
UPF1—From mRNA Degradation to Human Disorders

 , , , and
, , , and
Abstract
: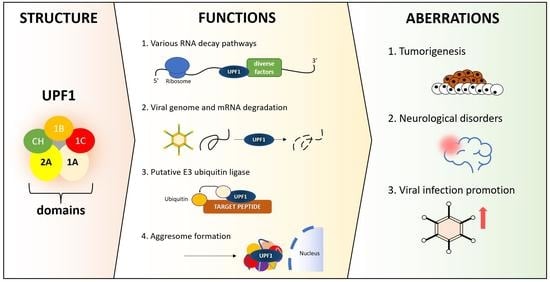
1. Introduction
2. UPF1 Structure
2.1. Helicase Core Structure
2.2. Nucleotide and RNA Binding
2.3. CH-Domain/RING-Like Domain
2.4. Importance of Phosphorylation and Dephosphorylation for UPF1 Function
2.5. eRF1/3 and UPF3 Binding, SURF and Surveillance Complex Formation in UPF1 Activation
3. UPF1 Functional Role and Importance
3.1. Nonsense-Mediated Decay Pathway (NMD)
3.2. Staufen1 (STAU1)-Mediated mRNA Decay (SMD)
3.3. Replication-Dependent Histone mRNA Decay (HMD)
3.4. Structure-Mediated RNA Decay (SRD)
3.5. Regnase-1-Mediated mRNA Decay (RMD)
3.6. Glucocorticoid-Receptor-Mediated mRNA Decay (GMD)
3.7. Tudor-Staphylococcal/Micrococcal-Like Nuclease (TSN)-Mediated MicroRNA Decay (TumiD)
3.8. Viral Targeting of Upf1
3.9. Ubiquitin Ligase Activity
3.10. Aggresome Formation
4. UPF1 in Human Disorders
4.1. UPF1 in Cancer
4.2. UPF1 in Neurological Disorders
4.3. UPF1 in Viral Infections
4.4. UPF1 in Antiprion Systems
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Culbertson, M.R.; Underbrink, K.M.; Fink, G.R. Frameshift suppression Saccharomyces cerevisiae. II. Genetic properties of group II suppressors. Genetics 1980, 95, 833–853. [Google Scholar] [CrossRef] [PubMed]
- Avery, P.; Vicente-Crespo, M.; Francis, D.; Nashchekina, O.; Alonso, C.R.; Palacios, I.M. Drosophila Upf1 and Upf2 loss of function inhibits cell growth and causes animal death in a Upf3-independent manner. RNA 2011, 17, 624–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittkopp, N.; Huntzinger, E.; Weiler, C.; Saulière, J.; Schmidt, S.; Sonawane, M.; Izaurralde, E. Nonsense-mediated mRNA decay effectors are essential for zebrafish embryonic development and survival. Mol. Cell Biol. 2009, 29, 3517–3528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medghalchi, S.M.; Frischmeyer, P.A.; Mendell, J.T.; Kelly, A.G.; Lawler, A.M.; Dietz, H.C. Rent1, a trans-effector of nonsense-mediated mRNA decay, is essential for mammalian embryonic viability. Hum. Mol. Genet. 2001, 10, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Riehs-Kearnan, N.; Gloggnitzer, J.; Dekrout, B.; Jonak, C.; Riha, K. Aberrant growth and lethality of Arabidopsis deficient in nonsense-mediated RNA decay factors is caused by autoimmune-like response. Nucleic Acids Res. 2012, 40, 5615–5624. [Google Scholar] [CrossRef] [Green Version]
- Weng, Y.; Czaplinski, K.; Peltz, S.W. Genetic and biochemical characterization of mutations in the ATPase and helicase regions of the Upf1 protein. Mol. Cell Biol. 1996, 16, 5477–5490. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.; Czaplinski, K.; Trifillis, P.; He, F.; Jacobson, A.; Peltz, S.W. Characterization of the biochemical properties of the human Upf1 gene product that is involved in nonsense-mediated mRNA decay. RNA 2000, 6, 1226–1235. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Muhlrad, D.; Lim, M.K.; Parker, R.; Song, H. Structural and functional insights into the human Upf1 helicase core. EMBO J. 2007, 26, 253–264. [Google Scholar] [CrossRef]
- Conti, E.; Izaurralde, E. Nonsense-mediated mRNA decay: Molecular insights and mechanistic variations across species. Curr. Opin. Cell Biol. 2005, 17, 316–325. [Google Scholar] [CrossRef]
- Gatfield, D.; Unterholzner, L.; Ciccarelli, F.D.; Bork, P.; Izaurralde, E. Nonsense-mediated mRNA decay in Drosophila: At the intersection of the yeast and mammalian pathways. EMBO J. 2003, 22, 3960–3970. [Google Scholar] [CrossRef]
- González, C.I.; Bhattacharya, A.; Wang, W.; Peltz, S.W. Nonsense-mediated mRNA decay in Saccharomyces cerevisiae. Gene 2001, 274, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Maquat, L. Nonsense-Mediated mRNA Decay: A Comparative Analysis of Different Species. Curr. Genom. 2004, 5, 175–190. [Google Scholar] [CrossRef]
- Pulak, R.; Anderson, P. mRNA surveillance by the Caenorhabditis elegans smg genes. Genes Dev. 1993, 7, 1885–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arciga-Reyes, L.; Wootton, L.; Kieffer, M.; Davies, B. UPF1 is required for nonsense-mediated mRNA decay (NMD) and RNAi in Arabidopsis. Plant J. Cell Mol. Biol. 2006, 47, 480–489. [Google Scholar] [CrossRef]
- Kim, Y.K.; Maquat, L.E. UPFront and center in RNA decay: UPF1 in nonsense-mediated mRNA decay and beyond. RNA 2019, 25, 407–422. [Google Scholar] [CrossRef] [Green Version]
- Feng, Q.; Jagannathan, S.; Bradley, R.K. The RNA Surveillance Factor UPF1 Represses Myogenesis via Its E3 Ubiquitin Ligase Activity. Mol. Cell 2017, 67, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Kuroha, K.; Tatematsu, T.; Inada, T. Upf1 stimulates degradation of the product derived from aberrant messenger RNA containing a specific nonsense mutation by the proteasome. EMBO Rep. 2009, 10, 1265–1271. [Google Scholar] [CrossRef] [Green Version]
- Zaepfel, B.L.; Zhang, Z.; Maulding, K.; Coyne, A.N.; Cheng, W.; Hayes, L.R.; Lloyd, T.E.; Sun, S.; Rothstein, J.D. UPF1 reduces C9orf72 HRE-induced neurotoxicity in the absence of nonsense-mediated decay dysfunction. Cell Rep. 2021, 34, 108925. [Google Scholar] [CrossRef]
- Chen, B.-L.; Wang, H.-M.; Lin, X.-S.; Zeng, Y.-M. UPF1: A potential biomarker in human cancers. Front. Biosci. 2021, 26, 76–84. [Google Scholar]
- May, J.P.; Simon, A.E. Targeting of viral RNAs by Upf1-mediated RNA decay pathways. Curr. Opin. Virol. 2021, 47, 1–8. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Jayachandran, U.; Bonneau, F.; Fiorini, F.; Basquin, C.; Domcke, S.; Le Hir, H.; Conti, E. Molecular mechanisms for the RNA-dependent ATPase activity of Upf1 and its regulation by Upf2. Mol. Cell 2011, 41, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Clerici, M.; Mourão, A.; Gutsche, I.; Gehring, N.H.; Hentze, M.W.; Kulozik, A.; Kadlec, J.; Sattler, M.; Cusack, S. Unusual bipartite mode of interaction between the nonsense-mediated decay factors, UPF1 and UPF2. EMBO J. 2009, 28, 2293–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadlec, J.; Guilligay, D.; Ravelli, R.B.; Cusack, S. Crystal structure of the UPF2-interacting domain of nonsense-mediated mRNA decay factor UPF1. RNA 2006, 12, 1817–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, P.V.; Gehring, N.H.; Kunz, J.B.; Hentze, M.W.; Kulozik, A.E. Interactions between UPF1, eRFs, PABP and the exon junction complex suggest an integrated model for mammalian NMD pathways. EMBO J. 2008, 27, 736–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, E.E.; Roy, B.; Amrani, N.; He, F.; Jacobson, A. Yeast Upf1 CH domain interacts with Rps26 of the 40S ribosomal subunit. RNA 2013, 19, 1105–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, S.; Araki, Y.; Ohya, Y.; Sakuno, T.; Hoshino, S.-I.; Kontani, K.; Nishina, H.; Katada, T. Upf1 potentially serves as a RING-related E3 ubiquitin ligase via its association with Upf3 in yeast. RNA 2008, 14, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Kashima, I.; Yamashita, A.; Izumi, N.; Kataoka, N.; Morishita, R.; Hoshino, S.; Ohno, M.; Dreyfuss, G.; Ohno, S. Binding of a novel SMG-1–Upf1–eRF1–eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev. 2006, 20, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, A.; Ohnishi, T.; Kashima, I.; Taya, Y.; Ohno, S. Human SMG-1, a novel phosphatidylinositol 3-kinase-related protein kinase, associates with components of the mRNA surveillance complex and is involved in the regulation of nonsense-mediated mRNA decay. Genes Dev. 2001, 15, 2215–2228. [Google Scholar] [CrossRef] [Green Version]
- Okada-Katsuhata, Y.; Yamashita, A.; Kutsuzawa, K.; Izumi, N.; Hirahara, F.; Ohno, S. N- and C-terminal Upf1 phosphorylations create binding platforms for SMG-6 and SMG-5:SMG-7 during NMD. Nucleic Acids Res. 2012, 40, 1251–1266. [Google Scholar] [CrossRef] [Green Version]
- Isken, O.; Kim, Y.K.; Hosoda, N.; Mayeur, G.L.; Hershey, J.W.B.; Maquat, L.E. Upf1 phosphorylation triggers translational repression during nonsense-mediated mRNA decay. Cell 2008, 133, 314–327. [Google Scholar] [CrossRef] [Green Version]
- Gehring, N.H.; Kunz, J.B.; Neu-Yilik, G.; Breit, S.; Viegas, M.H.; Hentze, M.W.; Kulozik, A.E. Exon-junction complex components specify distinct routes of nonsense-mediated mRNA decay with differential cofactor requirements. Mol. Cell 2005, 20, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, T.; Yamashita, A.; Kashima, I.; Schell, T.; Anders, K.R.; Grimson, A.; Hachiya, T.; Hentze, M.W.; Anderson, P.; Ohno, S. Phosphorylation of hUPF1 induces formation of mRNA surveillance complexes containing hSMG-5 and hSMG-7. Mol. Cell 2003, 12, 1187–1200. [Google Scholar] [CrossRef] [PubMed]
- Melero, R.; Uchiyama, A.; Castaño, R.; Kataoka, N.; Kurosawa, H.; Ohno, S.; Yamashita, A.; Llorca, O. Structures of SMG1-UPFs complexes: SMG1 contributes to regulate UPF2-dependent activation of UPF1 in NMD. Structure 2014, 22, 1105–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czaplinski, K.; Ruiz-Echevarria, M.J.; Paushkin, S.V.; Han, X.; Weng, Y.; Perlick, H.A.; Dietz, H.C.; Ter-Avanesyan, M.D.; Peltz, S.W. The surveillance complex interacts with the translation release factors to enhance termination and degrade aberrant mRNAs. Genes Dev. 1998, 12, 1665–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Applequist, S.E.; Selg, M.; Raman, C.; Jäck, H.M. Cloning and characterization of HUPF1, a human homolog of the Saccharomyces cerevisiae nonsense mRNA-reducing UPF1 protein. Nucleic Acids Res. 1997, 25, 814–821. [Google Scholar] [CrossRef] [Green Version]
- Leeds, P.; Wood, J.M.; Lee, B.S.; Culbertson, M.R. Gene products that promote mRNA turnover in Saccharomyces cerevisiae. Mol. Cell. Biol. 1992, 12, 2165–2177. [Google Scholar]
- Gowravaram, M.; Bonneau, F.; Kanaan, J.; Maciej, V.D.; Fiorini, F.; Raj, S.; Croquette, V.; Le Hir, H.; Chakrabarti, S. A conserved structural element in the RNA helicase UPF1 regulates its catalytic activity in an isoform-specific manner. Nucleic Acids Res. 2018, 46, 2648–2659. [Google Scholar] [CrossRef] [Green Version]
- Fritz, S.E.; Ranganathan, S.; Wang, C.D.; Hogg, J.R. An alternative UPF1 isoform drives conditional remodeling of nonsense-mediated mRNA decay. EMBO J. 2022, 41, e108898. [Google Scholar] [CrossRef]
- Czaplinski, K.; Weng, Y.; Hagan, K.W.; Peltz, S.W. Purification and characterization of the Upf1 protein: A factor involved in translation and mRNA degradation. RNA 1995, 1, 610–623. [Google Scholar]
- Fairman-Williams, M.E.; Guenther, U.-P.; Jankowsky, E. SF1 and SF2 helicases: Family matters. Curr. Opin. Struct. Biol. 2010, 20, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Weng, Y.; Czaplinski, K.; Peltz, S.W. Identification and characterization of mutations in the UPF1 gene that affect nonsense suppression and the formation of the Upf protein complex but not mRNA turnover. Mol. Cell. Biol. 1996, 16, 5491–5506. [Google Scholar] [CrossRef] [Green Version]
- Weng, Y.; Czaplinski, K.; Peltz, S.W. ATP is a cofactor of the Upf1 protein that modulates its translation termination and RNA binding activities. RNA 1998, 4, 205–214. [Google Scholar] [PubMed]
- Chamieh, H.; Ballut, L.; Bonneau, F.; Le Hir, H. NMD factors UPF2 and UPF3 bridge UPF1 to the exon junction complex and stimulate its RNA helicase activity. Nat. Struct. Mol. Biol. 2008, 15, 85–93. [Google Scholar] [CrossRef] [PubMed]
- de Pinto, B.; Lippolis, R.; Castaldo, R.; Altamura, N. Overexpression of Upf1p compensates for mitochondrial splicing deficiency independently of its role in mRNA surveillance. Mol. Microbiol. 2004, 51, 1129–1142. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Huang, S.; Jami-Alahmadi, Y.; McInerney, G.M.; Wohlschlegel, J.A.; Li, M.M.H. Elucidation of TRIM25 ubiquitination targets involved in diverse cellular and antiviral processes. PLoS Pathog. 2022, 18, e1010743. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.V.; Vermeulen, M.; Santamaria, A.; Kumar, C.; Miller, M.L.; Jensen, L.J.; Gnad, F.; Cox, J.; Jensen, T.S.; Nigg, E.A.; et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 2010, 3, ra3. [Google Scholar] [CrossRef]
- Luke, B.; Azzalin, C.M.; Hug, N.; Deplazes, A.; Peter, M.; Lingner, J. Saccharomyces cerevisiae Ebs1p is a putative ortholog of human Smg7 and promotes nonsense-mediated mRNA decay. Nucleic Acids Res. 2007, 35, 7688–7697. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Cajigas, I.J.; Peltz, S.W.; Wilkinson, M.F.; González, C.I. Role for Upf2p phosphorylation in Saccharomyces cerevisiae nonsense-mediated mRNA decay. Mol. Cell. Biol. 2006, 26, 3390–3400. [Google Scholar] [CrossRef] [Green Version]
- Lasalde, C.; Rivera, A.V.; León, A.J.; González-Feliciano, J.A.; Estrella, L.A.; Rodríguez-Cruz, E.N.; Correa, M.E.; Cajigas, I.J.; Bracho, D.P.; Vega, I.E.; et al. Identification and functional analysis of novel phosphorylation sites in the RNA surveillance protein Upf1. Nucleic Acids Res. 2014, 42, 1916–1929. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Funakoshi, Y.; Hoshino, S.-I.; Katada, T. The GTP-binding release factor eRF3 as a key mediator coupling translation termination to mRNA decay. J. Biol. Chem. 2004, 279, 45693–45700. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Czaplinski, K.; Rao, Y.; Peltz, S.W. The role of Upf proteins in modulating the translation read-through of nonsense-containing transcripts. EMBO J. 2001, 20, 880–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosson, B.; Berkova, N.; Couturier, A.; Chabelskaya, S.; Philippe, M.; Zhouravleva, G. Poly(A)-binding protein and eRF3 are associated in vivo in human and Xenopus cells. Biol. Cell 2002, 94, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Neu-Yilik, G.; Raimondeau, E.; Eliseev, B.; Yeramala, L.; Amthor, B.; Deniaud, A.; Huard, K.; Kerschgens, K.; Hentze, M.W.; Schaffitzel, C.; et al. Dual function of UPF3B in early and late translation termination. EMBO J. 2017, 36, 2968–2986. [Google Scholar] [CrossRef] [PubMed]
- Schuller, A.P.; Zinshteyn, B.; Enam, S.U.; Green, R. Directed hydroxyl radical probing reveals Upf1 binding to the 80S ribosomal E site rRNA at the L1 stalk. Nucleic Acids Res. 2018, 46, 2060–2073. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Wilkinson, M. An RNA decay factor wears a new coat: UPF3B modulates translation termination. F1000Research 2017, 6, 2159. [Google Scholar] [CrossRef] [Green Version]
- Melero, R.; Buchwald, G.; Castaño, R.; Raabe, M.; Gil, D.; Lázaro, M.; Urlaub, H.; Conti, E.; Llorca, O. The cryo-EM structure of the UPF-EJC complex shows UPF1 poised toward the RNA 3′ end. Nat. Struct. Mol. Biol. 2012, 19, 498–505. [Google Scholar] [CrossRef] [Green Version]
- Amrani, N.; Ganesan, R.; Kervestin, S.; Mangus, D.A.; Ghosh, S.; Jacobson, A. A faux 3′-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature 2004, 432, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ballut, L.; Barbosa, I.; Le Hir, H. Exon Junction Complexes can have distinct functional flavours to regulate specific splicing events. Sci. Rep. 2018, 8, 9509. [Google Scholar] [CrossRef] [Green Version]
- Popp, M.W.; Maquat, L.E. Nonsense-mediated mRNA Decay and Cancer. Curr. Opin. Genet. Dev. 2018, 48, 44–50. [Google Scholar] [CrossRef]
- Bühler, M.; Steiner, S.; Mohn, F.; Paillusson, A.; Mühlemann, O. EJC-independent degradation of nonsense immunoglobulin-mu mRNA depends on 3’ UTR length. Nat. Struct. Mol. Biol. 2006, 13, 462–464. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Jacobson, A. Nonsense-Mediated mRNA Decay: Degradation of Defective Transcripts Is Only Part of the Story. Annu. Rev. Genet. 2015, 49, 339–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karousis, E.D.; Nasif, S.; Mühlemann, O. Nonsense-mediated mRNA decay: Novel mechanistic insights and biological impact. Wiley Interdiscip. Rev. RNA 2016, 7, 661–682. [Google Scholar] [CrossRef] [Green Version]
- Bhuvanagiri, M.; Schlitter, A.M.; Hentze, M.W.; Kulozik, A.E. NMD: RNA biology meets human genetic medicine. Biochem. J. 2010, 430, 365–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Shum, E.Y.; Karam, R.; Nguyen, L.S.; Gecz, J.; Wilkinson, M.F. NMD-deficient Upf3b-null mice display behavioral and neuropathological defects. FASEB J. 2012, 26, 747-5. [Google Scholar] [CrossRef]
- Nguyen, L.S.; Wilkinson, M.F.; Gecz, J. Nonsense-mediated mRNA decay: Inter-individual variability and human disease. Neurosci. Biobehav. Rev. 2014, 46 Pt 2, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Kurosaki, T.; Popp, M.W.; Maquat, L.E. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol. 2019, 20, 406–420. [Google Scholar] [CrossRef]
- Azzalin, C.M.; Lingner, J. The human RNA surveillance factor UPF1 is required for S phase progression and genome stability. Curr. Biol. 2006, 16, 433–439. [Google Scholar] [CrossRef] [Green Version]
- Nelson, J.O.; Moore, K.A.; Chapin, A.; Hollien, J.; Metzstein, M.M. Degradation of Gadd45 mRNA by nonsense-mediated decay is essential for viability. Elife 2016, 5, e12876. [Google Scholar] [CrossRef]
- Gardner, L.B. Nonsense-mediated RNA decay regulation by cellular stress: Implications for tumorigenesis. Mol. Cancer Res. 2010, 8, 295–308. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Wengrod, J.; Gardner, L.B. Overexpression of the c-myc Oncogene Inhibits Nonsense-mediated RNA Decay in B Lymphocytes. J. Biol. Chem. 2011, 286, 40038–40043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broadus, J.; Fuerstenberg, S.; Doe, C.Q. Staufen-dependent localization of prospero mRNA contributes to neuroblast daughter-cell fate. Nature 1998, 391, 792–795. [Google Scholar] [CrossRef] [PubMed]
- Roegiers, F.; Jan, Y.N. Staufen: A common component of mRNA transport in oocytes and neurons? Trends Cell Biol. 2000, 10, 220–224. [Google Scholar] [CrossRef]
- Park, E.; Maquat, L.E. Staufen-mediated mRNA decay. Wiley Interdiscip. Rev. RNA 2013, 4, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Heraud-Farlow, J.E.; Kiebler, M.A. The multifunctional Staufen proteins: Conserved roles from neurogenesis to synaptic plasticity. Trends Neurosci. 2014, 37, 470–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharangdhar, T.; Sugimoto, Y.; Heraud-Farlow, J.; Fernández-Moya, S.M.; Ehses, J.; de los Mozos, I.R.; Ule, J.; Kiebler, M. A. A retained intron in the 3′- UTR of Calm3 mRNA mediates its Staufen2- and activity-dependent localization to neuronal dendrites. EMBO Rep. 2017, 18, 1762–1774. [Google Scholar] [CrossRef]
- Gong, C.; Kim, Y.K.; Woeller, C.F.; Tang, Y.; Maquat, L.E. SMD and NMD are competitive pathways that contribute to myogenesis: Effects on PAX3 and myogenin mRNAs. Genes Dev. 2009, 23, 54–66. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.J.; Meulemans, D.; Vazquez, L.; Colaco, N.; Schuman, E. A role for a rat homolog of staufen in the transport of RNA to neuronal dendrites. Neuron 2001, 32, 463–475. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Kim, K.M.; Han, S.; Choe, J.; Park, S.G.; Choi, S.S.; Kim, Y.K. Staufen1-mediated mRNA decay functions in adipogenesis. Mol. Cell 2012, 46, 495–506. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.; Maquat, L.E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3’ UTRs via Alu elements. Nature 2011, 470, 284–288. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Dansithong, W.; Figueroa, K.P.; Scoles, D.R.; Pulst, S.M. Staufen1 links RNA stress granules and autophagy in a model of neurodegeneration. Nat. Commun. 2018, 9, 3648. [Google Scholar] [CrossRef] [Green Version]
- Heintz, N.; Sive, H.L.; Roeder, R.G. Regulation of human histone gene expression: Kinetics of accumulation and changes in the rate of synthesis and in the half-lives of individual histone mRNAs during the HeLa cell cycle. Mol. Cell. Biol. 1983, 3, 539–550. [Google Scholar] [PubMed] [Green Version]
- Mei, Q.; Huang, J.; Chen, W.; Tang, J.; Xu, C.; Yu, Q.; Cheng, Y.; Ma, L.; Yu, X.; Li, S. Regulation of DNA replication-coupled histone gene expression. Oncotarget 2017, 8, 95005–95022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzluff, W.F.; Wagner, E.J.; Duronio, R.J. Metabolism and regulation of canonical histone mRNAs: Life without a poly(A) tail. Nat. Rev. Genet. 2008, 9, 843–854. [Google Scholar] [CrossRef] [Green Version]
- Jaeger, S.; Barends, S.; Giegé, R.; Eriani, G.; Martin, F. Expression of metazoan replication-dependent histone genes. Biochimie 2005, 87, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Marzluff, W.F.; Koreski, K.P. Birth and Death of Histone mRNAs. Trends Genet. 2017, 33, 745–759. [Google Scholar] [CrossRef]
- Hoefig, K.P.; Heissmeyer, V. Degradation of oligouridylated histone mRNAs: See UUUUU and goodbye. Wiley Interdiscip. Rev. RNA 2014, 5, 577–589. [Google Scholar] [CrossRef]
- Kaygun, H.; Marzluff, W.F. Regulated degradation of replication-dependent histone mRNAs requires both ATR and Upf1. Nat. Struct. Mol. Biol. 2005, 12, 794–800. [Google Scholar] [CrossRef]
- Mossanen-Parsi, A.; Parisi, D.; Browne-Marke, N.; Bharudin, I.; Connell, S.R.; Mayans, O.; Fucini, P.; Morozov, I.Y.; Caddick, M.X. Histone mRNA is subject to 3′ uridylation and re-adenylation in Aspergillus nidulans. Mol. Microbiol. 2021, 115, 238–254. [Google Scholar] [CrossRef]
- Meaux, S.A.; Holmquist, C.E.; Marzluff, W.F. Role of oligouridylation in normal metabolism and regulated degradation of mammalian histone mRNAs. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20180170. [Google Scholar] [CrossRef] [Green Version]
- Choe, J.; Ahn, S.H.; Kim, Y.K. The mRNP remodeling mediated by UPF1 promotes rapid degradation of replication-dependent histone mRNA. Nucleic Acids Res. 2014, 42, 9334–9349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Wei, X.; Peng, Y. Structure-Mediated Degradation of CircRNAs. Trends Cell Biol. 2020, 30, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, J.-D.; Novoa, E.M.; Vejnar, C.E.; Yartseva, V.; Takacs, C.M.; Kellis, M.; Giraldez, A.J. Analyses of mRNA structure dynamics identify embryonic gene regulatory programs. Nat. Struct. Mol. Biol. 2018, 25, 677–686. [Google Scholar] [CrossRef]
- Su, Z.; Tang, Y.; Ritchey, L.E.; Tack, D.C.; Zhu, M.; Bevilacqua, P.C.; Assmann, S.M. Genome-wide RNA structurome reprogramming by acute heat shock globally regulates mRNA abundance. Proc. Natl. Acad. Sci. USA 2018, 115, 12170–12175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, J.W.; Busa, V.F.; Shao, Y.; Leung, A.K.L. Structure-Mediated RNA Decay by UPF1 and G3BP1. Mol. Cell 2020, 78, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, K.; Takeuchi, O.; Standley, D.M.; Kumagai, Y.; Kawagoe, T.; Miyake, T.; Satoh, T.; Kato, H.; Tsujimura, T.; Nakamura, H.; et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature 2009, 458, 1185–1190. [Google Scholar] [CrossRef]
- Mino, T.; Murakawa, Y.; Fukao, A.; Vandenbon, A.; Wessels, H.-H.; Ori, D.; Uehata, T.; Tartey, S.; Akira, S.; Suzuki, Y.; et al. Regnase-1 and Roquin Regulate a Common Element in Inflammatory mRNAs by Spatiotemporally Distinct Mechanisms. Cell 2015, 161, 1058–1073. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Song, W.; Tromp, G.; Kolattukudy, P.E.; Fu, M. Genome-wide survey and expression profiling of CCCH-zinc finger family reveals a functional module in macrophage activation. PLoS ONE 2008, 3, e2880. [Google Scholar] [CrossRef]
- Xu, J.; Peng, W.; Sun, Y.; Wang, X.; Xu, Y.; Li, X.; Gao, G.; Rao, Z. Structural study of MCPIP1 N-terminal conserved domain reveals a PIN-like RNase. Nucleic Acids Res. 2012, 40, 6957–6965. [Google Scholar] [CrossRef] [Green Version]
- Yokogawa, M.; Tsushima, T.; Noda, N.N.; Kumeta, H.; Enokizono, Y.; Yamashita, K.; Standley, D.M.; Takeuchi, O.; Akira, S.; Inagaki, F. Structural basis for the regulation of enzymatic activity of Regnase-1 by domain-domain interactions. Sci. Rep. 2016, 6, 22324. [Google Scholar] [CrossRef] [Green Version]
- Mino, T.; Iwai, N.; Endo, M.; Inoue, K.; Akaki, K.; Hia, F.; Uehata, T.; Emura, T.; Hidaka, K.; Suzuki, Y. Translation-dependent unwinding of stem-loops by UPF1 licenses Regnase-1 to degrade inflammatory mRNAs. Nucleic Acids Res. 2019, 47, 8838–8859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leppek, K.; Schott, J.; Reitter, S.; Poetz, F.; Hammond, M.C.; Stoecklin, G. Roquin promotes constitutive mRNA decay via a conserved class of stem-loop recognition motifs. Cell 2013, 153, 869–881. [Google Scholar] [CrossRef] [Green Version]
- Nicolaides, N.C.; Galata, Z.; Kino, T.; Chrousos, G.P.; Charmandari, E. The human glucocorticoid receptor: Molecular basis of biologic function. Steroids 2010, 75, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Park, O.H.; Park, J.; Ryu, I.; Kim, J.; Ko, J.; Kim, Y.K. Glucocorticoid receptor interacts with PNRC2 in a ligand-dependent manner to recruit UPF1 for rapid mRNA degradation. Proc. Natl. Acad. Sci. USA 2015, 112, E1540–E1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishmael, F.T.; Fang, X.; Houser, K.R.; Pearce, K.; Abdelmohsen, K.; Zhan, M.; Gorospe, M.; Stellato, C. The human glucocorticoid receptor as an RNA-binding protein: Global analysis of glucocorticoid receptor-associated transcripts and identification of a target RNA motif. J. Immunol. 2011, 186, 1189–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, O.H.; Park, J.; Yu, M.; An, H.-T.; Ko, J.; Kim, Y.K. Identification and molecular characterization of cellular factors required for glucocorticoid receptor-mediated mRNA decay. Genes Dev. 2016, 30, 2093–2105. [Google Scholar] [CrossRef] [Green Version]
- Fabian, M.R.; Duchaine, T.F. MicroRNAs: The Bench and Beyond. Cell 2008, 135, 587–588. [Google Scholar] [CrossRef] [Green Version]
- Gebert, L.F.R.; MacRae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef]
- Elbarbary, R.A.; Miyoshi, K.; Hedaya, O.; Myers, J.R.; Maquat, L.E. UPF1 helicase promotes TSN-mediated miRNA decay. Genes Dev. 2017, 31, 1483–1493. [Google Scholar] [CrossRef] [Green Version]
- Pierouli, K.; Papakonstantinou, E.; Papageorgiou, L.; Diakou, I.; Mitsis, T.; Dragoumani, K.; Spandidos, D.A.; Bacopoulou, F.; Chrousos, G.P.; Goulielmos, G.Ν.; et al. Long non-coding RNAs and microRNAs as regulators of stress in cancer (Review). Mol. Med. Rep. 2022, 26, 361. [Google Scholar] [CrossRef] [PubMed]
- Kirchhof, L.; Fouani, Y.; Knau, A.; Aslan, G.S.; Heumüller, A.W.; Wittig, I.; Müller-McNicoll, M.; Dimmeler, S.; Jaé, N. The G3BP1-UPF1-Associated Long Non-Coding RNA CALA Regulates RNA Turnover in the Cytoplasm. Noncoding RNA 2022, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Imamachi, N.; Salam, K.A.; Suzuki, Y.; Akimitsu, N. A GC-rich sequence feature in the 3’ UTR directs UPF1-dependent mRNA decay in mammalian cells. Genome Res. 2017, 27, 407–418. [Google Scholar] [CrossRef] [Green Version]
- Lavysh, D.; Neu-Yilik, G. UPF1-Mediated RNA Decay-Danse Macabre in a Cloud. Biomolecules 2020, 10, 999. [Google Scholar] [CrossRef] [PubMed]
- Hyde, J.L.; Chen, R.; Trobaugh, D.W.; Diamond, M.S.; Weaver, S.C.; Klimstra, W.B.; Wilusz, J. The5 ′and 3′ ends of alphavirus RNAs--Non-coding is not non-functional. Virus Res. 2015, 206, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Balistreri, G.; Bognanni, C.; Mühlemann, O. Virus Escape and Manipulation of Cellular Nonsense-Mediated mRNA Decay. Viruses 2017, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Popp, M.W.-L.; Cho, H.; Maquat, L.E. Viral subversion of nonsense-mediated mRNA decay. RNA 2020, 26, 1509–1518. [Google Scholar] [CrossRef]
- Kuroha, K.; Ando, K.; Nakagawa, R.; Inada, T. The Upf Factor Complex Interacts with Aberrant Products Derived from mRNAs Containing a Premature Termination Codon and Facilitates Their Proteasomal Degradation. J. Biol. Chem. 2013, 288, 28630–28640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.; Park, J.; Hwang, H.J.; Kim, B.; Jeong, K.; Chang, J.; Lee, J.B.; Kim, Y.K. Nonsense-mediated mRNA decay factor UPF1 promotes aggresome formation. Nat. Commun. 2020, 11, 3106. [Google Scholar] [CrossRef]
- Johnston, J.A.; Ward, C.L.; Kopito, R.R. Aggresomes: A cellular response to misfolded proteins. J. Cell Biol. 1998, 143, 1883–1898. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Park, Y.; Ryu, I.; Choi, M.-H.; Lee, H.J.; Oh, N.; Kim, K.; Kim, K.M.; Choe, J.; Lee, C.; et al. Misfolded polypeptides are selectively recognized and transported toward aggresomes by a CED complex. Nat. Commun. 2017, 8, 15730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lelouard, H.; Ferrand, V.; Marguet, D.; Bania, J.; Camosseto, V.; David, A.; Gatti, E.; Pierre, P. Dendritic cell aggresome-like induced structures are dedicated areas for ubiquitination and storage of newly synthesized defective proteins. J. Cell Biol. 2004, 164, 667–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Gao, J.; Li, M.; Deng, Y.; Jiang, C. p38δ MAPK regulates aggresome biogenesis by phosphorylating SQSTM1 in response to proteasomal stress. J. Cell Sci. 2018, 131, jcs216671. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Park, J.; Kim, Y.K. Crosstalk between translation and the aggresome-autophagy pathway. Autophagy 2018, 14, 1079–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durand, S.; Franks, T.M.; Lykke-Andersen, J. Hyperphosphorylation amplifies UPF1 activity to resolve stalls in nonsense-mediated mRNA decay. Nat. Commun. 2016, 7, 12434. [Google Scholar] [CrossRef] [Green Version]
- Chin, L.-S.; Olzmann, J.A.; Li, L. Parkin-mediated ubiquitin signalling in aggresome formation and autophagy. Biochem. Soc. Trans. 2010, 38, 144–149. [Google Scholar] [CrossRef] [Green Version]
- Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000, 10, 524–530. [Google Scholar] [CrossRef]
- Kurosaki, T.; Imamachi, N.; Pröschel, C.; Mitsutomi, S.; Nagao, R.; Akimitsu, N.; Maquat, L.E. Loss of the fragile X syndrome protein FMRP results in misregulation of nonsense-mediated mRNA decay. Nat. Cell Biol. 2021, 23, 40–48. [Google Scholar] [CrossRef]
- Wickner, R.B.; Edskes, H.K.; Son, M.; Wu, S.; Niznikiewicz, M. Innate immunity to prions: Anti-prion systems turn a tsunami of prions into a slow drip. Curr. Genet. 2021, 67, 833–847. [Google Scholar] [CrossRef]
- Liu, S.; Chen, W.; Hu, H.; Zhang, T.; Wu, T.; Li, X.; Li, Y.; Kong, Q.; Lu, H.; Lu, Z. Long noncoding RNA PVT1 promotes breast cancer proliferation and metastasis by binding miR-128-3p and UPF1. Breast Cancer Res. 2021, 23, 115. [Google Scholar] [CrossRef]
- Zhu, C.; Zhang, L.; Zhao, S.; Dai, W.; Xu, Y.; Zhang, Y.; Zheng, H.; Sheng, W.; Xu, Y. UPF1 promotes chemoresistance to oxaliplatin through regulation of TOP2A activity and maintenance of stemness in colorectal cancer. Cell Death Dis. 2021, 12, 519. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wang, H.; Li, D.; Lin, X.; Ma, Z.; Zeng, Y. Up-frameshift Protein 1 Promotes Tumor Progression by Regulating Apoptosis and Epithelial-Mesenchymal Transition of Colorectal Cancer. Technol. Cancer Res. Treat. 2021, 20, 15330338211064438. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lai, Q.; He, J.; Li, Q.; Ding, J.; Lan, Z.; Gu, C.; Yan, Q.; Fang, Y.; Zhao, X.; et al. LncRNA SNHG6 promotes proliferation, invasion and migration in colorectal cancer cells by activating TGF-β/Smad signaling pathway via targeting UPF1 and inducing EMT via regulation of ZEB1. Int. J. Med. Sci. 2019, 16, 51–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokhari, A.; Jonchere, V.; Lagrange, A.; Bertrand, R.; Svrcek, M.; Marisa, L.; Buhard, O.; Greene, M.; Demidova, A.; Jia, J.; et al. Targeting nonsense-mediated mRNA decay in colorectal cancers with microsatellite instability. Oncogenesis 2018, 7, 70. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Ma, J.; Kong, F.; Song, N.; Wang, C.; Ma, X. UPF1 contributes to the maintenance of endometrial cancer stem cell phenotype by stabilizing LINC00963. Cell Death Dis. 2022, 13, 257. [Google Scholar] [CrossRef]
- Li, L.; Geng, Y.; Feng, R.; Zhu, Q.; Miao, B.; Cao, J.; Fei, S. The Human RNA Surveillance Factor UPF1 Modulates Gastric Cancer Progression by Targeting Long Non-Coding RNA MALAT1. Cell Physiol. Biochem. 2017, 42, 2194–2206. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zhang, G.; Hu, J.; Li, H.; Zhao, J.; Zong, S.; Guo, Z.; Jiang, Y.; Jing, Z. UPF1/circRPPH1/ATF3 feedback loop promotes the malignant phenotype and stemness of GSCs. Cell Death Dis. 2022, 13, 645. [Google Scholar] [CrossRef]
- Lv, Z.-H.; Wang, Z.-Y.; Li, Z.-Y. LncRNA PVT1 aggravates the progression of glioma via downregulating UPF1. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8956–8963. [Google Scholar]
- Chang, L.; Yuan, Y.; Li, C.; Guo, T.; Qi, H.; Xiao, Y.; Dong, X.; Liu, Z.; Liu, Q. Upregulation of SNHG6 regulates ZEB1 expression by competitively binding miR-101-3p and interacting with UPF1 in hepatocellular carcinoma. Cancer Lett. 2016, 383, 183–194. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, Y.; Wang, N.; Li, X.; Zheng, J.; Ge, L. UPF1 inhibits the hepatocellular carcinoma progression by targeting long non-coding RNA UCA1. Sci. Rep. 2019, 9, 6652. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Li, C.; Guo, T.; Wang, H.; Ma, W.; Yuan, Y.; Liu, Q.; Ye, Q.; Liu, Z. The human RNA surveillance factor UPF1 regulates tumorigenesis by targeting Smad7 in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2016, 35, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Plank, T.-D.; Su, F.; Shi, X.; Liu, C.; Ji, Y.; Li, S.; Huynh, A.; Shi, C.; Zhu, B.; et al. The nonsense-mediated RNA decay pathway is disrupted in inflammatory myofibroblastic tumors. J. Clin. Investig. 2016, 126, 3058–3062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, M.; Xu, L.; Suzuki, T.; Yoshikawa, T.; Sakamoto, H.; Uemura, H.; Yoshizawa, A.C.; Suzuki, Y.; Nakatsura, T.; Ishihama, Y.; et al. Aberrant splicing isoforms detected by full-length transcriptome sequencing as transcripts of potential neoantigens in non-small cell lung cancer. Genome Biol. 2021, 22, 9. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Cao, D.; Sha, J.; Zhu, X.; Chen, D. LncRNA ZFPM2-AS1 promotes lung adenocarcinoma progression by interacting with UPF1 to destabilize ZFPM2. Mol. Oncol. 2020, 14, 1074–1088. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Qi, L.; Zhang, L.; Song, W.; Yu, Y.; Xu, C.; Li, L.; Guo, Y.; Yang, L.; Liu, C.; et al. Human nonsense-mediated RNA decay regulates EMT by targeting the TGF-ß signaling pathway in lung adenocarcinoma. Cancer Lett. 2017, 403, 246–259. [Google Scholar] [CrossRef]
- Pei, C.-L.; Fei, K.-L.; Yuan, X.-Y.; Gong, X.-J. LncRNA DANCR aggravates the progression of ovarian cancer by downregulating UPF1. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 10657–10663. [Google Scholar]
- Liu, C.; Karam, R.; Zhou, Y.; Su, F.; Ji, Y.; Li, G.; Xu, G.; Lu, L.; Wang, C.; Song, M.; et al. The UPF1 RNA surveillance gene is commonly mutated in pancreatic adenosquamous carcinoma. Nat. Med. 2014, 20, 596–598. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Wang, Z.; Yang, S.; Lu, Y.; Li, G. The edited UPF1 is correlated with elevated asparagine synthetase in pancreatic ductal adenocarcinomas. Mol. Biol. Rep. 2022, 49, 3713–3720. [Google Scholar] [CrossRef]
- Yang, C.; Ströbel, P.; Marx, A.; Hofmann, I. Plakophilin-associated RNA-binding proteins in prostate cancer and their implications in tumor progression and metastasis. Virchows Arch. 2013, 463, 379–390. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Bai, J.; Qu, Y.; Cao, Y.; Yang, L.; Ge, L.; Jin, Y.; Wang, H.; Song, F. The SMN1 common variant c.22 dupA in Chinese patients causes spinal muscular atrophy by nonsense-mediated mRNA decay in humans. Gene 2018, 644, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Lin, Y.; Zhang, X.; Lan, F.; Zeng, J. Premature termination codons in SMN1 leading to spinal muscular atrophy trigger nonsense-mediated mRNA decay. Clin. Chim. Acta 2022, 530, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Mooney, C.M.; Jimenez-Mateos, E.M.; Engel, T.; Mooney, C.; Diviney, M.; Venø, M.T.; Kjems, J.; Farrell, M.A.; O’Brien, D.F.; Delanty, N.; et al. RNA sequencing of synaptic and cytoplasmic Upf1-bound transcripts supports contribution of nonsense-mediated decay to epileptogenesis. Sci. Rep. 2017, 7, 41517. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Pietzsch, C.; Tsaprailis, G.; Crynen, G.; Cho, K.F.; Ting, A.Y.; Bukreyev, A.; de la Torre, J.C.; Saphire, E.O. Functional interactomes of the Ebola virus polymerase identified by proximity proteomics in the context of viral replication. Cell Rep. 2022, 38, 110544. [Google Scholar] [CrossRef] [PubMed]
- Serquiña, A.K.P.; Das, S.R.; Popova, E.; Ojelabi, O.A.; Roy, C.K.; Göttlinger, H.G. UPF1 is crucial for the infectivity of human immunodeficiency virus type 1 progeny virions. J. Virol. 2013, 87, 8853–8861. [Google Scholar] [CrossRef] [Green Version]
- Son, M.; Wickner, R.B. Nonsense-mediated mRNA decay factors cure most [PSI+] prion variants. Proc. Natl. Acad. Sci. USA 2018, 115, E1184–E1193. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Petricca, J.; Ye, W.; Guan, J.; Zeng, Y.; Cheng, N.; Gong, L.; Shen, S.Y.; Hua, J.T.; Crumbaker, M.; et al. The cell-free DNA methylome captures distinctions between localized and metastatic prostate tumors. Nat. Commun. 2022, 13, 6467. [Google Scholar] [CrossRef]
- Baldi, S.; He, Y.; Ivanov, I.; Khamgan, H.; Safi, M.; Alradhi, M.; Shopit, A.; Al-Danakh, A.; Al-Nusaif, M.; Gao, Y.; et al. Aberrantly hypermethylated ARID1B is a novel biomarker and potential therapeutic target of colon adenocarcinoma. Front. Genet. 2022, 13, 914354. [Google Scholar] [CrossRef]
- Suzuki, A.; Makinoshima, H.; Wakaguri, H.; Esumi, H.; Sugano, S.; Kohno, T.; Tsuchihara, K.; Suzuki, Y. Aberrant transcriptional regulations in cancers: Genome, transcriptome and epigenome analysis of lung adenocarcinoma cell lines. Nucleic Acids Res. 2014, 42, 13557–13572. [Google Scholar] [CrossRef]
- Yamauchi, H.; Nishimura, K.; Yoshimi, A. Aberrant RNA splicing and therapeutic opportunities in cancers. Cancer Sci. 2022, 113, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Cui, S.; Bian, C.; Yu, X. Crosstalk between TGF-β/Smad3 and BMP/BMPR2 signaling pathways via miR-17-92 cluster in carotid artery restenosis. Mol. Cell. Biochem. 2014, 389, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, V.; Sixt, K.M.; Gao, S.; Xu, X.; Huang, J.; Weigert, R.; Zhou, M.; Zhang, Y.E. Direct regulation of alternative splicing by SMAD3 through PCBP1 is essential to the tumor-promoting role of TGF-β. Mol. Cell 2016, 64, 1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaowinn, S.; Kim, J.; Lee, J.; Shin, D.H.; Kang, C.-D.; Kim, D.-K.; Lee, S.; Kang, M.K.; Koh, S.S.; Kim, S.J.; et al. Cancer upregulated gene 2 induces epithelial-mesenchymal transition of human lung cancer cells via TGF-β signaling. Oncotarget 2017, 8, 5092–5110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Sun, Y.; Ji, P.; Kopetz, S.; Zhang, W. Topoisomerase IIα in chromosome instability and personalized cancer therapy. Oncogene 2015, 34, 4019–4031. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Brickner, J.R.; Majid, M.C.; Mosammaparast, N. Crosstalk between ubiquitin and other post-translational modifications on chromatin during double-strand break repair. Trends Cell Biol. 2014, 24, 426–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Guo, D.; Ren, M.; Zhao, Y.; Wang, X.; Chen, Y.; Liu, Y.; Lu, G.; He, S. Long non-coding RNA SNAI3-AS1 promotes the proliferation and metastasis of hepatocellular carcinoma by regulating the UPF1/Smad7 signalling pathway. J. Cell. Mol. Med. 2019, 23, 6271–6282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, Y.; Jiang, X.; Lee, S.T.; Karuturi, R.K.M.; Hooi, S.C.; Yu, Q. FOXQ1 regulates epithelial-mesenchymal transition in human cancers. Cancer Res. 2011, 71, 3076–3086. [Google Scholar] [CrossRef] [Green Version]
- Richter, J.D.; Bassell, G.J.; Klann, E. Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat. Rev. Neurosci. 2015, 16, 595–605. [Google Scholar] [CrossRef] [Green Version]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B., Jr.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X syndrome. Nat. Rev. Dis. Primers. 2017, 3, 17065. [Google Scholar] [CrossRef]
- Alsharafi, W.A.; Xiao, B.; Abuhamed, M.M.; Bi, F.-F.; Luo, Z.-H. Correlation Between IL-10 and microRNA-187 Expression in Epileptic Rat Hippocampus and Patients with Temporal Lobe Epilepsy. Front. Cell. Neurosci. 2015, 9, 466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, L.; Grigoryan, A.; Wang, D.; Wang, J.; Breda, L.; Rivella, S.; Cardozo, T.; Gardner, L.B. Identification and characterization of small molecules that inhibit nonsense-mediated RNA decay and suppress nonsense p53 mutations. Cancer Res. 2014, 74, 3104–3113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alper, T.; Haig, D.A.; Clarke, M.C. The exceptionally small size of the scrapie agent. Biochem. Biophys. Res. Commun. 1966, 22, 278–284. [Google Scholar] [CrossRef]
- Appleby, B.S.; Shetty, S.; Elkasaby, M. Genetic aspects of human prion diseases. Front. Neurol. 2022, 13, 1003056. [Google Scholar] [CrossRef] [PubMed]
- Vorberg, I.M. All the Same? The Secret Life of Prion Strains within Their Target Cells. Viruses 2019, 11, 334. [Google Scholar] [CrossRef] [Green Version]
- Oamen, H.P.; Lau, Y.; Caudron, F. Prion-like proteins as epigenetic devices of stress adaptation. Exp. Cell Res. 2020, 396, 112262. [Google Scholar] [CrossRef] [PubMed]
- Kushnirov, V.V.; Dergalev, A.A.; Alieva, M.K.; Alexandrov, A.I. Structural Bases of Prion Variation in Yeast. Int. J. Mol. Sci. 2022, 23, 5738. [Google Scholar] [CrossRef]
- Ganesan, R.; Mangkalaphiban, K.; Baker, R.; He, F.; Jacobson, A. Ribosome-bound Upf1 forms distinct 80S complexes and conducts mRNA surveillance. RNA 2022, 28, 1621–1642. [Google Scholar] [CrossRef]
- Tan, Y.; Jin, Y.; Wang, S.; Cao, J.; Ren, Z. The RNA surveillance factor UPF1 regulates the migration and adhesion of porcine skeletal muscle satellite cells. J. Muscle Res. Cell Motil. 2021, 42, 203–217. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Aberration | Effect | References |
|---|---|---|---|
| Cancer Types | |||
| bladder cancer | methylation of RISC components, leading to increased UPF1 binding | augmented TumiD, upregulated expression of proinvasive proteins and G1-to-S-phase transition | [109] |
| breast cancer (BC) | UPF1 downregulation by binding with lncRNA PVT1 | EMT, proliferation and metastasis | [130] |
| colorectal cancer (CRC) | UPF1 downregulation (microsatellite instable (MSI) CRC)/upregulation (microsatellite stable (MSS) CRC) | EMT, stemness maintenance and oxaliplatin chemoresistance | [131,132,133,134] |
| endometrial cancer (EC) | UPF1 upregulation | stem cell phenotype, metastasis, relapse, chemoresistance and interaction with lncRNA LINC00963 and miRNA miR-508-5p | [135] |
| gastric cancer (GC) | UPF1 downregulation and promoter hypermethylation | proliferation, cell cycle progression and interactions with lncRNA MALAT1 | [136] |
| glioblastoma multiforme (GBM) | elevated UPF1 transcriptional levels by ATF3 | malignant phenotype, cell stemness and self-renewal | [137] |
| glioma | UPF1 downregulation by binding with lncRNA PVT1 | tumor progression and proliferation | [138] |
| hepatocellular carcinoma (HCC) | UPF1 downregulation and promoter hypermethylation | lower interaction with suppressive lncRNAs—UCA1; SNHG6, migration, proliferation and EMT | [139,140,141] |
| inflammatory myofibroblastic tumor (IMT) | UPF1 downregulation, somatic mutations and aberrant splicing | NMD downregulation, immune infiltration, elevated chemokines and IgE levels—IMT characteristics | [142] |
| nonsmall cell lung cancer (NSCLC) | UPF1 downregulation and splice site mutations | neoantigenic aberrant splicing isoforms of proteins | [143] |
| lung adenocarcinoma (LUAD) | UPF1 downregulation/upregulation | EMT, proliferation, invasion and interactions with lncRNA ZFPM2-AS1 | [144,145] |
| ovarian cancer (OC) | UPF1 downregulation by binding with lncRNA DANCR | metastasis, proliferation and migration | [146] |
| pancreatic adenosquamous carcinoma (PASC) | UPF1 downregulation, genomic point mutations and aberrant splicing | disruption of exonic and intronic splicing enhancers and NMD target accumulation | [147] |
| pancreatic ductal adenocarcinoma (PDAC) | UPF1 mRNA editing | elevated asparagine synthetase (NMD target) and tumor growth caused by asparagine uptake | [148] |
| prostate cancer | UPF1 cytoplasmic localization instead of nuclear | progression, metastasis, proliferation, cell growth and interactions with plakophilins (PKP) 1 and 3 (cell–cell contacts) | [149] |
| Neurological Disorders | |||
| fragile X syndrome (FXS) | UPF1 upregulation through loss of its repressor—fragile X mental retardation protein (FMRP) | FXS phenotype, intellectual disability and autism spectrum disorders, NMD misregulation and molecular abnormalities | [128] |
| amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) | - - - | mitigated neurotoxicity of a G4C2 hexanucleotide repeat expansion in the C9orf72 gene, the most common factor leading to ALS and FTD | [18,150,151] |
| spinal muscular atrophy (SMA) | extensive UPF1 targeting of partially functional mutated SMN1 (survival motor neuron) mRNA with premature stop codon | complete loss of SMN1 leading to haploinsufficiency and neurodegeneration | [152,153] |
| epilepsy | UPF1 upregulation | increased NMD, more frequent seizures and epileptogenesis | [154] |
| Viral Infections | |||
| Ebola | UPF1 hijacked by Ebola genome | promotes viral replication | [155] |
| HIV | UPF1 hijacked by HIV genome | increased infectivity crucial for virion assemble | [156] |
| RNA and DNA viral infections | - - - | viral genomes as targets to UPF1-mediated SMD and SRD due to their policistronic organization and high GC content | [20] |
| Antiprion Systems | |||
| prion infections | - - - | proposed yeast model of antiprion system depending on Upf1 activity for studying human prion infections | [157] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Staszewski, J.; Lazarewicz, N.; Konczak, J.; Migdal, I.; Maciaszczyk-Dziubinska, E. UPF1—From mRNA Degradation to Human Disorders. Cells 2023, 12, 419. https://doi.org/10.3390/cells12030419
Staszewski J, Lazarewicz N, Konczak J, Migdal I, Maciaszczyk-Dziubinska E. UPF1—From mRNA Degradation to Human Disorders. Cells. 2023; 12(3):419. https://doi.org/10.3390/cells12030419
Chicago/Turabian StyleStaszewski, Jacek, Natalia Lazarewicz, Julia Konczak, Iwona Migdal, and Ewa Maciaszczyk-Dziubinska. 2023. "UPF1—From mRNA Degradation to Human Disorders" Cells 12, no. 3: 419. https://doi.org/10.3390/cells12030419
APA StyleStaszewski, J., Lazarewicz, N., Konczak, J., Migdal, I., & Maciaszczyk-Dziubinska, E. (2023). UPF1—From mRNA Degradation to Human Disorders. Cells, 12(3), 419. https://doi.org/10.3390/cells12030419