
Flavonoids Regulate Redox-Responsive Transcription Factors in Glioblastoma and Microglia
 ,
, 
Abstract
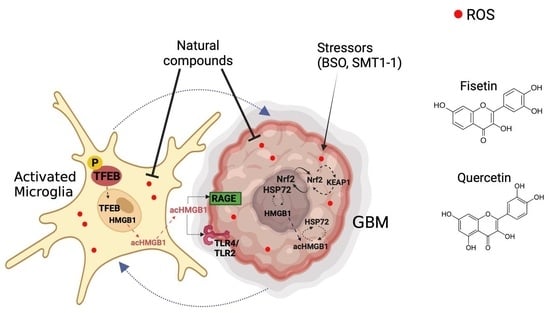
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Mitochondrial Metabolic Activity (MTT)
2.3. Lactose Dehydrogenase (LDH) Assay
2.4. Cell Counting
2.5. CellROX Assay
2.6. Immunocytochemistry
2.7. GBM Tumoroid Preparation
2.8. Molecular Docking
2.9. Proximity Ligation Assay
2.10. Western Blot
2.11. Statistical Analysis
3. Results
3.1. GBM Cells Are More Sensitive to Fisetin and Quercetin Compared to Microglia
3.2. Combination of Fisetin and Temozolomide Synergistically Inhibits GBM Survival
3.3. Fisetin Reduces Oxidative Stress in GBM and Microglia
3.4. Fisetin Increases TFEB Abundance in Stressed Microglia
3.5. Fisetin Decreases Cytosolic acHMGB1 Abundance in Microglia, but Not in GBM
3.6. Molecular Docking
3.7. Fisetin and Quercetin Can Bind to KEAP1, HSP72 and HMGB1
3.8. Modulation of Protein Targets in Microglia and Glioblastoma Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matias, D.; Balça-Silva, J.; da Graça, G.C.; Wanjiru, C.M.; Macharia, L.W.; Nascimento, C.P.; Roque, N.R.; Coelho-Aguiar, J.M.; Pereira, C.M.; Dos Santos, M.F.; et al. Microglia/Astrocytes–Glioblastoma Crosstalk: Crucial Molecular Mechanisms and Microenvironmental Factors. Front. Cell. Neurosci. 2018, 12, 235. [Google Scholar] [CrossRef] [PubMed]
- Shergalis, A.; Bankhead, A., 3rd; Luesakul, U.; Muangsin, N.; Neamati, N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef] [PubMed]
- Geribaldi-Doldán, N.; Fernández-Ponce, C.; Quiroz, R.N.; Sánchez-Gomar, I.; Escorcia, L.G.; Velásquez, E.P.; Quiroz, E.N. The Role of Microglia in Glioblastoma. Front. Oncol. 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, D.H.; Kettenmann, H. Microglia/Brain Macrophages as Central Drivers of Brain Tumor Pathobiology. Neuron 2019, 104, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef]
- Gülke, E.; Gelderblom, M.; Magnus, T. Danger signals in stroke and their role on microglia activation after ischemia. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418774254. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef]
- Donat, C.K.; Scott, G.; Gentleman, S.M.; Sastre, M. Microglial Activation in Traumatic Brain Injury. Front. Aging Neurosci. 2017, 9, 208. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Liu, H.; Sun, Y.; Zhang, Q.; Jin, W.; Gordon, R.E.; Zhang, Y.; Wang, J.; Sun, C.; Wang, Z.J.; Qi, X.; et al. Pro-inflammatory and proliferative microglia drive progression of glioblastoma. Cell Rep. 2021, 36, 109718. [Google Scholar] [CrossRef] [PubMed]
- Rosciszewski, G.; Cadena, V.; Auzmendi, J.; Cieri, M.B.; Lukin, J.; Rossi, A.R.; Murta, V.; Villarreal, A.; Reinés, A.; Gomes, F.C.A.; et al. Detrimental Effects of HMGB-1 Require Microglial-Astroglial Interaction: Implications for the Status Epilepticus -Induced Neuroinflammation. Front. Cell. Neurosci. 2019, 13, 380. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Tracey, K.J.; Yang, H. Post-Translational Modification of HMGB1 Disulfide Bonds in Stimulating and Inhibiting Inflammation. Cells 2021, 10, 3323. [Google Scholar] [CrossRef] [PubMed]
- Ou, A.; Ott, M.; Fang, D.; Heimberger, A.B. The Role and Therapeutic Targeting of JAK/STAT Signaling in Glioblastoma. Cancers 2021, 13, 437. [Google Scholar] [CrossRef]
- Rapoport, B.L.; Steel, H.C.; Theron, A.J.; Heyman, L.; Smit, T.; Ramdas, Y.; Anderson, R. High Mobility Group Box 1 in Human Cancer. Cells 2020, 9, 1664. [Google Scholar] [CrossRef]
- Campisi, J.; Leem, T.H.; Fleshner, M. Stress-induced extracellular Hsp72 is a functionally significant danger signal to the immune system. Cell Stress Chaperones 2003, 8, 272–286. [Google Scholar] [CrossRef]
- Fujii, K.; Idogawa, M.; Suzuki, N.; Iwatsuki, K.; Kanekura, T. Functional Depletion of HSP72 by siRNA and Quercetin Enhances Vorinostat-Induced Apoptosis in an HSP72-Overexpressing Cutaneous T-Cell Lymphoma Cell Line, Hut78. Int. J. Mol. Sci. 2021, 22, 11258. [Google Scholar] [CrossRef]
- Matokanovic, M.; Barisic, K.; Filipovic-Grcic, J.; Maysinger, D. Hsp70 silencing with siRNA in nanocarriers enhances cancer cell death induced by the inhibitor of Hsp90. Eur. J. Pharm. Sci. 2013, 50, 149–158. [Google Scholar] [CrossRef]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef]
- Ngo, V.; Duennwald, M.L. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants 2022, 11, 2345. [Google Scholar] [CrossRef]
- Batra, P.; Sharma, A.K. Anti-cancer potential of flavonoids: Recent trends and future perspectives. 3 Biotech 2013, 3, 439–459. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.-M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Moquin, A.; Bomal, E.; Na, L.; Maysinger, D.; Kakkar, A. Telodendrimers for Physical Encapsulation and Covalent Linking of Individual or Combined Therapeutics. Mol. Pharm. 2017, 14, 2607–2615. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Berman, A.Y.; Motechin, R.A.; Wiesenfeld, M.Y.; Holz, M.K. The therapeutic potential of resveratrol: A review of clinical trials. NPJ Precis. Oncol. 2017, 1, 35. [Google Scholar] [CrossRef]
- Jia, S.; Xu, X.; Zhou, S.; Chen, Y.; Ding, G.; Cao, L. Fisetin induces autophagy in pancreatic cancer cells via endoplasmic reticulum stress- and mitochondrial stress-dependent pathways. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Maysinger, D.; Moquin, A.; Choi, J.; Kodiha, M.; Stochaj, U. Gold nanourchins and celastrol reorganize the nucleo- and cytoskeleton of glioblastoma cells. Nanoscale 2017, 10, 1716–1726. [Google Scholar] [CrossRef]
- Rauf, A.; Imran, M.; Khan, I.A.; Ur-Rehman, M.; Gilani, S.A.; Mehmood, Z.; Mubarak, M.S. Anticancer potential of quercetin: A comprehensive review. Phytother. Res. 2018, 32, 2109–2130. [Google Scholar] [CrossRef]
- Aalinkeel, R.; Bindukumar, B.; Reynolds, J.L.; Sykes, D.E.; Mahajan, S.D.; Chadha, K.C.; Schwartz, S.A. The dietary bioflavonoid, quercetin, selectively induces apoptosis of prostate cancer cells by down-regulating the expression of heat shock protein. Prostate 2008, 68, 1773–1789. [Google Scholar] [CrossRef] [PubMed]
- Anhê, F.F.; Desjardins, Y.; Pilon, G.; Dudonné, S.; Genovese, M.I.; Lajolo, F.M.; Marette, A. Polyphenols and type 2 diabetes: A prospective review. PharmaNutrition 2013, 1, 105–114. [Google Scholar] [CrossRef]
- Cao, H.; Ou, J.; Chen, L.; Zhang, Y.; Szkudelski, T.; Delmas, D.; Daglia, M.; Xiao, J. Dietary polyphenols and type 2 diabetes: Human Study and Clinical Trial. Crit. Rev. Food Sci. Nutr. 2018, 59, 3371–3379. [Google Scholar] [CrossRef] [PubMed]
- Del Duca, D.; Werbowetski, T.; Del Maestro, R.F. Spheroid Preparation from Hanging Drops: Characterization of a Model of Brain Tumor Invasion. J. Neuro-Oncol. 2004, 67, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.W.; Tian, M.; Song, F.; Venereau, E.; Preti, A.; Park, S.-W.; Hamilton, K.; Swapna, G.V.T.; Manohar, M.; Moreau, M.; et al. Aspirin’s Active Metabolite Salicylic Acid Targets High Mobility Group Box 1 to Modulate Inflammatory Responses. Mol. Med. 2015, 21, 526–535. [Google Scholar] [CrossRef]
- Mollica, L.; De Marchis, F.; Spitaleri, A.; Dallacosta, C.; Pennacchini, D.; Zamai, M.; Agresti, A.; Trisciuoglio, L.; Musco, G.; Bianchi, M.E. Glycyrrhizin Binds to High-Mobility Group Box 1 Protein and Inhibits Its Cytokine Activities. Chem. Biol. 2007, 14, 431–441. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Khan, S.U.; Othman, I.; Shaikh, M.F. Naturally Occurring HMGB1 Inhibitor, Glycyrrhizin, Modulates Chronic Seizures-Induced Memory Dysfunction in Zebrafish Model. ACS Chem. Neurosci. 2021, 12, 3288–3302. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera?A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Zeng, Y.; Song, J.; Zhang, M.; Wang, H.; Zhang, Y.; Suo, H. Comparison of In Vitro and In Vivo Antioxidant Activities of Six Flavonoids with Similar Structures. Antioxidants 2020, 9, 732. [Google Scholar] [CrossRef] [PubMed]
- Olivier, C.; Oliver, L.; Lalier, L.; Vallette, F.M. Drug Resistance in Glioblastoma: The Two Faces of Oxidative Stress. Front. Mol. Biosci. 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Scholer-Dahirel, A.; Mechta-Grigoriou, F. The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin. Cancer Biol. 2014, 25, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Baghbanbashi, M.; Yong, H.W.; Zhang, I.; Lotocki, V.; Yuan, Z.; Pazuki, G.; Maysinger, D.; Kakkar, A. Stimuli-Responsive Miktoarm Polymer-Based Formulations for Fisetin Delivery and Regulatory Effects in Hyperactive Human Microglia. Macromol. Biosci. 2022, 22, e2200174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, I.; Cui, Y.; Amiri, A.; Ding, Y.; Campbell, R.E.; Maysinger, D. Pharmacological inhibition of lipid droplet formation enhances the effectiveness of curcumin in glioblastoma. Eur. J. Pharm. Biopharm. 2016, 100, 66–76. [Google Scholar] [CrossRef]
- Martina, J.A.; Puertollano, R. TFEB and TFE3: The art of multi-tasking under stress conditions. Transcription 2016, 8, 48–54. [Google Scholar] [CrossRef]
- Yu, Y.; Tang, D.; Kang, R. Oxidative stress-mediated HMGB1 biology. Front. Physiol. 2015, 6, 93. [Google Scholar] [CrossRef]
- Ong, A.J.S.; Bladen, C.E.; Tigani, T.A.; Karamalakis, A.P.; Evason, K.J.; Brown, K.K.; Cox, A.G. The KEAP1–NRF2 pathway regulates TFEB/TFE3-dependent lysosomal biogenesis. Proc. Natl. Acad. Sci. USA 2023, 120. [Google Scholar] [CrossRef]
- Jones, A.M.; Westwood, I.M.; Osborne, J.D.; Matthews, T.P.; Cheeseman, M.D.; Rowlands, M.G.; Jeganathan, F.; Burke, R.; Lee, D.; Kadi, N.; et al. A fragment-based approach applied to a highly flexible target: Insights and challenges towards the inhibition of HSP70 isoforms. Sci. Rep. 2016, 6, 34701. [Google Scholar] [CrossRef]
- Fukutomi, T.; Takagi, K.; Mizushima, T.; Ohuchi, N.; Yamamoto, M. Kinetic, Thermodynamic, and Structural Characterizations of the Association between Nrf2-DLGex Degron and Keap1. Mol. Cell. Biol. 2014, 34, 832–846. [Google Scholar] [CrossRef]
- Hörer, S.; Reinert, D.; Ostmann, K.; Hoevels, Y.; Nar, H. Crystal-contact engineering to obtain a crystal form of the Kelch domain of human Keap1 suitable for ligand-soaking experiments. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2013, 69, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Ontoria, J.M.; Biancofiore, I.; Fezzardi, P.; Ferrigno, F.; Torrente, E.; Colarusso, S.; Bianchi, E.; Andreini, M.; Patsilinakos, A.; Kempf, G.; et al. Combined Peptide and Small-Molecule Approach toward Nonacidic THIQ Inhibitors of the KEAP1/NRF2 Interaction. ACS Med. Chem. Lett. 2020, 11, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-Y.; Watabe, K. The roles of microglia macrophages in tumor progression of brain cancer and metastatic disease. Front. Biosci. 2017, 22, 1805–1829. [Google Scholar] [CrossRef] [PubMed]
- Zhong, M.; Lynch, A.; Muellers, S.N.; Jehle, S.; Luo, L.; Hall, D.R.; Iwase, R.; Carolan, J.P.; Egbert, M.; Wakefield, A.; et al. Interaction Energetics and Druggability of the Protein–Protein Interaction between Kelch-like ECH-Associated Protein 1 (KEAP1) and Nuclear Factor Erythroid 2 Like 2 (Nrf2). Biochemistry 2019, 59, 563–581. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.I.; Fareed, M.I.; Rashid, H.; Aziz, H.; Ehsan, N.; Khalid, S.; Ghaffar, I.; Ali, R.; Gul, A.; Hakeem, K.R. Flavonoids and Their Biological Secrets. Plant Hum. Health 2019, 2, 579–605. [Google Scholar] [CrossRef]
- Manach, C.; Mazur, A.; Scalbert, A. Polyphenols and prevention of cardiovascular diseases. Curr. Opin. Infect. Dis. 2005, 16, 77–84. [Google Scholar] [CrossRef]
- Michalska, M.; Gluba, A.; Mikhailidis, D.P.; Nowak, P.; Bielecka-Dabrowa, A.; Rysz, J.; Banach, M. The role of polyphenols in cardiovascular disease. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2010, 16, RA110–RA119. [Google Scholar]
- Pandey, K.B.; Rizvi, S.I. Plant polyphenols as dietary antioxidants in human health and disease. Oxid. Med. Cell. Longev. 2009, 2, 270–278. [Google Scholar] [CrossRef]
- Panzella, L. Natural Phenolic Compounds for Health, Food and Cosmetic Applications. Antioxidants 2020, 9, 427. [Google Scholar] [CrossRef]
- Quiñones, M.; Miguel, M.; Aleixandre, A. Beneficial effects of polyphenols on cardiovascular disease. Pharmacol. Res. 2013, 68, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Zillich, O.V.; Schweiggert-Weisz, U.; Eisner, P.; Kerscher, M. Polyphenols as active ingredients for cosmetic products. Int. J. Cosmet. Sci. 2015, 37, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, A.; Schluesener, H. Natural polyphenols against neurodegenerative disorders: Potentials and pitfalls. Ageing Res. Rev. 2012, 11, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.F.M.; Pogačnik, L. Polyphenols from Food and Natural Products: Neuroprotection and Safety. Antioxidants 2020, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.; Rahu, N. Oxidative Stress and Inflammation: What Polyphenols Can Do for Us? Oxid. Med. Cell. Longev. 2016, 2016, 7432797. [Google Scholar] [CrossRef] [PubMed]
- Shakoor, H.; Feehan, J.; Apostolopoulos, V.; Platat, C.; Al Dhaheri, A.S.; Ali, H.I.; Ismail, L.C.; Bosevski, M.; Stojanovska, L. Immunomodulatory Effects of Dietary Polyphenols. Nutrients 2021, 13, 728. [Google Scholar] [CrossRef] [PubMed]
- Asensi, M.; Ortega, A.; Mena, S.; Feddi, F.; Estrela, J.M. Natural polyphenols in cancer therapy. Crit. Rev. Clin. Lab. Sci. 2011, 48, 197–216. [Google Scholar] [CrossRef]
- Mahbub, A.; Maitre, C.; Haywood-Small, S.L.; McDougall, G.J.; Cross, N.A.; Jordan-Mahy, N. Differential Effects of Polyphenols on Proliferation and Apoptosis in Human Myeloid and Lymphoid Leukemia Cell Lines. Anti-Cancer Agents Med. Chem. 2013, 13, 1601–1613. [Google Scholar] [CrossRef]
- Maiuolo, J.; Gliozzi, M.; Carresi, C.; Musolino, V.; Oppedisano, F.; Scarano, F.; Nucera, S.; Scicchitano, M.; Bosco, F.; Macri, R.; et al. Nutraceuticals and Cancer: Potential for Natural Polyphenols. Nutrients 2021, 13, 3834. [Google Scholar] [CrossRef]
- Cháirez-Ramírez, M.H.; de la Cruz-López, K.G.; García-Carrancá, A. Polyphenols as Antitumor Agents Targeting Key Players in Cancer-Driving Signaling Pathways. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef]
- Miccadei, S.; Di Venere, D.; Cardinali, A.; Romano, F.; Durazzo, A.; Foddai, M.S.; Fraioli, R.; Mobarhan, S.; Maiani, G. Antioxidative and Apoptotic Properties of Polyphenolic Extracts from Edible Part of Artichoke (Cynara scolymus L.) on Cultured Rat Hepatocytes and on Human Hepatoma Cells. Nutr. Cancer 2008, 60, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Mileo, A.M.; Di Venere, D.; Abbruzzese, C.; Miccadei, S. Long Term Exposure to Polyphenols of Artichoke (Cynara scolymus L.) Exerts Induction of Senescence Driven Growth Arrest in the MDA-MB231 Human Breast Cancer Cell Line. Oxidative Med. Cell. Longev. 2015, 2015, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Prakash, M.D.; Stojanovska, L.; Feehan, J.; Nurgali, K.; Donald, E.L.; Plebanski, M.; Flavel, M.; Kitchen, B.; Apostolopoulos, V. Anti-cancer effects of polyphenol-rich sugarcane extract. PLOS ONE 2021, 16, e0247492. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zheng, W.; Feng, X.; Yang, F.; Qin, H.; Wu, S.; Hou, D.-X.; Chen, J. Nrf2–ARE Signaling Acts as Master Pathway for the Cellular Antioxidant Activity of Fisetin. Molecules 2019, 24, 708. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.I.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [PubMed]
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Cellular senescence and senolytics: The path to the clinic. Nat. Med. 2022, 28, 1556–1568. [Google Scholar] [CrossRef]
- Schapira, M.; Tyers, M.; Torrent, M.; Arrowsmith, C.H. WD40 repeat domain proteins: A novel target class? Nat. Rev. Drug Discov. 2017, 16, 773–786. [Google Scholar] [CrossRef]
- Lo, S.-C.; Li, X.; Henzl, M.T.; Beamer, L.J.; Hannink, M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006, 25, 3605–3617. [Google Scholar] [CrossRef]
- Ma, B.; Lucas, B.; Capacci, A.; Lin, E.Y.-S.; Jones, J.H.; Dechantsreiter, M.; Enyedy, I.; Marcotte, D.; Xiao, G.; Li, B.; et al. Design, synthesis and identification of novel, orally bioavailable non-covalent Nrf2 activators. Bioorganic Med. Chem. Lett. 2019, 30, 126852. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, B.; Tong, K.I.; Ohta, T.; Nakamura, Y.; Scharlock, M.; Ohtsuji, M.; Kang, M.-I.; Kobayashi, A.; Yokoyama, S.; Yamamoto, M. Structural Basis for Defects of Keap1 Activity Provoked by Its Point Mutations in Lung Cancer. Mol. Cell 2006, 21, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Pallesen, J.S.; Narayanan, D.; Tran, K.T.; Solbak, S.M.; Marseglia, G.; Sørensen, L.M.E.; Høj, L.J.; Munafò, F.; Carmona, R.M.C.; Garcia, A.D.; et al. Deconstructing Noncovalent Kelch-like ECH-Associated Protein 1 (Keap1) Inhibitors into Fragments to Reconstruct New Potent Compounds. J. Med. Chem. 2021, 64, 4623–4661. [Google Scholar] [CrossRef] [PubMed]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [PubMed]
- Satta, S.; Mahmoud, A.M.; Wilkinson, F.L.; Yvonne Alexander, M.; White, S.J. The role of Nrf2 in cardiovascular function and disease. Oxid. Med. Cell. Longev. 2017, 2017, 9237263. [Google Scholar] [CrossRef] [PubMed]
- Clifford, T.; Acton, J.P.; Cocksedge, S.P.; Davies, K.A.B.; Bailey, S.J. The effect of dietary phytochemicals on nuclear factor erythroid 2-related factor 2 (Nrf2) activation: A systematic review of human intervention trials. Mol. Biol. Rep. 2021, 48, 1745–1761. [Google Scholar] [CrossRef]
- Khan, J.; Deb, P.K.; Priya, S.; Medina, K.D.; Devi, R.; Walode, S.G.; Rudrapal, M. Dietary Flavonoids: Cardioprotective Potential with Antioxidant Effects and Their Pharmacokinetic, Toxicological and Therapeutic Concerns. Molecules 2021, 26, 4021. [Google Scholar] [CrossRef]
- Andersson, U.; Yang, H.; Harris, H. High-mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. In Seminars in Immunology; Academic Press: New York, NY, USA, 2018; pp. 40–48. [Google Scholar]
- Andersson, U.; Tracey, K.J. HMGB1 Is a Therapeutic Target for Sterile Inflammation and Infection. Annu. Rev. Immunol. 2011, 29, 139–162. [Google Scholar] [CrossRef]
- Chen, R.; Kang, R.; Tang, D. The mechanism of HMGB1 secretion and release. Exp. Mol. Med. 2022, 54, 91–102. [Google Scholar] [CrossRef]
- Deng, M.; Scott, M.J.; Fan, J.; Billiar, T.R. Location is the key to function: HMGB1 in sepsis and trauma-induced inflammation. J. Leukoc. Biol. 2019, 106, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhao, X.; Antoine, D.J.; Xiao, X.; Wang, H.; Andersson, U.; Billiar, T.R.; Tracey, K.J.; Lu, B.; Papatheodorou, A.; et al. Regulation of Posttranslational Modifications of HMGB1 During Immune Responses. Antioxid. Redox Signal. 2016, 24, 620–634. [Google Scholar] [CrossRef]
- Bianchi, M.E. Significant (re)location: How to use chromatin and/or abundant proteins as messages of life and death. Trends Cell Biol. 2004, 14, 287–293. [Google Scholar] [CrossRef]
- Müller, S.; Ronfani, L.; Bianchi, M.E. Regulated expression and subcellular localization of HMGB1, a chromatin protein with a cytokine function. J. Intern. Med. 2004, 255, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Raucci, A.; Palumbo, R.; Bianchi, M.E. HMGB1: A signal of necrosis. Autoimmunity 2007, 40, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Ulloa, L.; Messmer, D. High-mobility group box 1 (HMGB1) protein: Friend and foe. Cytokine Growth Factor Rev. 2006, 17, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Czura, C.J.; Tracey, K.J. The cytokine activity of HMGB1. J. Leukoc. Biol. 2005, 78, 1–8. [Google Scholar] [CrossRef]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Bassi, R.; Giussani, P.; Anelli, V.; Colleoni, T.; Pedrazzi, M.; Patrone, M.; Viani, P.; Sparatore, B.; Melloni, E.; Riboni, L. HMGB1 as an autocrine stimulus in human T98G glioblastoma cells: Role in cell growth and migration. J. Neuro-Oncol. 2007, 87, 23–33. [Google Scholar] [CrossRef]
- Ambrose, A.J.; Chapman, E. Function, Therapeutic Potential, and Inhibition of Hsp70 Chaperones. J. Med. Chem. 2021, 64, 7060–7082. [Google Scholar] [CrossRef]
- Zhao, K.; Zhou, G.; Liu, Y.; Zhang, J.; Chen, Y.; Liu, L.; Zhang, G. HSP70 Family in Cancer: Signaling Mechanisms and Therapeutic Advances. Biomolecules 2023, 13, 601. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Xiao, W.; Jiang, L.; Liu, M.; Shi, Y.; Wang, K.; Wang, H.; Xiao, X. Nuclear Heat Shock Protein 72 as a Negative Regulator of Oxidative Stress (Hydrogen Peroxide)-Induced HMGB1 Cytoplasmic Translocation and Release. J. Immunol. 2007, 178, 7376–7384. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Sharma, D. Evolution of Magnetic Hyperthermia for Glioblastoma Multiforme Therapy. ACS Chem. Neurosci. 2019, 10, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Hermisson, M.; Strik, H.; Rieger, J.; Dichgans, J.; Meyermann, R.; Weller, M. Expression and functional activity of heat shock proteins in human glioblastoma multiforme. Neurology 2000, 54, 1357–1365. [Google Scholar] [CrossRef] [PubMed]
- Iglesia, R.P.; Fernandes, C.F.d.L.; Coelho, B.P.; Prado, M.B.; Escobar, M.I.M.; Almeida, G.H.D.R.; Lopes, M.H. Heat Shock Proteins in Glioblastoma Biology: Where Do We Stand? Int. J. Mol. Sci. 2019, 20, 5794. [Google Scholar] [CrossRef] [PubMed]
- Sha, G.; Jiang, Z.; Zhang, W.; Jiang, C.; Wang, D.; Tang, D. The multifunction of HSP70 in cancer: Guardian or traitor to the survival of tumor cells and the next potential therapeutic target. Int. Immunopharmacol. 2023, 122, 110492. [Google Scholar] [CrossRef]
- Dobrzynska, M.; Napierala, M.; Florek, E. Flavonoid Nanoparticles: A Promising Approach for Cancer Therapy. Biomolecules 2020, 10, 1268. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, H.R.; Patravale, V.B.; Joshi, M.D. Polymeric nanoparticles for targeted treatment in oncology: Current insights. Int. J. Nanomed. 2015, 10, 1001–1018. [Google Scholar]
- Kothamasu, P.; Kanumur, H.; Ravur, N.; Maddu, C.; Parasuramrajam, R.; Thangavel, S. Nanocapsules: The Weapons for Novel Drug Delivery Systems. Bioimpacts 2012, 2, 71–81. [Google Scholar] [CrossRef]
- Jain, S.; Hirst, D.G.; O’Sullivan, J.M. Gold nanoparticles as novel agents for cancer therapy. Br. J. Radiol. 2012, 85, 101–113. [Google Scholar] [CrossRef]
- Mu, H.; Holm, R. Solid lipid nanocarriers in drug delivery: Characterization and design. Expert Opin. Drug Deliv. 2018, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Dierge, E.; Debock, E.; Guilbaud, C.; Corbet, C.; Mignolet, E.; Mignard, L.; Bastien, E.; Dessy, C.; Larondelle, Y.; Feron, O. Peroxidation of n-3 and n-6 polyunsaturated fatty acids in the acidic tumor environment leads to ferroptosis-mediated anticancer effects. Cell Metab. 2021, 33, 1701–1715.e5. [Google Scholar] [CrossRef] [PubMed]
- Yeo, A.T.; Rawal, S.; Delcuze, B.; Christofides, A.; Atayde, A.; Strauss, L.; Balaj, L.; Rogers, V.A.; Uhlmann, E.J.; Varma, H.; et al. Single-cell RNA sequencing reveals evolution of immune landscape during glioblastoma progression. Nat. Immunol. 2022, 23, 971–984. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Mass (g mol−1) | Water Solubility (mg mL−1) | Topological Polar Surface Area (Å2) | Number of H-Bond Donors | Number of H-Bond Acceptors | λmax in Water (nm) | |
|---|---|---|---|---|---|---|
| Fisetin (C15H10O6) | 286.24 | 0.01 | 107 | 4 | 6 | 360 |
| Quercetin (C15H10O7) | 302.23 | 0.06 | 127 | 5 | 7 | 557 |
| Target | ΔG (Kcal mol−1) | Hydrophobic Contacts | Hydrogen Bonds | Number of Contacting Residues | Residues in Common with Known Ligand |
|---|---|---|---|---|---|
| KEAP 1 | −9.1 | 5 | 5 | 10 | 9 |
| HSP72 | −7.9 | 9 | 1 | 10 | 9 |
| HMGB1 | −8.1 | 3 | 1 | 4 | 2 |
| Target | ΔG (Kcal mol−1) | Hydrophobic Contacts | Hydrogen Bonds | Number of Contacting Residues | Residues in Common with Known Ligand |
|---|---|---|---|---|---|
| KEAP 1 | −9.1 | 8 | 7 | 13 | 10 |
| HSP72 | −7.2 | 5 | 2 | 7 | 5 |
| HMGB1 | −6.2 | 6 | 1 | 7 | 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joma, N.; Zhang, I.; Righetto, G.L.; McKay, L.; Gran, E.R.; Kakkar, A.; Maysinger, D. Flavonoids Regulate Redox-Responsive Transcription Factors in Glioblastoma and Microglia. Cells 2023, 12, 2821. https://doi.org/10.3390/cells12242821
Joma N, Zhang I, Righetto GL, McKay L, Gran ER, Kakkar A, Maysinger D. Flavonoids Regulate Redox-Responsive Transcription Factors in Glioblastoma and Microglia. Cells. 2023; 12(24):2821. https://doi.org/10.3390/cells12242821
Chicago/Turabian StyleJoma, Natali, Issan Zhang, Germanna L. Righetto, Laura McKay, Evan Rizzel Gran, Ashok Kakkar, and Dusica Maysinger. 2023. "Flavonoids Regulate Redox-Responsive Transcription Factors in Glioblastoma and Microglia" Cells 12, no. 24: 2821. https://doi.org/10.3390/cells12242821
APA StyleJoma, N., Zhang, I., Righetto, G. L., McKay, L., Gran, E. R., Kakkar, A., & Maysinger, D. (2023). Flavonoids Regulate Redox-Responsive Transcription Factors in Glioblastoma and Microglia. Cells, 12(24), 2821. https://doi.org/10.3390/cells12242821