
PGNneo: A Proteogenomics-Based Neoantigen Prediction Pipeline in Noncoding Regions

 ,
,
Abstract
:1. Introduction
2. Methods
2.1. Data Collection
2.2. RNA-Seq Data Processing
2.3. Mutation Annotation and Peptide Extraction
2.4. Database Construction and Peptide Identification
2.5. Neoantigen Prediction and Selection
2.6. PGNneo Pipeline Implementation
3. Results
3.1. The Workflow of the PGNneo Pipeline
3.2. Evaluation of PGNneo Pipeline Results
3.3. Neoantigen Prediction, Selection, and Cross-Comparison from HCC Cohorts
3.4. The Sharing of Noncoding Neoantigens and Genes in Different Samples
3.5. Function Verification Analysis of Frequently Mutated Genes and Neoantigens in HCC
3.6. Extended Application of PGNneo to Other Tumor Types
3.7. Comparing PGNneo with Other Tools
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 2018, 18, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hundal, J.; Kiwala, S.; McMichael, J.; Miller, C.A.; Xia, H.; Wollam, A.T.; Liu, C.J.; Zhao, S.; Feng, Y.Y.; Graubert, A.P.; et al. pVACtools: A Computational Toolkit to Identify and Visualize Cancer Neoantigens. Cancer Immunol. Res. 2020, 8, 409–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenck, R.O.; Lakatos, E.; Gatenbee, C.; Graham, T.A.; Anderson, A.R.A. NeoPredPipe: High-throughput neoantigen prediction and recognition potential pipeline. BMC Bioinform. 2019, 20, 264. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kim, H.S.; Kim, E.; Lee, M.G.; Shin, E.C.; Paik, S.; Kim, S. Neopepsee: Accurate genome-level prediction of neoantigens by harnessing sequence and amino acid immunogenicity information. Ann. Oncol. 2018, 29, 1030–1036. [Google Scholar] [CrossRef]
- Blass, E.; Ott, P.A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 2021, 18, 215–229. [Google Scholar] [CrossRef]
- Müller, M.; Gfeller, D.; Coukos, G.; Bassani-Sternberg, M. ‘Hotspots’ of Antigen Presentation Revealed by Human Leukocyte Antigen Ligandomics for Neoantigen Prioritization. Front. Immunol. 2017, 8, 1367. [Google Scholar] [CrossRef]
- Lei, J.T.; Zhang, B. Proteogenomics drives therapeutic hypothesis generation for precision oncology. Br. J. Cancer 2021, 125, 1–3. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, J.; Wang, X.; Zhu, J.; Liu, Q.; Shi, Z.; Chambers, M.C.; Zimmerman, L.J.; Shaddox, K.F.; Kim, S.; et al. Proteogenomic characterization of human colon and rectal cancer. Nature 2014, 513, 382–387. [Google Scholar] [CrossRef] [Green Version]
- Creech, A.L.; Ting, Y.S.; Goulding, S.P.; Sauld, J.F.K.; Barthelme, D.; Rooney, M.S.; Addona, T.A.; Abelin, J.G. The Role of Mass Spectrometry and Proteogenomics in the Advancement of HLA Epitope Prediction. Proteomics 2018, 18, e1700259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassani-Sternberg, M.; Braunlein, E.; Klar, R.; Engleitner, T.; Sinitcyn, P.; Audehm, S.; Straub, M.; Weber, J.; Slotta-Huspenina, J.; Specht, K.; et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 2016, 7, 13404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Qi, Y.; Zhang, Q.; Liu, W. Application of mass spectrometry-based MHC immunopeptidome profiling in neoantigen identification for tumor immunotherapy. Biomed. Pharmacother. 2019, 120, 109542. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, G.; Tan, X.; Ouyang, J.; Zhang, M.; Song, X.; Liu, Q.; Leng, Q.; Chen, L.; Xie, L. ProGeo-neo: A customized proteogenomic workflow for neoantigen prediction and selection. BMC Med. Genom. 2020, 13, 52. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Zhang, Y.; Jian, X.; Tan, X.; Lu, M.; Ouyang, J.; Liu, Z.; Li, Y.; Xu, L.; Chen, L.; et al. ProGeo-Neo v2.0: A One-Stop Software for Neoantigen Prediction and Filtering Based on the Proteogenomics Strategy. Genes 2022, 13, 783. [Google Scholar] [CrossRef]
- Wen, B.; Li, K.; Zhang, Y.; Zhang, B. Cancer neoantigen prioritization through sensitive and reliable proteogenomics analysis. Nat. Commun. 2020, 11, 1759. [Google Scholar] [CrossRef] [Green Version]
- Khurana, E.; Fu, Y.; Chakravarty, D.; Demichelis, F.; Rubin, M.A.; Gerstein, M. Role of non-coding sequence variants in cancer. Nat. Rev. Genet. 2016, 17, 93–108. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Liao, K.; Xu, J.; Yang, W.; You, X.; Zhong, Q.; Wang, X. The research progress of LncRNA involved in the regulation of inflammatory diseases. Mol. Immunol. 2018, 101, 182–188. [Google Scholar] [CrossRef]
- Ruiz-Orera, J.; Messeguer, X.; Subirana, J.A.; Alba, M.M. Long non-coding RNAs as a source of new peptides. eLife 2014, 3, e03523. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Zhang, J.; Lian, X.; Sun, L.; Meng, K.; Chen, Y.; Sun, Z.; Yin, X.; Li, Y.; Zhao, J.; et al. A hidden human proteome encoded by ‘non-coding’ genes. Nucleic Acids Res. 2019, 47, 8111–8125. [Google Scholar] [CrossRef] [PubMed]
- Laumont, C.M.; Daouda, T.; Laverdure, J.P.; Bonneil, E.; Caron-Lizotte, O.; Hardy, M.P.; Granados, D.P.; Durette, C.; Lemieux, S.; Thibault, P.; et al. Global proteogenomic analysis of human MHC class I-associated peptides derived from non-canonical reading frames. Nat. Commun. 2016, 7, 10238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laumont, C.M.; Perreault, C. Exploiting non-canonical translation to identify new targets for T cell-based cancer immunotherapy. Cell. Mol. Life Sci. 2018, 75, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Ehx, G.; Larouche, J.D.; Durette, C.; Laverdure, J.P.; Hesnard, L.; Vincent, K.; Hardy, M.P.; Thériault, C.; Rulleau, C.; Lanoix, J.; et al. Atypical acute myeloid leukemia-specific transcripts generate shared and immunogenic MHC class-I-associated epitopes. Immunity 2021, 54, 737–752.e710. [Google Scholar] [CrossRef] [PubMed]
- Laumont, C.M.; Vincent, K.; Hesnard, L.; Audemard, E.; Bonneil, E.; Laverdure, J.P.; Gendron, P.; Courcelles, M.; Hardy, M.P.; Cote, C.; et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci. Transl. Med. 2018, 10, 470. [Google Scholar] [CrossRef] [Green Version]
- Xiang, R.; Ma, L.; Yang, M.; Zheng, Z.; Chen, X.; Jia, F.; Xie, F.; Zhou, Y.; Li, F.; Wu, K.; et al. Increased expression of peptides from non-coding genes in cancer proteomics datasets suggests potential tumor neoantigens. Commun. Biol. 2021, 4, 496. [Google Scholar] [CrossRef]
- Hu, B.; Li, H.; Guo, W.; Sun, Y.F.; Zhang, X.; Tang, W.G.; Yang, L.X.; Xu, Y.; Tang, X.Y.; Ding, G.H.; et al. Establishment of a hepatocellular carcinoma patient-derived xenograft platform and its application in biomarker identification. Int. J. Cancer 2020, 146, 1606–1617. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Sun, A.; Zhao, Y.; Ying, W.; Sun, H.; Yang, X.; Xing, B.; Sun, W.; Ren, L.; Hu, B.; et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature 2019, 567, 257–261. [Google Scholar] [CrossRef]
- Cleyle, J.; Hardy, M.P.; Minati, R.; Courcelles, M.; Durette, C.; Lanoix, J.; Laverdure, J.P.; Vincent, K.; Perreault, C.; Thibault, P. Immunopeptidomic analyses of colorectal cancers with and without microsatellite instability. Mol. Cell. Proteom. MCP 2022, 21, 100228. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- “Picard Toolkit” Broad Institute. GitHub Repository. 2019. Available online: https://broadinstitute.github.io/picard/ (accessed on 1 September 2020).
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szolek, A.; Schubert, B.; Mohr, C.; Sturm, M.; Feldhahn, M.; Kohlbacher, O. OptiType: Precision HLA typing from next-generation sequencing data. Bioinformatics 2014, 30, 3310–3316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, J.; Chen, L.; Xiao, Y.; Zhao, Z.; Su, X. Investigations of sequencing data and sample type on HLA class Ia typing with different computational tools. Brief. Bioinform. 2021, 22, bbaa143. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Nesvizhskii, A.I. Proteogenomics: Concepts, applications and computational strategies. Nat. Methods 2014, 11, 1114–1125. [Google Scholar] [CrossRef]
- Zickmann, F.; Renard, B.Y. MSProGene: Integrative proteogenomics beyond six-frames and single nucleotide polymorphisms. Bioinformatics 2015, 31, i106–i115. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Andreatta, M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 2016, 8, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balachandran, V.P.; Luksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGinnis, S.; Madden, T.L. BLAST: At the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004, 32, W20–W25. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Xu, L.; Jian, X.; Tan, X.; Zhao, J.; Liu, Z.; Zhang, Y.; Liu, C.; Chen, L.; Lin, Y.; et al. dbPepNeo2.0: A Database for Human Tumor Neoantigen Peptides From Mass Spectrometry and TCR Recognition. Front. Immunol. 2022, 13, 855976. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Kosaloglu-Yalcin, Z.; Lanka, M.; Frentzen, A.; Logandha Ramamoorthy Premlal, A.; Sidney, J.; Vaughan, K.; Greenbaum, J.; Robbins, P.; Gartner, J.; Sette, A.; et al. Predicting T cell recognition of MHC class I restricted neoepitopes. Oncoimmunology 2018, 7, e1492508. [Google Scholar] [CrossRef] [Green Version]
- Migdal, M.; Ruan, D.F.; Forrest, W.F.; Horowitz, A.; Hammer, C. MiDAS-Meaningful Immunogenetic Data at Scale. PLoS Comput. Biol. 2021, 17, e1009131. [Google Scholar] [CrossRef]
- Rao, C.V.; Asch, A.S.; Yamada, H.Y. Frequently mutated genes/pathways and genomic instability as prevention targets in liver cancer. Carcinogenesis 2017, 38, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Sharpnack, M.F.; Johnson, T.S.; Chalkley, R.; Han, Z.; Carbone, D.; Huang, K.; He, K. TSAFinder: Exhaustive tumor-specific antigen detection with RNAseq. Bioinformatics 2022, 38, 2422–2427. [Google Scholar] [CrossRef]
- Li, L.; Goedegebuure, S.P.; Gillanders, W.E. Preclinical and clinical development of neoantigen vaccines. Ann. Oncol. 2017, 28, xii11–xii17. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Jiang, J.; Zhan, M.; Zhang, H.; Wang, Q.T.; Sun, S.N.; Guo, X.K.; Yin, H.; Wei, Y.; Liu, J.O.; et al. Targeting Neoantigens in Hepatocellular Carcinoma for Immunotherapy: A Futile Strategy? Hepatology 2021, 73, 414–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Ramadori, P.; Pfister, D.; Seehawer, M.; Zender, L.; Heikenwalder, M. The immunological and metabolic landscape in primary and metastatic liver cancer. Nat. Rev. Cancer 2021, 21, 541–557. [Google Scholar] [CrossRef] [PubMed]
- Conde de la Rosa, L.; Garcia-Ruiz, C.; Vallejo, C.; Baulies, A.; Nuñez, S.; Monte, M.J.; Marin, J.J.G.; Baila-Rueda, L.; Cenarro, A.; Civeira, F.; et al. STARD1 promotes NASH-driven HCC by sustaining the generation of bile acids through the alternative mitochondrial pathway. J. Hepatol. 2021, 74, 1429–1441. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, K.; Zou, X.; Hua, Z.; Wang, H.; Bian, W.; Wang, H.; Chen, F.; Dai, T. LncRNA DHRS4-AS1 ameliorates hepatocellular carcinoma by suppressing proliferation and promoting apoptosis via miR-522-3p/SOCS5 axis. Bioengineered 2021, 12, 10862–10877. [Google Scholar] [CrossRef]
- Coudray, A.; Battenhouse, A.M.; Bucher, P.; Iyer, V.R. Detection and benchmarking of somatic mutations in cancer genomes using RNA-seq data. PeerJ 2018, 6, e5362. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Lv, D.; Li, D.; Yin, J.; Ma, Y.; Luo, Y.; Fu, L.; Ding, N.; Li, Y.; Pan, Z.; et al. IEAtlas: An atlas of HLA-presented immune epitopes derived from non-coding regions. Nucleic Acids Res. 2022, 51, D409–D417. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
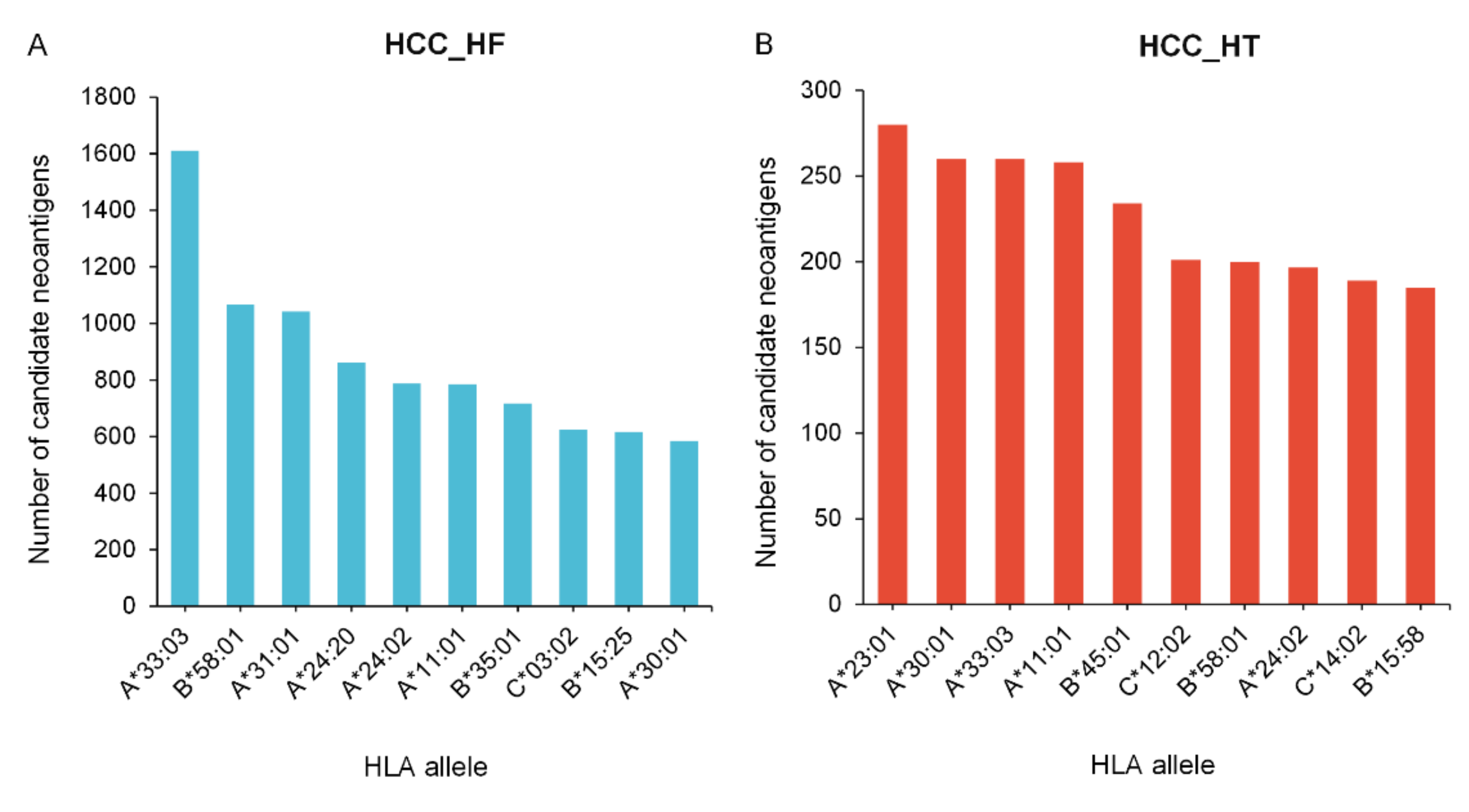{kind=link}
{kind=link}
| Module | Software | Function |
|---|---|---|
| (1) Noncoding somatic variant calling and HLA typing | Trimmomatic-0.39 [30] | Trims adapters and filters low-quality reads |
| BWA-0.7.17 [31] | Sequence alignment | |
| SAMtools(V1.7) [32] | Converts .sam files to .bam, sort, and index files | |
| GATK4.2.0.0 [34] | Call somatic mutation | |
| Picard-2.23.9 [33] | Modifies the headers of .bam files | |
| OptiType-1.3.5 [35] | Predicts HLA typing | |
| (2) Peptide extraction and customized database construction | Annovar [37] | Mutation annotation |
| Bedtools(v2.29.2) [38] | Sources the nucleotide sequence | |
| (3) Variant peptide identification | MaxQuant [47] | Peptide identification |
| (4) Neoantigen prediction and selection | NetMHCpan-4.1 [42] | Calculates the binding affinity of peptides to patient-specific HLA alleles |
| Blast-2.11.0+ [45] | Sequence similarity analysis |
| Gene | Neoantigen | HLA | %Rank | Bind Level |
|---|---|---|---|---|
| WWP1 | VSHDGATAL | HLA-C*03:04 | 0.009 | SB |
| NFE2L2 | KTDAQAISL | HLA-C*04:03 | 0.025 | SB |
| TP53 | TMAGQLLHV | HLA-A*02:06 | 0.052 | SB |
| NFE2L2 | SSRPAWPTR | HLA-A*33:03 | 0.08 | SB |
| KMT2D | QQKNPSLFL | HLA-B*13:02 | 0.126 | SB |
| NFE2L2 | GQHSETPSL | HLA-B*15:01 | 0.154 | SB |
| NFE2L2 | WPGHQFFKY | HLA-B*35:01 | 0.155 | SB |
| KMT2C | IVSSRFCTR | HLA-A*31:01 | 0.167 | SB |
| KMT2D | QQKNPSLFLI | HLA-B*13:02 | 0.185 | SB |
| NFE2L2 | GIWPGHQFF | HLA-B*15:25 | 0.206 | SB |
| NFE2L2 | LFFETRSRF | HLA-A*24:02 | 0.226 | SB |
| ATM | AEAGEPLEP | HLA-B*40:06 | 0.232 | SB |
| WWP1 | YRCVPPHPANF | HLA-C*06:02 | 0.258 | SB |
| KMT2C | KLGDNHFFM | HLA-A*02:01 | 0.293 | SB |
| NFE2L2 | ATRTGRLWWR | HLA-A*31:01 | 0.323 | SB |
| NFE2L2 | HPKSKQISCTW | HLA-B*58:01 | 0.362 | SB |
| NFE2L2 | IWPGHQFF | HLA-A*24:02 | 0.376 | SB |
| KMT2D | QKNPSLFLI | HLA-B*13:02 | 0.379 | SB |
| NFE2L2 | RMPVIQAAW | HLA-A*24:20 | 0.385 | SB |
| NFE2L2 | RMPVIQAAW | HLA-B*58:01 | 0.396 | SB |
| WWP1 | FSCLSLSGGW | HLA-B*58:01 | 0.432 | SB |
| ATM | RACQRQAVGIK | HLA-A*30:01 | 0.433 | SB |
| NFE2L2 | KTDAQAISL | HLA-C*03:04 | 0.442 | SB |
| NFE2L2 | GQHSETPSLLK | HLA-A*11:01 | 0.46 | SB |
| TP53 | ATMAGQLLHV | HLA-A*02:06 | 0.474 | SB |
| WWP1 | VSHDGATAL | HLA-C*04:03 | 0.48 | SB |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, X.; Xu, L.; Jian, X.; Ouyang, J.; Hu, B.; Yang, X.; Wang, T.; Xie, L. PGNneo: A Proteogenomics-Based Neoantigen Prediction Pipeline in Noncoding Regions. Cells 2023, 12, 782. https://doi.org/10.3390/cells12050782
Tan X, Xu L, Jian X, Ouyang J, Hu B, Yang X, Wang T, Xie L. PGNneo: A Proteogenomics-Based Neoantigen Prediction Pipeline in Noncoding Regions. Cells. 2023; 12(5):782. https://doi.org/10.3390/cells12050782
Chicago/Turabian StyleTan, Xiaoxiu, Linfeng Xu, Xingxing Jian, Jian Ouyang, Bo Hu, Xinrong Yang, Tao Wang, and Lu Xie. 2023. "PGNneo: A Proteogenomics-Based Neoantigen Prediction Pipeline in Noncoding Regions" Cells 12, no. 5: 782. https://doi.org/10.3390/cells12050782
APA StyleTan, X., Xu, L., Jian, X., Ouyang, J., Hu, B., Yang, X., Wang, T., & Xie, L. (2023). PGNneo: A Proteogenomics-Based Neoantigen Prediction Pipeline in Noncoding Regions. Cells, 12(5), 782. https://doi.org/10.3390/cells12050782