

Molecular Defense Response of Bursaphelenchus xylophilus to the Nematophagous Fungus Arthrobotrys robusta

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation and RNA Sequencing
2.2. Analysis of Raw Data
2.3. Analysis of Differentially Expressed Gene (DEG)
2.4. Construction of the Weighted Gene Co-Expression Network Analysis (WGCNA)
2.5. Validation of Gene Expression by qRT-PCR
2.6. Data Analysis
3. Results
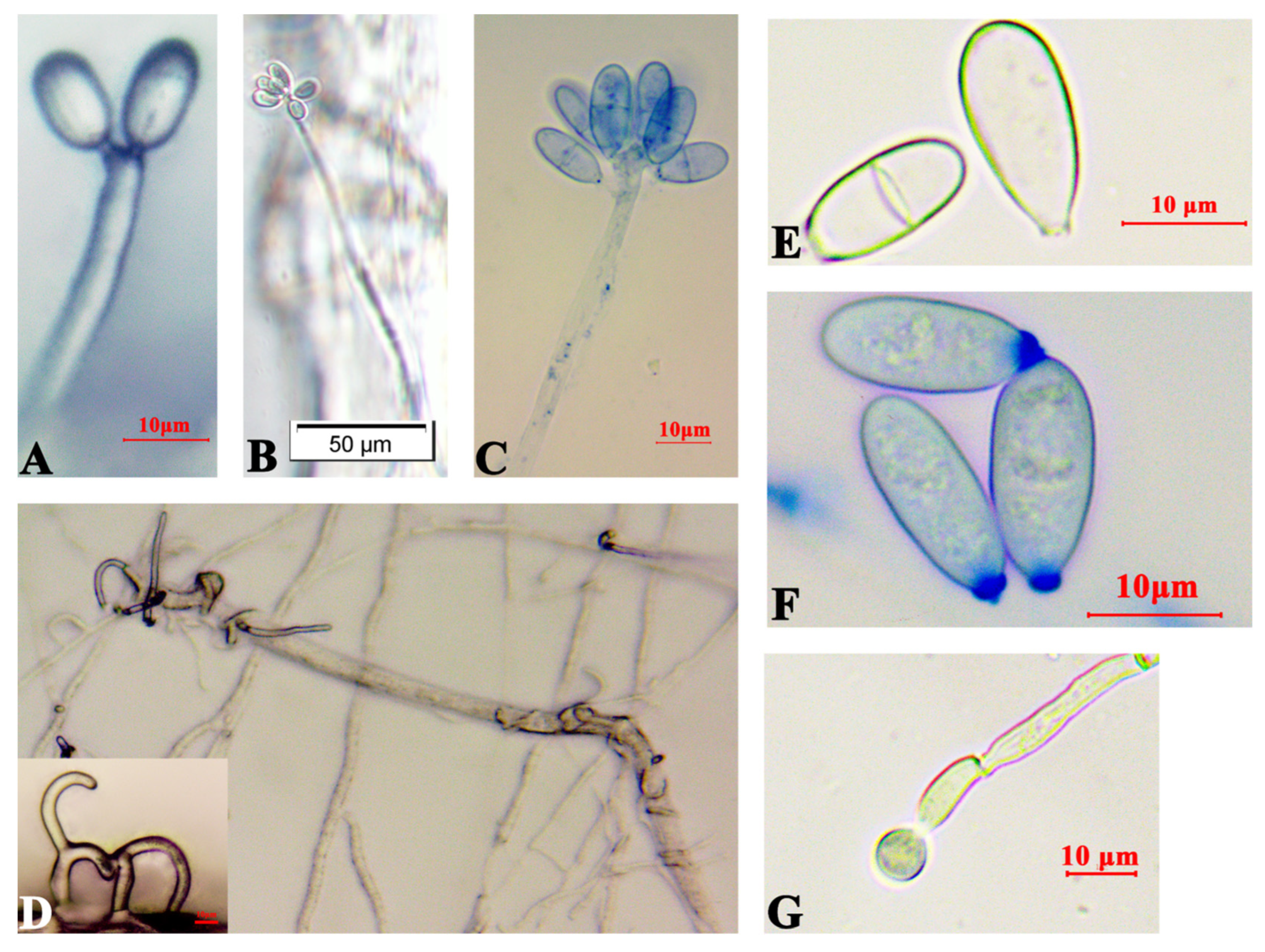
3.1. A. robusta Identification and Interactions with B. xylophilus
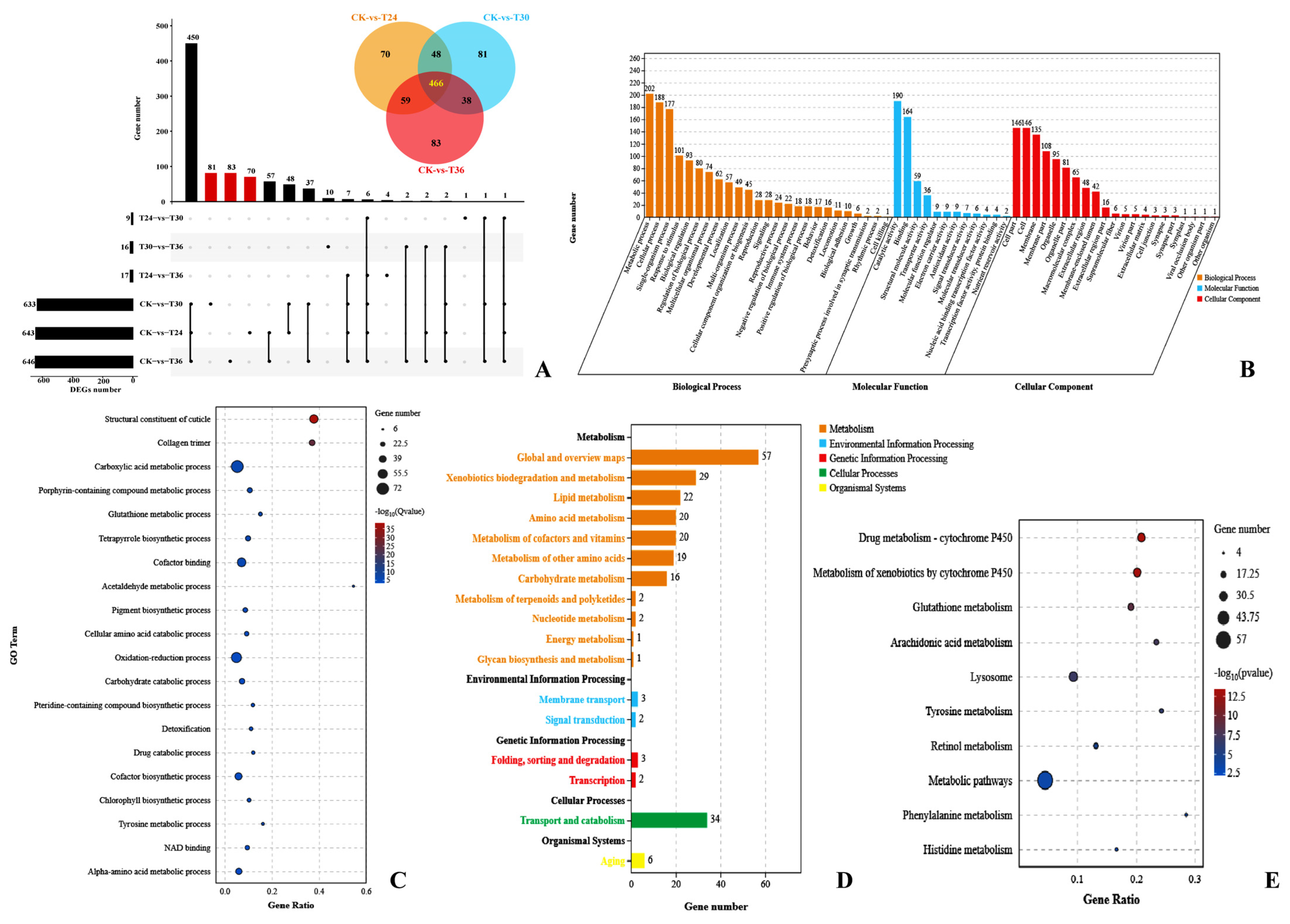
3.2. RNA Sequencing and Gene Expression Patterns
3.3. Gene Set Enrichment Analysis
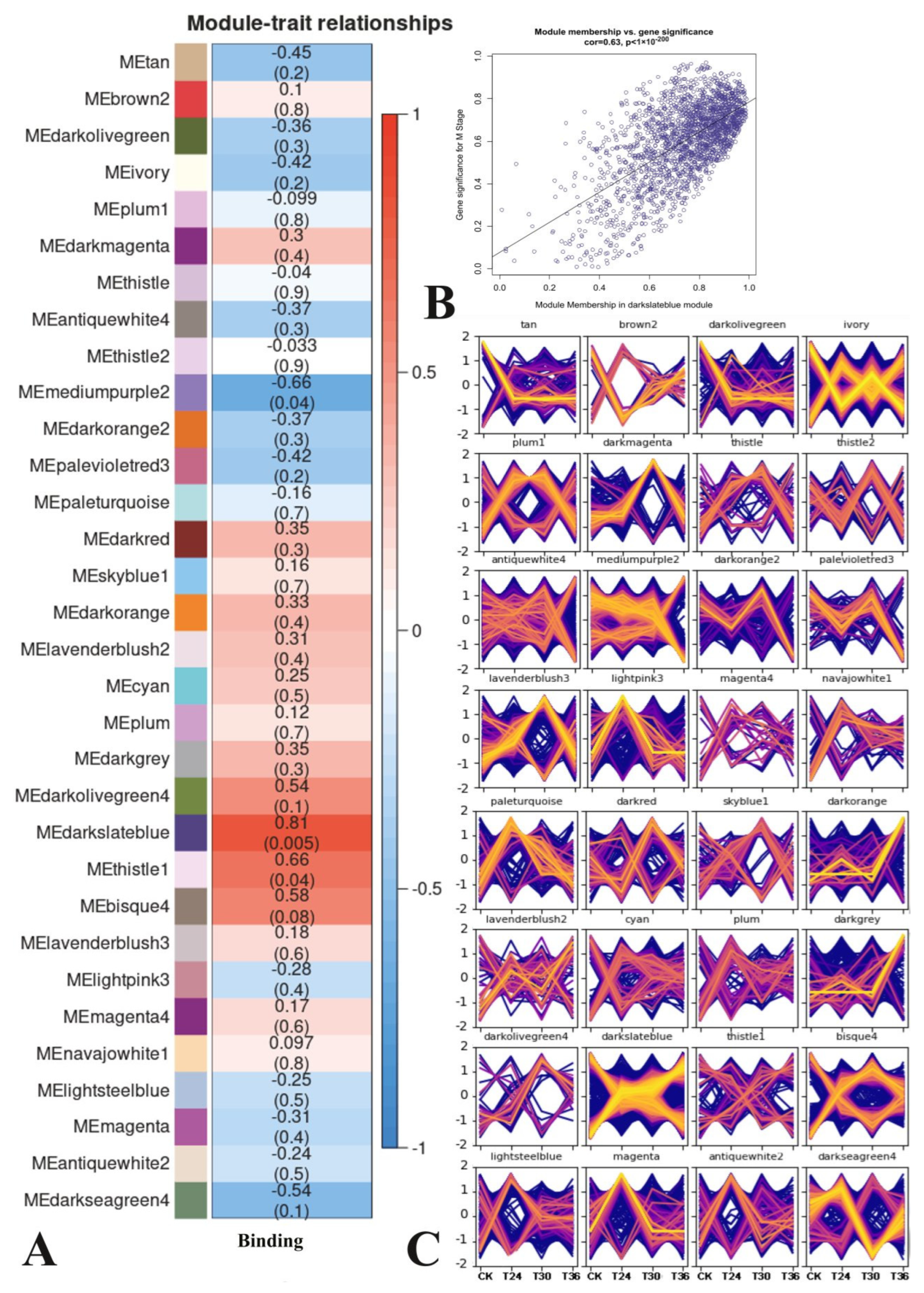
3.4. Weighted Gene Co-Expression Network Analysis
3.5. Analysis of Darkslateblue Module
3.6. Validation of Transcriptome Data by RT-qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Phani, V.; Khan, M.R.; Dutta, T.K. Plant-parasitic nematodes as a potential threat to protected agriculture: Current status and management options. Crop Prot. 2021, 144, 105573. [Google Scholar] [CrossRef]
- Jones, J.T.; Haegeman, A.; Danchin, E.G.J.; Gaur, H.S.; Helder, J.; Jones, M.G.K.; Kikuchi, T.; Manzanilla-Lopez, R.; Palomares-Rius, J.E.; Wesemael, W.M.L.; et al. Top 10 plant-parasitic nematodes in molecular plant pathology. Mol. Plant Pathol. 2013, 14, 946–961. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.; Huang, J.; Li, X.; Sun, H.; Fang, G. A multi-point aggregation trend of the outbreak of pine wilt disease in China over the past 20 years. For. Ecol. Manag. 2022, 505, 119890. [Google Scholar] [CrossRef]
- Li, M.; Li, H.; Ding, X.; Wang, L.; Wang, X.; Chen, F. The Detection of Pine Wilt Disease: A Literature Review. Int. J. Mol. Sci. 2022, 23, 10797. [Google Scholar] [CrossRef] [PubMed]
- Takai, K.; Suzuki, T.; Kawazu, K. Distribution and persistence of emamectin benzoate at efficacious concentrations in pine tissues after injection of a liquid formulation. Pest Manag. Sci. 2004, 60, 42–48. [Google Scholar] [CrossRef]
- Sousa, E.; Naves, P.; Vieira, M. Prevention of pine wilt disease induced by Bursaphelenchus xylophilus and Monochamus galloprovincialis by trunk injection of emamectin benzoate. Phytoparasitica 2013, 41, 143–148. [Google Scholar] [CrossRef]
- Park, M.; Ren, Y.; Lee, B. Preliminary study to evaluate ethanedinitrile (C2N2) for quarantine treatment of four wood destroying pests. Pest Manag. Sci. 2021, 77, 5213–5219. [Google Scholar] [CrossRef]
- Zhang, H.; Wei, Z.; Zhang, J.; Liu, X. Classification of dendrocola nematode-trapping fungi. J. For. Res. 2021, 32, 1295–1304. [Google Scholar] [CrossRef]
- Dou, G.; Yan, D. Research Progress on Biocontrol of Pine Wilt Disease by Microorganisms. Forests 2022, 13, 1047. [Google Scholar] [CrossRef]
- Hsueh, Y.; Mahanti, P.; Schroeder, F.C.; Sternberg, P.W. Nematode-Trapping Fungi Eavesdrop on Nematode Pheromones. Curr. Biol. 2013, 23, 83–86. [Google Scholar] [CrossRef] [Green Version]
- Kassam, R.; Yadav, J.; Chawla, G.; Kundu, A.; Hada, A.; Jaiswal, N.; Bollinedi, H.; Kamil, D.; Devi, P.; Rao, U. Identification, Characterization, and Evaluation of Nematophagous Fungal Species of Arthrobotrys and Tolypocladium for the Management of Meloidogyne incognita. Front. Microbiol. 2021, 12, 790223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Boonmee, S.; Bhat, J.D.; Xiao, W.; Yang, X.-Y. New Arthrobotrys Nematode-Trapping Species (Orbiliaceae) from Terrestrial Soils and Freshwater Sediments in China. J. Fungi 2022, 8, 671. [Google Scholar] [CrossRef]
- Perrine-Walker, F.M. Use of calcofluor white to study the trapping of Pratylenchus coffeae by nematophagous fungi Arthrobotrys musiformis and Arthrobotrys oligospora. Australas. Plant Pathol. 2021, 50, 357–364. [Google Scholar] [CrossRef]
- Vieira, I.S.; Oliveira, I.d.C.; Campos, A.K.; Araujo, J.V.d. In vitrobiological control of bovine parasitic nematodes by Arthrobotrys cladodes, Duddingtonia flagrans and Pochonia chlamydosporiaunder different temperature conditions. J. Helminthol. 2020, 94, E194. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, J.B.; Sanyal, P.K.; Subramanian, R.B. The nematode-trapping efficacy of two chlamydospore-forming fungi against Haemonchus contortus in sheep. J. Helminthol. 2005, 79, 315–319. [Google Scholar] [CrossRef]
- Soliman, M.S.; El-Deriny, M.M.; Ibrahim, D.S.S.; Zakaria, H.; Ahmed, Y. Suppression of root-knot nematode Meloidogyne incognita on tomato plants using the nematode trapping fungus Arthrobotrys oligospora Fresenius. J. Appl. Microbiol. 2021, 131, 2402–2415. [Google Scholar] [CrossRef]
- Braxton, S.M.; Onstad, D.W.; Dockter, D.E.; Giordano, R.; Larsson, R.; Humber, R.A. Description and analysis of two internet-based databases of insect pathogens: EDWIP and VIDIL. J. Invertebr. Pathol. 2003, 83, 185–195. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, Y.; Xu, J.; Zhang, K.-Q.; Zhang, Y. The complete mitochondrial genomes of the nematode-trapping fungus Arthrobotrys oligospora. Mitochondrial DNA Part B-Resour. 2018, 3, 968–969. [Google Scholar] [CrossRef]
- Zhang, Y.-Q.; Yu, Z.-F. The complete mitochondrial genomes of the nematode-trapping fungus Arthrobotrys musiformis. Mitochondrial DNA Part B-Resour. 2019, 4, 979–980. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, G.; Fang, M.; Deng, C.; Zhang, K.-Q.; Yu, Z.; Xu, J. Comparative Analyses of Mitochondrial Genomes Provide Evolutionary Insights Into Nematode-Trapping Fungi. Front. Microbiol. 2020, 11, 617. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Wang, L.; Ji, X.; Feng, Y.; Li, X.; Zou, C.; Xu, J.; Ren, Y.; Mi, Q.; Wu, J.; et al. Genomic and Proteomic Analyses of the Fungus Arthrobotrys oligospora Provide Insights into Nematode-Trap Formation. PLoS Pathog. 2011, 7, e1002179. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.; Vicente, C.S.L.; Menendez, E.; Faria, J.M.S.; Rusinque, L.; Camacho, M.J.; Inacio, M.L. The Fight against Plant-Parasitic Nematodes: Current Status of Bacterial and Fungal Biocontrol Agents. Pathogens 2022, 11, 1178. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.; Vicente, C.S.L.; Inacio, M.L.; Mota, M. The Potential of Esteya spp. for the Biocontrol of the Pinewood Nematode, Bursaphelenchus xylophilus. Microorganisms 2022, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.Y.; Koh, E.; Wong, A.; March, J.C.; Bentley, W.E.; Lee, Y.S.; Chang, M.W. Engineered probiotic Escherichia coli can eliminate and prevent Pseudomonas aeruginosa gut infection in animal models. Nat. Commun. 2017, 8, 15028. [Google Scholar] [CrossRef]
- Zhang, H.; Wei, Z.; Liu, X.; Zhang, J.; Diao, G. Growth and decline of arboreal fungi that prey on Bursaphelenchus xylophilus and their predation rate. J. For. Res. 2022, 33, 699–709. [Google Scholar] [CrossRef]
- Tan, M.; Xue, J.; Wang, L.; Huang, J.; Fu, C.; Yan, X. Transcriptomic Analysis for Different Sex Types of Ricinus communis L. during Development from Apical Buds to Inflorescences by Digital Gene Expression Profiling. Front. Plant Sci. 2016, 6, 1208. [Google Scholar] [CrossRef]
- Yang, Y.; Chou, H.; Crofts, A.J.; Zhang, L.; Tian, L.; Washida, H.; Fukuda, M.; Kumamaru, T.; Oviedo, O.J.; Starkenburg, S.R.; et al. Selective sets of mRNAs localize to extracellular paramural bodies in a rice glup6 mutant. J. Exp. Bot. 2018, 69, 5045–5058. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, W.; Wang, H.; Du, Q.; Fu, Z.; Li, W.; Tang, J. ZmEHD1 Is Required for Kernel Development and Vegetative Growth through Regulating Auxin Homeostasis(1). Plant Physiol. 2020, 182, 1467–1480. [Google Scholar] [CrossRef]
- Kodo, K.; Ong, S.; Jahanbani, F.; Termglinchan, V.; Hirono, K.; InanlooRahatloo, K.; Ebert, A.D.; Shukla, P.; Abilez, O.J.; Churko, J.M.; et al. iPSC-derived cardiomyocytes reveal abnormal TGF-beta signalling in left ventricular non-compaction cardiomyopathy. Nat. Cell Biol. 2016, 18, 1031–1042. [Google Scholar] [CrossRef]
- Kong, K.E.; Hung, T.F.; Man, P.M.; Wong, T.; Cheng, T.; Jin, D. Post-transcriptional negative feedback regulation of proteostasis through the Dis3 ribonuclease and its disruption by polyQ-expanded Huntingtin. Nucleic Acids Res. 2019, 47, 10040–10058. [Google Scholar] [CrossRef]
- Zhou, T.; Zheng, Y.; Sun, L.; Badea, S.R.; Jin, Y.; Liu, Y.; Rolfe, A.J.; Sun, H.; Wang, X.; Cheng, Z.; et al. Microvascular endothelial cells engulf myelin debris and promote macrophage recruitment and fibrosis after neural injury. Nat. Neurosci. 2019, 22, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Biddy, B.A.; Kong, W.; Kamimoto, K.; Guo, C.; Waye, S.E.; Sun, T.; Morris, S.A. Single-cell mapping of lineage and identity in direct reprogramming. Nature 2018, 564, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Onuma, R.; Hirooka, S.; Kanesaki, Y.; Fujiwara, T.; Yoshikawa, H.; Miyagishima, S. Changes in the transcriptome, ploidy, and optimal light intensity of a cryptomonad upon integration into a kleptoplastic dinoflagellate. Isme J. 2020, 14, 2407–2423. [Google Scholar] [CrossRef]
- Ding, N.; Yuan, Z.; Zhang, X.; Chen, J.; Zhou, S.; Deng, Y. Programmable cross-ribosome-binding sites to fine-tune the dynamic range of transcription factor-based biosensor. Nucleic Acids Res. 2020, 48, 10602–10613. [Google Scholar] [CrossRef]
- Sultana, T.; Kim, J.; Lee, S.; Han, H.; Kim, S.; Min, G.-S.; Nadler, S.A.; Park, J. Comparative analysis of complete mitochondrial genome sequences confirms independent origins of plant-parasitic nematodes. BMC Evol. Biol. 2013, 13, 12. [Google Scholar] [CrossRef]
- Zhan, C.; Li, X.; Zhao, Z.; Yang, T.; Wang, X.; Luo, B.; Zhang, Q.; Hu, Y.; Hu, X. Comprehensive Analysis of the Triterpenoid Saponins Biosynthetic Pathway in Anemone flaccida by Transcriptome and Proteome Profiling. Front. Plant Sci. 2016, 7, 1094. [Google Scholar] [CrossRef]
- Hu, G.; Yue, X.; Song, J.; Xing, G.; Chen, J.; Wang, H.; Su, N.; Cui, J. Calcium Positively Mediates Blue Light-Induced Anthocyanin Accumulation in Hypocotyl of Soybean Sprouts. Front. Plant Sci. 2021, 12, 983. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Bonavita, E.; Bromley, C.P.; Jonsson, G.; Pelly, V.S.; Sahoo, S.; Walwyn-Brown, K.; Mensurado, S.; Moeini, A.; Flanagan, E.; Bell, C.R.; et al. Antagonistic Inflammatory Phenotypes Dictate Tumor Fate and Response to Immune Checkpoint Blockade. Immunity 2020, 53, 1215–1229.e8. [Google Scholar] [CrossRef]
- Hou, J.; Ye, X.; Feng, W.; Zhang, Q.; Han, Y.; Liu, Y.; Li, Y.; Wei, Y. Distance correlation application to gene co-expression network analysis. BMC Bioinform. 2022, 23, 81. [Google Scholar] [CrossRef]
- Qin, P.; Lu, H.; Du, H.; Wang, H.; Chen, W.; Chen, Z.; He, Q.; Ou, S.; Zhang, H.; Li, X.; et al. Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations. Cell 2021, 184, 3542–3558.e16. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, Y.; Yang, X.O.; Nurieva, R.I.; Chang, S.H.; Ojeda, S.S.; Kang, H.S.; Schluns, K.S.; Gui, J.; Jetten, A.M.; et al. Transcription of Il17 and Il17f Is Controlled by Conserved Noncoding Sequence 2. Immunity 2012, 36, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Morton, C.O.; Hirsch, P.R.; Kerry, B.R. Infection of plant-parasitic nematodes by nematophagous fungi—A review of the application of molecular biology to understand infection processes and to improve biological control. Nematology 2004, 6, 161–170. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, S.; Li, H.; Wang, R.; Zhang, K.; Xu, J. Fungi-Nematode Interactions: Diversity, Ecology, and Biocontrol Prospects in Agriculture. J. Fungi 2020, 6, 206. [Google Scholar] [CrossRef]
- Braga, F.R.; Carvalho, R.O.; Silva, A.R.; Araujo, J.V.; Frassy, L.N.; Lafisca, A.; Soares, F.E.F. Predatory capability of the nematophagous fungus Arthrobotrys robusta preserved in silica gel on infecting larvae of Haemonchus contortus. Trop. Anim. Health Prod. 2014, 46, 571–574. [Google Scholar] [CrossRef]
- Araujo, J.V.; Santos, M.A.; Ferraz, S.; Maia, A.S. Antagonistic effect of predacious Arthrobotrys fungi on infective Haemonchus placei larvae. J. Helminthol. 1993, 67, 136–138. [Google Scholar] [CrossRef]
- Gonzalez Cruz, M.E.; Mendoza de Gives, P.; Quiroz Romero, H. Comparison of the trapping ability of Arthrobotrys robusta and Monacrosporium gephyropagum on infective larvae of Strongyloides papillosus. J. Helminthol. 1998, 72, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Braga, F.R.; e Silva, A.R.; Araujo, J.M.; Carvalho, R.O.; de Araujo, J.V.; Frassy, L.N. Predatory activity of the nematophagous fungi Duddingtonia flagrans, Monacrosporium thaumasium and Arthrobotrys robusta on Strongyloides stercoralis infective larvae. Rev. Soc. Bras. Med. Trop. 2010, 43, 588–590. [Google Scholar] [CrossRef]
- Braga, F.R.; Araujo, J.M.; Ricardo e Silva, A.; de Araujo, J.V.; Carvalho, R.O.; Tavela, A.d.O.; da Silva, M.E.; Fernandes, F.M.; de Melo, A.L. Destruction of Strongyloides venezuelensis infective larvae by fungi Duddingtonia flagrans, Arthrobotrys robusta and Monacrosporium sinense. Rev. Soc. Bras. Med. Trop. 2011, 44, 389–391. [Google Scholar] [CrossRef] [Green Version]
- Ahamefule, C.S.; Ezeuduji, B.C.; Ogbonna, J.C.; Moneke, A.N.; Ike, A.C.; Jin, C.; Wang, B.; Fang, W. Caenorhabditis elegans as an Infection Model for Pathogenic Mold and Dimorphic Fungi: Applications and Challenges. Front. Cell. Infect. Microbiol. 2021, 11, 1004. [Google Scholar] [CrossRef]
- Kuo, T.; Yang, C.; Chang, H.; Hsueh, Y.; Hsu, C. Nematode-Trapping Fungi Produce Diverse Metabolites during Predator-Prey Interaction. Metabolites 2020, 10, 117. [Google Scholar] [CrossRef]
- Zhu, M.; Li, X.; Zhao, N.; Yang, L.; Zhang, K.; Yang, J. Regulatory Mechanism of Trap Formation in the Nematode-Trapping Fungi. J. Fungi 2022, 8, 406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Dong, X.; Si, J.; Sheng, K.; Wang, J.; Kong, X.; Zha, X.; Wang, Y. The role of WSC domain-containing protein encoding gene AOL_s00043g401 in the growth and nematode trapping of Arthrobotrys oligospora. Arch. Microbiol. 2022, 204, 500. [Google Scholar] [CrossRef]
- Teng, L.; Song, T.; Chen, Y.; Chen, Y.; Zhang, K.; Li, S.; Niu, X. Novel Polyketide-Terpenoid Hybrid Metabolites from a Potent Nematicidal Arthrobotrys oligospora Mutant Delta AOL_s00215g278. J. Agric. Food Chem. 2020, 68, 11449–11458. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, X.; Dai, R.; Ou, X.; Xu, Z.; Zhang, K.; Niu, X. Novel Polyketide-Terpenoid Hybrid Metabolites and Increased Fungal Nematocidal Ability by Disruption of Genes 277 and 279 in Nematode-Trapping Fungus Arthrobotrys oligospora. J. Agric. Food Chem. 2020, 68, 7870–7879. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, Y.; Song, T.; Zeng, Z.; Yan, N.; Zhang, K.; Niu, X. Nematicidal Key Precursors for the Biosynthesis of Morphological Regulatory Arthrosporols in the Nematode-Trapping Fungus Arthrobotrys oligospora. J. Agric. Food Chem. 2016, 64, 7949–7956. [Google Scholar] [CrossRef]
- Mei, X.; Wang, X.; Li, G. Pathogenicity and Volatile Nematicidal Metabolites from Duddingtonia flagrans against Meloidogyne incognita. Microorganisms 2021, 9, 2268. [Google Scholar] [CrossRef]
- Otarigho, B.; Aballay, A. Cholesterol Regulates Innate Immunity via Nuclear Hormone Receptor NHR-8. Iscience 2020, 23, 101068. [Google Scholar] [CrossRef]
- Hartman, J.H.; Widmayer, S.J.; Bergemann, C.M.; King, D.E.; Morton, K.S.; Romersi, R.F.; Jameson, L.E.; Leung, M.C.K.; Andersen, E.C.; Taubert, S.; et al. Xenobiotic metabolism and transport in Caenorhabditis elegans. J. Toxicol. Environ. Health-Part B-Crit. Rev. 2021, 24, 51–94. [Google Scholar] [CrossRef]
- Chen, J.; Hao, X.; Wang, B.; Ma, L. Transcriptomics and coexpression network profiling of the effects of levamisole hydrochloride on Bursaphelenchus xylophilus. Pestic. Biochem. Physiol. 2022, 181, 105019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yu, H.; Lv, Y.; Bushley, K.E.; Wickham, J.D.; Gao, S.; Hu, S.; Zhao, L.; Sun, J. Gene family expansion of pinewood nematode to detoxify its host defence chemicals. Mol. Ecol. 2020, 29, 940–955. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.W.; Lizzappi, M.C.; Magemizoglu, E.; Hur, H.; Liang, Y.; Shaham, S. Nuclear hormone receptors promote gut and glia detoxifying enzyme induction and protect C. elegans from the mold P. brevicompactum. Cell Rep. 2021, 37, 110166. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.M.; Alshagga, M.; Kong, C.; Alshawsh, M.A.; Alshehade, S.A.; Pan, Y. CYP35 family in Caenorhabditis elegans biological processes: Fatty acid synthesis, xenobiotic metabolism, and stress responses. Arch. Toxicol. 2022, 96, 3163–3174. [Google Scholar] [CrossRef]
- Huang, S.; Howington, M.B.; Dobry, C.J.; Evans, C.R.; Leiser, S.F. Flavin-Containing Monooxygenases Are Conserved Regulators of Stress Resistance and Metabolism. Front. Cell Dev. Biol. 2021, 9, 630188. [Google Scholar] [CrossRef]
- Stasiuk, S.J.; MacNevin, G.; Workentine, M.L.; Gray, D.; Redman, E.; Bartley, D.; Morrison, A.; Sharma, N.; Colwell, D.; Ro, D.K.; et al. Similarities and differences in the biotransformation and transcriptomic responses of Caenorhabditis elegans and Haemonchus contortus to five different benzimidazole drugs. Int. J. Parasitol.-Drugs Drug Resist. 2019, 11, 13–29. [Google Scholar] [CrossRef]
- Dimunova, D.; Matouskova, P.; Podlipna, R.; Bousova, I.; Skalova, L. The role of UDP-glycosyltransferases in xenobioticresistance. Drug Metab. Rev. 2022, 54, 282–298. [Google Scholar] [CrossRef] [PubMed]
- Kooliyottil, R.; Rao Gadhachanda, K.; Solo, N.; Dandurand, L.-M. ATP-Binding Cassette (ABC) Transporter Genes in Plant-Parasitic Nematodes: An Opinion for Development of Novel Control Strategy. Front. Plant Sci. 2020, 11, 582424. [Google Scholar] [CrossRef] [PubMed]
- Pechincha, C.; Groessl, S.; Kalis, R.; de Almeida, M.; Zanotti, A.; Wittmann, M.; Schneider, M.; de Campos, R.P.; Rieser, S.; Brandstetter, M.; et al. Lysosomal enzyme trafficking factor LYSET enables nutritional usage of extracellular proteins. Science 2022, 378, eabn5637. [Google Scholar] [CrossRef]
- Richards, C.M.; Jabs, S.; Qiao, W.; Varanese, L.D.; Schweizer, M.; Mosen, P.R.; Riley, N.M.; Klüssendorf, M.; Zengel, J.R.; Flynn, R.A.; et al. The human disease gene LYSET is essential for lysosomal enzyme transport and viral infection. Science 2022, 378, eabn5648. [Google Scholar] [CrossRef]
- O’Donnell, M.P.; Fox, B.W.; Chao, P.-H.; Schroeder, F.C.; Sengupta, P. A neurotransmitter produced by gut bacteria modulates host sensory behaviour. Nature 2020, 583, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Wani, K.A.; Goswamy, D.; Irazoqui, J.E. Nervous system control of intestinal host defense in C. elegans. Curr. Opin. Neurobiol. 2020, 62, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Flavell, S.W. Host-microbe interactions and the behavior of Caenorhabditis elegans. J. Neurogenet. 2020, 34, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Aballay, A. Neural control of behavioral and molecular defenses in C. elegans. Curr. Opin. Neurobiol. 2020, 62, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Meng, Y.; Wang, Y.; Zhang, T.; Yang, G.; Mo, M.; Ji, K.; Liang, L.; Zou, C.; Zhang, K. Survival and infectivity of second-stage root-knot nematode Meloidogyne incognita juveniles depend on lysosome-mediated lipolysis. J. Biol. Chem. 2022, 298, 101637. [Google Scholar] [CrossRef]
- Schmidt, M.F.; Gan, Z.Y.; Komander, D.; Dewson, G. Ubiquitin signalling in neurodegeneration: Mechanisms and therapeutic opportunities. Cell Death Differ. 2021, 28, 570–590. [Google Scholar] [CrossRef]
- Jha, S.; Holmberg, C.I. Tissue-Specific Impact of Autophagy Genes on the Ubiquitin-Proteasome System in C. elegans. Cells 2020, 9, 1858. [Google Scholar] [CrossRef]
- Koltowska, K.; Paterson, S.; Bower, N.I.; Baillie, G.J.; Lagendijk, A.K.; Astin, J.W.; Chen, H.; Francois, M.; Crosier, P.S.; Taft, R.J.; et al. mafba is a downstream transcriptional effector of Vegfc signaling essential for embryonic lymphangiogenesis in zebrafish. Genes Dev. 2015, 29, 1618–1630. [Google Scholar] [CrossRef]
- Bai, X.; Grewal, P.S.; Hogenhout, S.A.; Adams, B.J.; Ciche, T.A.; Gaugler, R.; Sternberg, P.W. Expressed sequence tag analysis of gene representation in insect parasitic nematode Heterorhabditis bacteriophora. J. Parasitol. 2007, 93, 1343–1349. [Google Scholar] [CrossRef]
- Tang, L.T.H.; Trivedi, M.; Freund, J.; Salazar, C.J.; Rahman, M.; Ramirez-Suarez, N.J.; Lee, G.; Wang, Y.; Grant, B.D.; Bulow, H.E. The CATP-8/P5A-type ATPase functions in multiple pathways during neuronal patterning. PLoS Genet. 2021, 17, e1009475. [Google Scholar] [CrossRef]
- Hall, D.H. The role of gap junctions in the C. elegans connectome. Neurosci. Lett. 2019, 695, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Perez-Varela, M.; Corral, J.; Aranda, J.; Barbe, J. Roles of Efflux Pumps from Different Superfamilies in the Surface-Associated Motility and Virulence of Acinetobacter baumannii ATCC 17978. Antimicrob. Agents Chemother. 2019, 63, e02190–e02218. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hao, X.; Tan, R.; Li, Y.; Wang, B.; Pan, J.; Ma, W.; Ma, L. Functional Study on Cytochrome P450 in Response to L(-)-Carvone Stress in Bursaphelenchus xylophilus. Genes 2022, 13, 1956. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, X.; Chen, J.; Li, Y.; Liu, X.; Li, Y.; Wang, B.; Cao, J.; Gu, Y.; Ma, W.; Ma, L. Molecular Defense Response of Bursaphelenchus xylophilus to the Nematophagous Fungus Arthrobotrys robusta. Cells 2023, 12, 543. https://doi.org/10.3390/cells12040543
Hao X, Chen J, Li Y, Liu X, Li Y, Wang B, Cao J, Gu Y, Ma W, Ma L. Molecular Defense Response of Bursaphelenchus xylophilus to the Nematophagous Fungus Arthrobotrys robusta. Cells. 2023; 12(4):543. https://doi.org/10.3390/cells12040543
Chicago/Turabian StyleHao, Xin, Jie Chen, Yongxia Li, Xuefeng Liu, Yang Li, Bowen Wang, Jingxin Cao, Yaru Gu, Wei Ma, and Ling Ma. 2023. "Molecular Defense Response of Bursaphelenchus xylophilus to the Nematophagous Fungus Arthrobotrys robusta" Cells 12, no. 4: 543. https://doi.org/10.3390/cells12040543
APA StyleHao, X., Chen, J., Li, Y., Liu, X., Li, Y., Wang, B., Cao, J., Gu, Y., Ma, W., & Ma, L. (2023). Molecular Defense Response of Bursaphelenchus xylophilus to the Nematophagous Fungus Arthrobotrys robusta. Cells, 12(4), 543. https://doi.org/10.3390/cells12040543