hypertrophic cardiomyopathy. Clin. Endocrinol. 2008, 69, 61–68. [Google Scholar] [CrossRef]
- Gambineri, A.; Semple, R.K.; Forlani, G.; Genghini, S.; Grassi, I.; Hyden, C.S.; Pagotto, U.; O’Rahilly, S.; Pasquali, R. Monogenic polycystic ovary syndrome due to a mutation in the lamin A/C gene is sensitive to thiazolidinediones but not to metformin. Eur. J. Endocrinol. 2008, 159, 347–353. [Google Scholar] [CrossRef]
- Mosbah, H.; Donadille, B.; Vatier, C.; Janmaat, S.; Atlan, M.; Badens, C.; Barat, P.; Beliard, S.; Beltrand, J.; Ben Yaou, R.; et al. Dunnigan lipodystrophy syndrome: French National Diagnosis and Care Protocol (PNDS; Protocole National de Diagnostic et de Soins). Orphanet J. Rare Dis. 2022, 17, 170. [Google Scholar] [CrossRef]
- Araujo-Vilar, D.; Santini, F. Diagnosis and treatment of lipodystrophy: A step-by-step approach. J. Endocrinol. Investig. 2019, 42, 61–73. [Google Scholar] [CrossRef]
- Araujo-Vilar, D.; Lattanzi, G.; Gonzalez-Mendez, B.; Costa-Freitas, A.T.; Prieto, D.; Columbaro, M.; Mattioli, E.; Victoria, B.; Martinez-Sanchez, N.; Ramazanova, A.; et al. Site-dependent differences in both prelamin A and adipogenic genes in subcutaneous adipose tissue of patients with type 2 familial partial lipodystrophy. J. Med. Genet. 2009, 46, 40–48. [Google Scholar] [CrossRef]
- Capanni, C.; Mattioli, E.; Columbaro, M.; Lucarelli, E.; Parnaik, V.K.; Novelli, G.; Wehnert, M.; Cenni, V.; Maraldi, N.M.; Squarzoni, S.; et al. Altered pre-lamin A processing is a common mechanism leading to lipodystrophy. Hum. Mol. Genet. 2005, 14, 1489–1502. [Google Scholar] [CrossRef] [PubMed]
- Afonso, P.; Auclair, M.; Boccara, F.; Vantyghem, M.C.; Katlama, C.; Capeau, J.; Vigouroux, C.; Caron-Debarle, M. LMNA mutations resulting in lipodystrophy and HIV protease inhibitors trigger vascular smooth muscle cell senescence and calcification: Role of ZMPSTE24 downregulation. Atherosclerosis 2016, 245, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Infante, A.; Gago, A.; de Eguino, G.R.; Calvo-Fernandez, T.; Gomez-Vallejo, V.; Llop, J.; Schlangen, K.; Fullaondo, A.; Aransay, A.M.; Martin, A.; et al. Prelamin A accumulation and stress conditions induce impaired Oct-1 activity and autophagy in prematurely aged human mesenchymal stem cell. Aging 2014, 6, 264–280. [Google Scholar] [CrossRef]
- Czapiewski, R.; Batrakou, D.G.; de Las Heras, J.I.; Carter, R.N.; Sivakumar, A.; Sliwinska, M.; Dixon, C.R.; Webb, S.; Lattanzi, G.; Morton, N.M.; et al. Genomic loci mispositioning in Tmem120a knockout mice yields latent lipodystrophy. Nat. Commun. 2022, 13, 321. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, A.R.; Delbarre, E.; Thiede, B.; Vigouroux, C.; Collas, P. Deregulation of Fragile X-related protein 1 by the lipodystrophic lamin A p.R482W mutation elicits a myogenic gene expression program in preadipocytes. Hum. Mol. Genet. 2014, 23, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, A.; Briand, N.; Sorensen, A.L.; Cahyani, I.; Shah, A.; Moskaug, J.O.; Collas, P. A lipodystrophy-causing lamin A mutant alters conformation and epigenetic regulation of the anti-adipogenic MIR335 locus. J. Cell Biol. 2017, 216, 2731–2743. [Google Scholar] [CrossRef]
- Hartinger, R.; Lederer, E.M.; Schena, E.; Lattanzi, G.; Djabali, K. Impact of Combined Baricitinib and FTI Treatment on Adipogenesis in Hutchinson–Gilford Progeria Syndrome and Other Lipodystrophic Laminopathies. Cells 2023, 12, 1350. [Google Scholar] [CrossRef]
- Kwok, K.H.; Lam, K.S.; Xu, A. Heterogeneity of white adipose tissue: Molecular basis and clinical implications. Exp. Mol. Med. 2016, 48, e215. [Google Scholar] [CrossRef]
- Giordano, A.; Smorlesi, A.; Frontini, A.; Barbatelli, G.; Cinti, S. White, brown and pink adipocytes: The extraordinary plasticity of the adipose organ. Eur. J. Endocrinol. 2014, 170, R159–R171. [Google Scholar] [CrossRef]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef]
- Cinti, S. Adipose Organ Development and Remodeling. Compr. Physiol. 2018, 8, 1357–1431. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, C.E. What Is the Role of the Adipocyte Mineralocorticoid Receptor in the Metabolic Syndrome? Hypertension 2015, 66, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Armani, A.; Marzolla, V.; Fabbri, A.; Caprio, M. Cellular mechanisms of MR regulation of adipose tissue physiology and pathophysiology. J. Mol. Endocrinol. 2015, 55, R1–R10. [Google Scholar] [CrossRef] [PubMed]
- Armani, A.; Cinti, F.; Marzolla, V.; Morgan, J.; Cranston, G.A.; Antelmi, A.; Carpinelli, G.; Canese, R.; Pagotto, U.; Quarta, C.; et al. Mineralocorticoid receptor antagonism induces browning of white adipose tissue through impairment of autophagy and prevents adipocyte dysfunction in high-fat-diet-fed mice. FASEB J. 2014, 28, 3745–3757. [Google Scholar] [CrossRef] [PubMed]
- Hirata, A.; Maeda, N.; Hiuge, A.; Hibuse, T.; Fujita, K.; Okada, T.; Kihara, S.; Funahashi, T.; Shimomura, I. Blockade of mineralocorticoid receptor reverses adipocyte dysfunction and insulin resistance in obese mice. Cardiovasc. Res. 2009, 84, 164–172. [Google Scholar] [CrossRef]
- Feraco, A.; Marzolla, V.; Scuteri, A.; Armani, A.; Caprio, M. Mineralocorticoid Receptors in Metabolic Syndrome: From Physiology to Disease. Trends Endocrinol. Metab. 2020, 31, 205–217. [Google Scholar] [CrossRef]
- Infante, M.; Armani, A.; Mammi, C.; Fabbri, A.; Caprio, M. Impact of Adrenal Steroids on Regulation of Adipose Tissue. Compr. Physiol. 2017, 7, 1425–1447. [Google Scholar] [CrossRef]
- Corbould, A. Effects of spironolactone on glucose transport and interleukin-6 secretion in adipose cells of women. Horm. Metab. Res. 2007, 39, 915–918. [Google Scholar] [CrossRef]
- Marzolla, V.; Feraco, A.; Limana, F.; Kolkhof, P.; Armani, A.; Caprio, M. Class-specific responses of brown adipose tissue to steroidal and nonsteroidal mineralocorticoid receptor antagonists. J. Endocrinol. Investig. 2022, 45, 215–220. [Google Scholar] [CrossRef]
- Liu, K.; Czaja, M.J. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 2013, 20, 3–11. [Google Scholar] [CrossRef]
- Bereziat, V.; Cervera, P.; Le Dour, C.; Verpont, M.C.; Dumont, S.; Vantyghem, M.C.; Capeau, J.; Vigouroux, C.; Lipodystrophy Study, G. LMNA mutations induce a non-inflammatory fibrosis and a brown fat-like dystrophy of enlarged cervical adipose tissue. Am. J. Pathol. 2011, 179, 2443–2453. [Google Scholar] [CrossRef] [PubMed]
- Mattioli, E.; Columbaro, M.; Capanni, C.; Santi, S.; Maraldi, N.M.; D’Apice, M.R.; Novelli, G.; Riccio, M.; Squarzoni, S.; Foisner, R.; et al. Drugs affecting prelamin A processing: Effects on heterochromatin organization. Exp. Cell Res. 2008, 314, 453–462. [Google Scholar] [CrossRef]
- Capanni, C.; Schena, E.; Di Giampietro, M.L.; Montecucco, A.; Mattioli, E.; Lattanzi, G. The role of prelamin A post-translational maturation in stress response and 53BP1 recruitment. Front. Cell Dev. Biol. 2022, 10, 1018102. [Google Scholar] [CrossRef] [PubMed]
- Santi, S.; Cenni, V.; Capanni, C.; Lattanzi, G.; Mattioli, E. PCAF Involvement in Lamin A/C-HDAC2 Interplay during the Early Phase of Muscle Differentiation. Cells 2020, 9, 1735. [Google Scholar] [CrossRef]
- Caron, M.; Auclair, M.; Donadille, B.; Bereziat, V.; Guerci, B.; Laville, M.; Narbonne, H.; Bodemer, C.; Lascols, O.; Capeau, J.; et al. Human lipodystrophies linked to mutations in A-type lamins and to HIV protease inhibitor therapy are both associated with prelamin A accumulation, oxidative stress and premature cellular senescence. Cell Death Differ. 2007, 14, 1759–1767. [Google Scholar] [CrossRef]
- Dominici, S.; Fiori, V.; Magnani, M.; Schena, E.; Capanni, C.; Camozzi, D.; D’Apice, M.R.; Le Dour, C.; Auclair, M.; Caron, M.; et al. Different prelamin A forms accumulate in human fibroblasts: A study in experimental models and progeria. Eur. J. Histochem. 2009, 53, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Le Dour, C.; Schneebeli, S.; Bakiri, F.; Darcel, F.; Jacquemont, M.L.; Maubert, M.A.; Auclair, M.; Jeziorowska, D.; Reznik, Y.; Bereziat, V.; et al. A homozygous mutation of prelamin-A preventing its farnesylation and maturation leads to a severe lipodystrophic phenotype: New insights into the pathogenicity of nonfarnesylated prelamin-A. J. Clin. Endocrinol. Metab. 2011, 96, E856–E862. [Google Scholar] [CrossRef]
- Andre, P.; Schneebeli, S.; Vigouroux, C.; Lascols, O.; Schaaf, M.; Chevalier, P. Metabolic and cardiac phenotype characterization in 37 atypical Dunnigan patients with nonfarnesylated mutated prelamin A. Am. Heart J. 2015, 169, 587–593. [Google Scholar] [CrossRef]
- Camozzi, D.; D’Apice, M.R.; Schena, E.; Cenni, V.; Columbaro, M.; Capanni, C.; Maraldi, N.M.; Squarzoni, S.; Ortolani, M.; Novelli, G.; et al. Altered chromatin organization and SUN2 localization in mandibuloacral dysplasia are rescued by drug treatment. Histochem. Cell Biol. 2012, 138, 643–651. [Google Scholar] [CrossRef]
- Sawada, T.; Miyoshi, H.; Shimada, K.; Suzuki, A.; Okamatsu-Ogura, Y.; Perfield, J.W., II; Kondo, T.; Nagai, S.; Shimizu, C.; Yoshioka, N.; et al. Perilipin overexpression in white adipose tissue induces a brown fat-like phenotype. PLoS ONE 2010, 5, e14006. [Google Scholar] [CrossRef]
- Souza, S.C.; Christoffolete, M.A.; Ribeiro, M.O.; Miyoshi, H.; Strissel, K.J.; Stancheva, Z.S.; Rogers, N.H.; D’Eon, T.M.; Perfield, J.W., II; Imachi, H.; et al. Perilipin regulates the thermogenic actions of norepinephrine in brown adipose tissue. J. Lipid Res. 2007, 48, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Ricchiuti, V.; Lian, B.Q.; Yao, T.M.; Coutinho, P.; Romero, J.R.; Li, J.; Williams, G.H.; Adler, G.K. Mineralocorticoid receptor blockade reverses obesity-related changes in expression of adiponectin, peroxisome proliferator-activated receptor-gamma, and proinflammatory adipokines. Circulation 2008, 117, 2253–2261. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Sanchez, N. There and Back Again: Leptin Actions in White Adipose Tissue. Int. J. Mol. Sci. 2020, 21, 6039. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Canino, R.; Lorenzo-Diaz, F.; Jaisser, F.; Farman, N.; Giraldez, T.; Alvarez de la Rosa, D. Histone Deacetylase 6-Controlled Hsp90 Acetylation Significantly Alters Mineralocorticoid Receptor Subcellular Dynamics But Not its Transcriptional Activity. Endocrinology 2016, 157, 2515–2532. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Q.; Wu, D.; Chen, Q.; Gong, G.; He, L.; Wu, X. Lamin-A interacting protein Hsp90 is required for DNA damage repair and chemoresistance of ovarian cancer cells. Cell Death Dis. 2021, 12, 786. [Google Scholar] [CrossRef]
- Thanomkitti, K.; Fong-Ngern, K.; Sueksakit, K.; Thuangtong, R.; Thongboonkerd, V. Molecular functional analyses revealed essential roles of HSP90 and lamin A/C in growth, migration, and self-aggregation of dermal papilla cells. Cell Death Discov. 2018, 4, 53. [Google Scholar] [CrossRef]
- Hamczyk, M.R.; Villa-Bellosta, R.; Quesada, V.; Gonzalo, P.; Vidak, S.; Nevado, R.M.; Andres-Manzano, M.J.; Misteli, T.; Lopez-Otin, C.; Andres, V. Progerin accelerates atherosclerosis by inducing endoplasmic reticulum stress in vascular smooth muscle cells. EMBO Mol. Med. 2019, 11, e9736. [Google Scholar] [CrossRef]
- Galigniana, M.D.; Erlejman, A.G.; Monte, M.; Gomez-Sanchez, C.; Piwien-Pilipuk, G. The hsp90-FKBP52 complex links the mineralocorticoid receptor to motor proteins and persists bound to the receptor in early nuclear events. Mol. Cell. Biol. 2010, 30, 1285–1298. [Google Scholar] [CrossRef]
- Vahabikashi, A.; Sivagurunathan, S.; Nicdao, F.A.S.; Han, Y.L.; Park, C.Y.; Kittisopikul, M.; Wong, X.; Tran, J.R.; Gundersen, G.G.; Reddy, K.L.; et al. Nuclear lamin isoforms differentially contribute to LINC complex-dependent nucleocytoskeletal coupling and whole-cell mechanics. Proc. Natl. Acad. Sci. USA 2022, 119, e2121816119. [Google Scholar] [CrossRef]
- Verstraeten, V.L.; Renes, J.; Ramaekers, F.C.; Kamps, M.; Kuijpers, H.J.; Verheyen, F.; Wabitsch, M.; Steijlen, P.M.; van Steensel, M.A.; Broers, J.L. Reorganization of the nuclear lamina and cytoskeleton in adipogenesis. Histochem. Cell Biol. 2011, 135, 251–261. [Google Scholar] [CrossRef]
- Ong, G.S.; Young, M.J. Mineralocorticoid regulation of cell function: The role of rapid signalling and gene transcription pathways. J. Mol. Endocrinol. 2017, 58, R33–R57. [Google Scholar] [CrossRef] [PubMed]
- Cenni, V.; Capanni, C.; Mattioli, E.; Schena, E.; Squarzoni, S.; Bacalini, M.G.; Garagnani, P.; Salvioli, S.; Franceschi, C.; Lattanzi, G. Lamin A involvement in ageing processes. Ageing Res. Rev. 2020, 62, 101073. [Google Scholar] [CrossRef] [PubMed]
- Squarzoni, S.; Schena, E.; Sabatelli, P.; Mattioli, E.; Capanni, C.; Cenni, V.; D’Apice, M.R.; Andrenacci, D.; Sarli, G.; Pellegrino, V.; et al. Interleukin-6 neutralization ameliorates symptoms in prematurely aged mice. Aging Cell 2021, 20, e13285. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schena, E.; Mattioli, E.; Peres, C.; Zanotti, L.; Morselli, P.; Iozzo, P.; Guzzardi, M.A.; Bernardini, C.; Forni, M.; Nesci, S.; et al. Mineralocorticoid Receptor Antagonism Prevents Type 2 Familial Partial Lipodystrophy Brown Adipocyte Dysfunction. Cells 2023, 12, 2586. https://doi.org/10.3390/cells12222586
Schena E, Mattioli E, Peres C, Zanotti L, Morselli P, Iozzo P, Guzzardi MA, Bernardini C, Forni M, Nesci S, et al. Mineralocorticoid Receptor Antagonism Prevents Type 2 Familial Partial Lipodystrophy Brown Adipocyte Dysfunction. Cells. 2023; 12(22):2586. https://doi.org/10.3390/cells12222586
Chicago/Turabian StyleSchena, Elisa, Elisabetta Mattioli, Chiara Peres, Laura Zanotti, Paolo Morselli, Patricia Iozzo, Maria Angela Guzzardi, Chiara Bernardini, Monica Forni, Salvatore Nesci, and et al. 2023. "Mineralocorticoid Receptor Antagonism Prevents Type 2 Familial Partial Lipodystrophy Brown Adipocyte Dysfunction" Cells 12, no. 22: 2586. https://doi.org/10.3390/cells12222586
APA StyleSchena, E., Mattioli, E., Peres, C., Zanotti, L., Morselli, P., Iozzo, P., Guzzardi, M. A., Bernardini, C., Forni, M., Nesci, S., Caprio, M., Cecchetti, C., Pagotto, U., Gabusi, E., Cattini, L., Lisignoli, G., Blalock, W., Gambineri, A., & Lattanzi, G. (2023). Mineralocorticoid Receptor Antagonism Prevents Type 2 Familial Partial Lipodystrophy Brown Adipocyte Dysfunction. Cells, 12(22), 2586. https://doi.org/10.3390/cells12222586