


Molecular Analysis of the Superior Efficacy of a Dual Epidermal Growth Factor Receptor (EGFR)-DNA-Targeting Combi-Molecule in Comparison with Its Putative Prodrugs 6-Mono-Alkylamino- and 6,6-Dialkylaminoquinazoline in a Human Osteosarcoma Xenograft Model

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Sulforhodamine B (SRB) Growth Inhibition Assay
2.3. In Vivo Experiments
2.4. Effects on 2′-Deoxy-2′-[18F] Fluoro-D-Glucose (18F-FDG) on In Vivo Cellular Metabolisms Using PET/CT Imaging
2.5. Ex Vivo Analysis
2.5.1. Immunostaining
2.5.2. Western Blots
2.5.3. LC-MS Analysis
Standards and Solutions Preparations
Calibration Standards and Quality Control (QC) Sample Preparation
Plasma Sample Preparation
Tumor Sample Preparation
Metabolites Identification with LC-MS
Quantification by LC-MS/MS
2.6. Statistical Analysis
3. Results
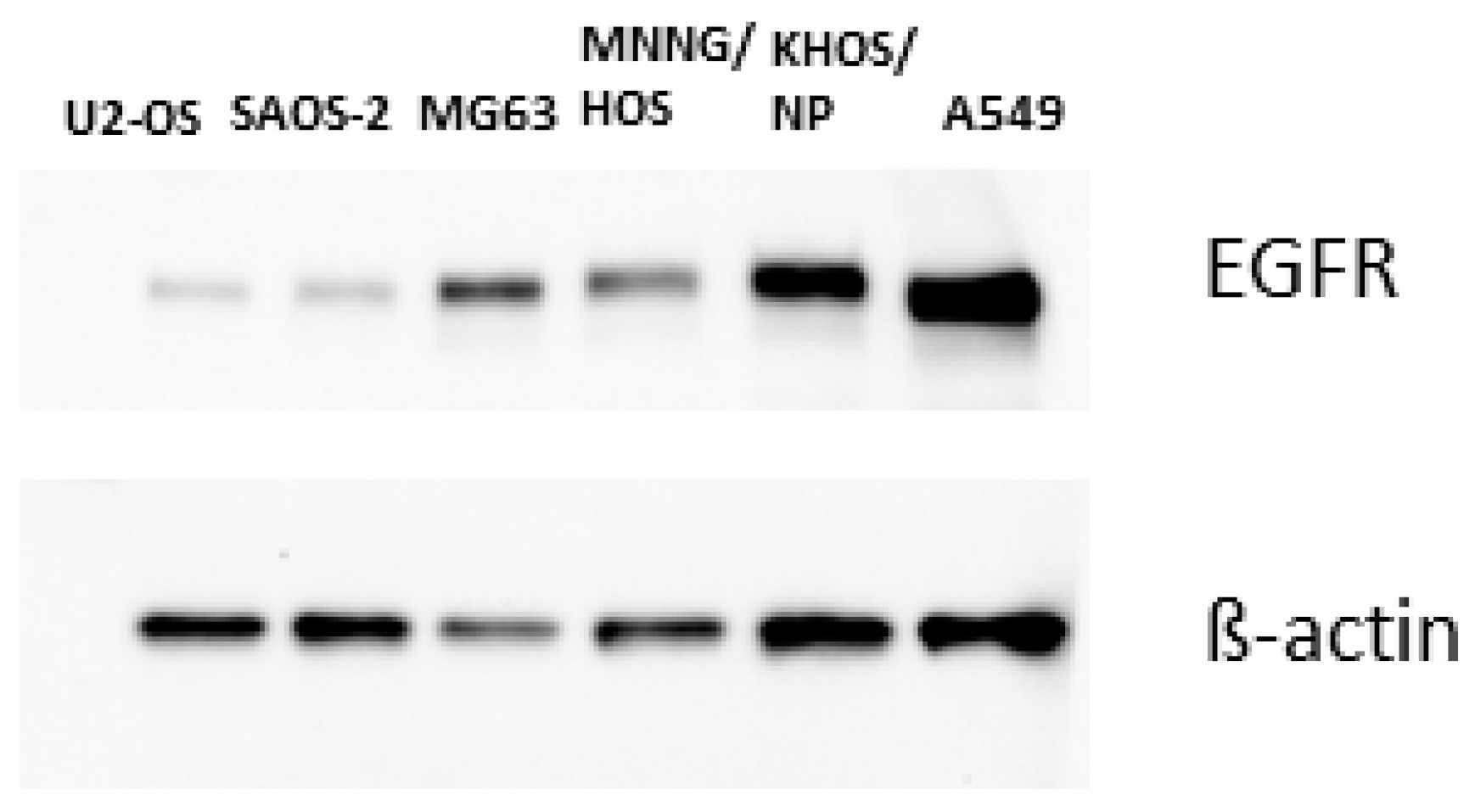
3.1. Growth Inhibition
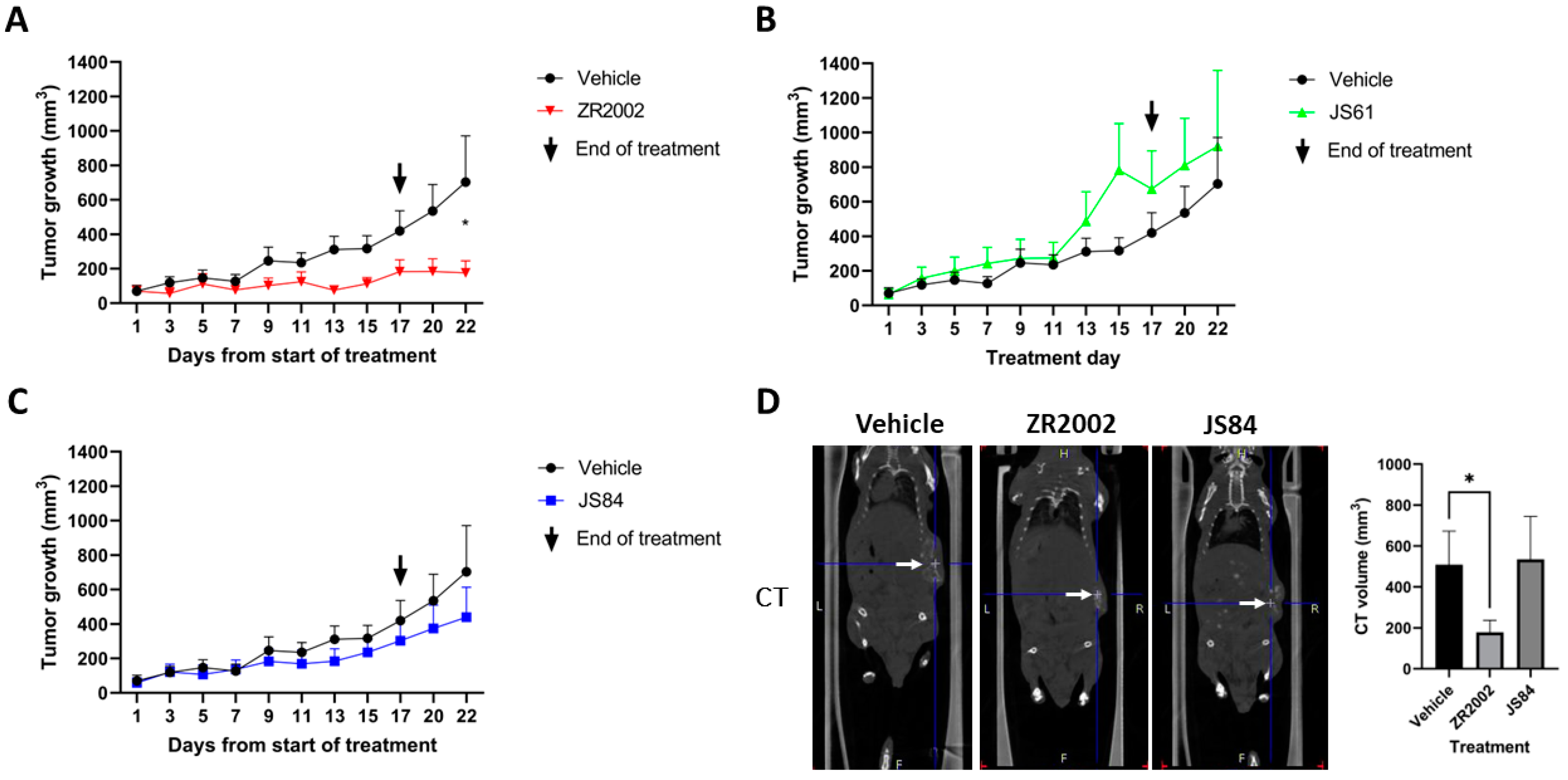
3.2. In Vivo Efficacy
3.3. Pharmacodynamics
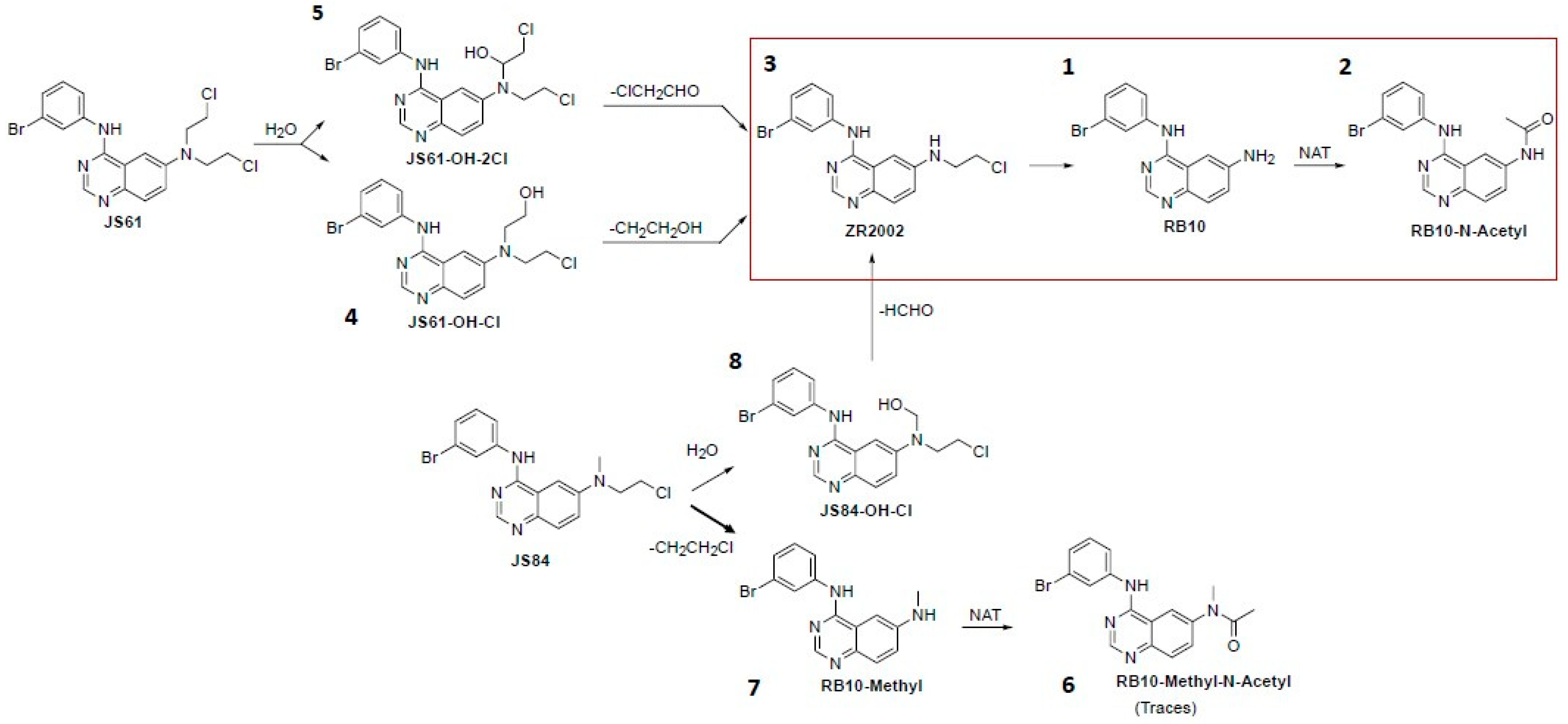
3.4. Pharmacokinetics: Plasma and Tumor Metabolism
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A



References
- Rao, S.; Larroque-Lombard, A.-L.; Peyrard, L.; Thauvin, C.; Rachid, Z.; Williams, C.; Jean-Claude, B.J. Target Modulation by a Kinase Inhibitor Engineered to Induce a Tandem Blockade of the Epidermal Growth Factor Receptor (EGFR) and c-Src: The Concept of Type III Combi-Targeting. PLoS ONE 2015, 10, e0117215. [Google Scholar] [CrossRef] [Green Version]
- Rachid, Z.; MacPhee, M.; Williams, C.; Todorova, M.; Jean-Claude, B.J. Design and Synthesis of New Stabilized Combi-Triazenes for Targeting Solid Tumors Expressing the Epidermal Growth Factor Receptor (EGFR) or Its Closest Homologue HER2. Bioorg. Med. Chem. Lett. 2009, 19, 5505–5509. [Google Scholar] [CrossRef]
- Brahimi, F.; Rachid, Z.; McNamee, J.P.; Alaoui-Jamali, M.A.; Tari, A.M.; Jean-Claude, B.J. Mechanism of Action of a Novel “Combi-Triazene” Engineered to Possess a Polar Functional Group on the Alkylating Moiety: Evidence for Enhancement of Potency. Biochem. Pharmacol. 2005, 70, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Matheson, S.L.; McNamee, J.P.; Wang, T.; Alaoui-Jamali, M.A.; Tari, A.M.; Jean-Claude, B.J. The Combi-Targeting Concept: Dissection of the Binary Mechanism of Action of the Combi-Triazene SMA41 In Vitro and Antitumor Activity In Vivo. J. Pharmacol. Exp. Ther. 2004, 311, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Matheson, S.L.; Brahimi, F.; Jean-Claude, B.J. The Combi-Targeting Concept: Intracellular Fragmentation of the Binary Epidermal Growth Factor (EGFR)/DNA Targeting “Combi-Triazene” SMA41. Biochem. Pharmacol. 2004, 67, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Rachid, Z.; Qiu, Q.; McNamee, J.P.; Tari, A.M.; Jean-Claude, B.J. Sustained Antiproliferative Mechanisms by RB24, a Targeted Precursor of Multiple Inhibitors of Epidermal Growth Factor Receptor and a DNA Alkylating Agent in the A431 Epidermal Carcinoma of the Vulva Cell Line. Br. J. Cancer 2004, 91, 1066–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Q.; Dudouit, F.; Banerjee, R.; McNamee, J.P.; Jean-Claude, B.J. Inhibition of Cell Signaling by the Combi-Nitrosourea FD137 in the Androgen Independent DU145 Prostate Cancer Cell Line. Prostate 2004, 59, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Katsoulas, A.; Rachid, Z.; Brahimi, F.; McNamee, J.; Jean-Claude, B.J. Engineering 3-Alkyltriazenes to Block Bcr-Abl Kinase: A Novel Strategy for the Therapy of Advanced Bcr-Abl Expressing Leukemias. Leuk. Res. 2005, 29, 693–700. [Google Scholar] [CrossRef]
- Banerjee, R.; Huang, Y.; McNamee, J.P.; Todorova, M.; Jean-Claude, B.J. The Combi-Targeting Concept: Selective Targeting of the Epidermal Growth Factor Receptor- and Her2-Expressing Cancer Cells by the Complex Combi-Molecule RB24. J. Pharmacol. Exp. Ther. 2010, 334, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, R.; Huang, Y.; Qiu, Q.; McNamee, J.P.; Belinsky, G.; Jean-Claude, B.J. The Combi-Targeting Concept: Mechanism of Action of the Pleiotropic Combi-Molecule RB24 and Discovery of a Novel Cell Signaling-Based Combination Principle. Cell. Signal. 2011, 23, 630–640. [Google Scholar] [CrossRef]
- Todorova, M.I.; Larroque, A.-L.; Dauphin-Pierre, S.; Fang, Y.-Q.; Jean-Claude, B.J. Subcellular Distribution of a Fluorescence-Labeled Combi-Molecule Designed to Block Epidermal Growth Factor Receptor Tyrosine Kinase and Damage DNA with a Green Fluorescent Species. Mol. Cancer Ther. 2010, 9, 869–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Q.; Domarkas, J.; Banerjee, R.; Merayo, N.; Brahimi, F.; McNamee, J.P.; Gibbs, B.F.; Jean-Claude, B.J. The Combi-Targeting Concept: In Vitro and In Vivo Fragmentation of a Stable Combi-Nitrosourea Engineered to Interact with the Epidermal Growth Factor Receptor While Remaining DNA Reactive. Clin. Cancer Res. 2007, 13, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Q.; Domarkas, J.; Banerjee, R.; Katsoulas, A.; McNamee, J.P.; Jean-Claude, B.J. Type II Combi-Molecules: Design and Binary Targeting Properties of the Novel Triazolinium-Containing Molecules JDD36 and JDE05. Anticancer. Drugs 2007, 18, 171–177. [Google Scholar] [CrossRef]
- Golabi, N.; Brahimi, F.; Huang, Y.; Rachid, Z.; Qiu, Q.; Larroque-Lombard, A.-L.; Jean-Claude, B.J. A Bioanalytical Investigation on the Exquisitely Strong In Vitro Potency of the EGFR-DNA Targeting Type II Combi-Molecule ZR2003 and Its Mitigated In Vivo Antitumour Activity. J. Pharm. Biomed. Anal. 2011, 56, 592–599. [Google Scholar] [CrossRef]
- Brahimi, F.; Rachid, Z.; Qiu, Q.; McNamee, J.P.; Li, Y.-J.; Tari, A.M.; Jean-Claude, B.J. Multiple Mechanisms of Action of ZR2002 in Human Breast Cancer Cells: A Novel Combi-Molecule Designed to Block Signaling Mediated by the ERB Family of Oncogenes and to Damage Genomic DNA. Int. J. Cancer 2004, 112, 484–491. [Google Scholar] [CrossRef]
- Schmitt, J.; Goodfellow, E.; Huang, S.; Williams, C.; Gomes, I.N.F.; Rosa, M.N.; Reis, R.M.; Yang, R.; Titi, H.M.; Jean-Claude, B.J. Comparative Analysis of the Dual EGFR-DNA Targeting and Growth Inhibitory Properties of 6-Mono-Alkylamino- and 6,6-Dialkylaminoquinazoline-Based Type II Combi-Molecules. Eur. J. Med. Chem. 2020, 192, 112185. [Google Scholar] [CrossRef] [PubMed]
- Rachid, Z.; Brahimi, F.; Domarkas, J.; Jean-Claude, B.J. Synthesis of Half-Mustard Combi-Molecules with Fluorescence Properties: Correlation with EGFR Status. Bioorg. Med. Chem. Lett. 2005, 15, 1135–1138. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, Z.; Abdulkarim, B.; Meehan, B.; Rak, J.; Daniel, P.; Schmitt, J.; Lauzon, N.; Eppert, K.; Duncan, H.M.; Petrecca, K.; et al. Mechanisms and Antitumor Activity of a Binary EGFR/DNA–Targeting Strategy Overcomes Resistance of Glioblastoma Stem Cells to Temozolomide. Clin. Cancer Res. 2019, 25, 7594–7608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domarkas, J.; Dudouit, F.; Williams, C.; Qiyu, Q.; Banerjee, R.; Brahimi, F.; Jean-Claude, B.J. The Combi-Targeting Concept: Synthesis of Stable Nitrosoureas Designed to Inhibit the Epidermal Growth Factor Receptor (EGFR). J. Med. Chem. 2006, 49, 3544–3552. [Google Scholar] [CrossRef]
- Wang, S.-L.; Zhong, G.-X.; Wang, X.-W.; Yu, F.-Q.; Weng, D.-F.; Wang, X.-X.; Lin, J.-H. Prognostic Significance of the Expression of HER Family Members in Primary Osteosarcoma. Oncol. Lett. 2018, 16, 2185–2194. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.H.; Koeppen, H.; Garcia, R.; Chiriboga, L.; Tarlow, B.D.; Peters, B.A.; Eigenbrot, C.; Yee, H.; Steiner, G.; Greco, M.A. Epidermal Growth Factor Receptor in Osteosarcoma: Expression and Mutational Analysis. Hum. Pathol. 2007, 38, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Boulytcheva, I.V.; Soloviev, Y.N.; Kushlinskii, N.E.; Mahson, A.N. Expression of Molecular Markers in the Tumor and Survival Prognosis in Osteosarcoma. Bull. Exp. Biol. Med. 2010, 150, 237–242. [Google Scholar] [CrossRef]
- Kersting, C.; Gebert, C.; Agelopoulos, K.; Schmidt, H.; van Diest, P.J.; Juergens, H.; Winkelmann, W.; Kevric, M.; Gosheger, G.; Brandt, B.; et al. Epidermal Growth Factor Receptor Expression in High-Grade Osteosarcomas Is Associated with a Good Clinical Outcome. Clin. Cancer Res. 2007, 13, 2998–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kersting, C.; Agelopoulos, K.; Schmidt, H.; Korsching, E.; August, C.; Gosheger, G.; Dirksen, U.; Juergens, H.; Winkelmann, W.; Brandt, B.; et al. Biological Importance of a Polymorphic CA Sequence within Intron 1 of the Epidermal Growth Factor Receptor Gene (EGFR) in High Grade Central Osteosarcomas. Genes. Chromosomes Cancer 2008, 47, 657–664. [Google Scholar] [CrossRef]
- Hughes, D.P.M.; Thomas, D.G.; Giordano, T.J.; Baker, L.H.; McDonagh, K.T. Cell Surface Expression of Epidermal Growth Factor Receptor and Her-2 with Nuclear Expression of Her-4 in Primary Osteosarcoma. Cancer Res. 2004, 64, 2047–2053. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.J.; Livingston, J.A.; Patel, S.R.; Benjamin, R.S. Chemotherapy for Bone Sarcoma in Adults. J. Oncol. Pract. 2016, 12, 208–216. [Google Scholar] [CrossRef]
- Hassan, S.E.; Bekarev, M.; Kim, M.Y.; Lin, J.; Piperdi, S.; Gorlick, R.; Geller, D.S. Cell Surface Receptor Expression Patterns in Osteosarcoma. Cancer 2012, 118, 740–749. [Google Scholar] [CrossRef] [PubMed]
- De Vita, A.; Recine, F.; Miserocchi, G.; Pieri, F.; Spadazzi, C.; Cocchi, C.; Vanni, S.; Liverani, C.; Farnedi, A.; Fabbri, F.; et al. The Potential Role of the Extracellular Matrix in the Activity of Trabectedin in UPS and L-Sarcoma: Evidences from a Patient-derived Primary Culture Case Series in Tridimensional and Zebrafish Models. J. Exp. Clin. Cancer Res. 2021, 40, 165. [Google Scholar] [CrossRef]
- Lin, Y.; Yang, Y.; Yuan, K.; Yang, S.; Zhang, S.; Li, H.; Tang, T. Multi-Omics Analysis Based on 3D-Bioprinted Models Innovates Therapeutic Target Discovery of Osteosarcoma. Bioact. Mater. 2022, 18, 459–470. [Google Scholar] [CrossRef]
- Lee, H.; Paeng, J.C.; Hong, S.H.; Yoo, H.J.; Cheon, G.J.; Lee, D.S.; Chung, J.-K.; Kang, K.W. Appropriate Margin Thresholds for Isocontour Metabolic Volumetry of Fluorine-18 Fluorodeoxyglucose PET in Sarcoma: A Hybrid PET/MRI Study. Nucl. Med. Commun. 2016, 37, 1088. [Google Scholar] [CrossRef]
- Lee, J.A.; Ko, Y.; Kim, D.H.; Lim, J.S.; Kong, C.-B.; Cho, W.H.; Jeon, D.-G.; Lee, S.-Y.; Koh, J.-S. Epidermal Growth Factor Receptor: Is It a Feasible Target for the Treatment of Osteosarcoma? Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2012, 44, 202–209. [Google Scholar] [CrossRef]
- Sevelda, F.; Mayr, L.; Kubista, B.; Lötsch, D.; van Schoonhoven, S.; Windhager, R.; Pirker, C.; Micksche, M.; Berger, W. EGFR Is Not a Major Driver for Osteosarcoma Cell Growth In Vitro but Contributes to Starvation and Chemotherapy Resistance. J. Exp. Clin. Cancer Res. CR 2015, 34, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldarella, C.; Salsano, M.; Isgrò, M.A.; Treglia, G. The Role of Fluorine-18-Fluorodeoxyglucose Positron Emission Tomography in Assessing the Response to Neoadjuvant Treatment in Patients with Osteosarcoma. Int. J. Mol. Imaging 2012, 2012, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, R.; Rachid, Z.; McNamee, J.; Jean-Claude, B.J. Synthesis of a Prodrug Designed to Release Multiple Inhibitors of the Epidermal Growth Factor Receptor Tyrosine Kinase and an Alkylating Agent: A Novel Tumor Targeting Concept. J. Med. Chem. 2003, 46, 5546–5551. [Google Scholar] [CrossRef] [PubMed]
- Rachid, Z.; Brahimi, F.; Katsoulas, A.; Teoh, N.; Jean-Claude, B.J. The Combi-Targeting Concept: Chemical Dissection of the Dual Targeting Properties of a Series of “Combi-Triazenes”. J. Med. Chem. 2003, 46, 4313–4321. [Google Scholar] [CrossRef]
- Marshall, S.K.; Saelim, B.; Taweesap, M.; Pachana, V.; Panrak, Y.; Makchuchit, N.; Jaroenpakdee, P. Anti-EGFR Targeted Multifunctional I-131 Radio-Nanotherapeutic for Treating Osteosarcoma: In Vitro 3D Tumor Spheroid Model. Nanomaterials 2022, 12, 3517. [Google Scholar] [CrossRef]
- Lim, S.-O.; Li, C.-W.; Xia, W.; Lee, H.-H.; Chang, S.-S.; Shen, J.; Hsu, J.L.; Raftery, D.; Djukovic, D.; Gu, H.; et al. EGFR Signaling Enhances Aerobic Glycolysis in Triple-Negative Breast Cancer Cells to Promote Tumor Growth and Immune Escape. Cancer Res. 2016, 76, 1284–1296. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Ou, Y.; Hao, L. The Roles of Glycolysis in Osteosarcoma. Front. Pharmacol. 2022, 13, 950886. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Cao, Y.; Zheng, Y.; Bu, W.; Zhang, L.; You, M.J.; Koh, M.Y.; Cote, G.; Aldape, K.; et al. EGFR-Induced and PKCε Monoubiquitylation-Dependent NF-ΚB Activation Upregulates PKM2 Expression and Promotes Tumorigenesis. Mol. Cell 2012, 48, 771–784. [Google Scholar] [CrossRef] [Green Version]
- Keremu, A.; Aini, A.; Maimaitirexiati, Y.; Liang, Z.; Aila, P.; Xierela, P.; Tusun, A.; Moming, H.; Yusufu, A. Overcoming Cisplatin Resistance in Osteosarcoma through the MiR-199a-Modulated Inhibition of HIF-1α. Biosci. Rep. 2019, 39, BSR20170080. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Kang, Y.; Chen, L.; Wang, H.; Liu, J.; Zeng, S.; Yu, L. The Drug-Resistance Mechanisms of Five Platinum-Based Antitumor Agents. Front. Pharmacol. 2020, 11, 343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasin, N.F.; Abdul Rashid, M.L.; Ajit Singh, V. Survival Analysis of Osteosarcoma Patients: A 15-Year Experience. J. Orthop. Surg. 2020, 28, 2309499019896662. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) a | ||||
|---|---|---|---|---|---|
| U2-OS | Saos-2 | MG-63 | MNNG/HOS | KHOS/NP | |
| JS61 | 1.39 ± 0.02 | 1.59 ± 0.10 | 12.53 ± 0.61 | 5.86 ± 0.15 | 6.83 ± 0.50 |
| JS84 | 0.18 ± 0.03 | 0.25 ± 0.04 | 5.04 ± 0.98 | 0.88 ± 0.43 | 1.38 ± 0.30 |
| ZR2002 | 0.08 ± 0.01 | 0.08 ± 0.01 | 0.45 ± 0.08 | 0.31 ± 0.09 | 0.19 ± 0.03 |
| Gefitinib | 22.16 ± 4.42 | 20.62 ± 4.20 | 13.40 ± 0.08 | 10.64 ± 0.56 | 13.41 ± 0.45 |
| Chlorambucil | 9.59 ± 0.55 | 7.53 ± 0.75 | 13.05 ± 1.28 | 12.10 ± 1.01 | 9.25 ± 1.35 |
| Gefitinib + chlorambucil b | 3.97 ± 0.84 | 1.96 ± 0.28 | 13.38 ± 4.68 | 10.29 ± 3.21 | 6.60 ± 0.19 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Facchin, C.; Fraga-Timiraos, A.B.; Schmitt, J.; Babaa, N.; Pannu, N.; Aliaga, A.; Larroque, A.-L.; Jean-Claude, B.J. Molecular Analysis of the Superior Efficacy of a Dual Epidermal Growth Factor Receptor (EGFR)-DNA-Targeting Combi-Molecule in Comparison with Its Putative Prodrugs 6-Mono-Alkylamino- and 6,6-Dialkylaminoquinazoline in a Human Osteosarcoma Xenograft Model. Cells 2023, 12, 914. https://doi.org/10.3390/cells12060914
Facchin C, Fraga-Timiraos AB, Schmitt J, Babaa N, Pannu N, Aliaga A, Larroque A-L, Jean-Claude BJ. Molecular Analysis of the Superior Efficacy of a Dual Epidermal Growth Factor Receptor (EGFR)-DNA-Targeting Combi-Molecule in Comparison with Its Putative Prodrugs 6-Mono-Alkylamino- and 6,6-Dialkylaminoquinazoline in a Human Osteosarcoma Xenograft Model. Cells. 2023; 12(6):914. https://doi.org/10.3390/cells12060914
Chicago/Turabian StyleFacchin, Caterina, Ana B. Fraga-Timiraos, Julie Schmitt, Nadia Babaa, Naveet Pannu, Antonio Aliaga, Anne-Laure Larroque, and Bertrand J. Jean-Claude. 2023. "Molecular Analysis of the Superior Efficacy of a Dual Epidermal Growth Factor Receptor (EGFR)-DNA-Targeting Combi-Molecule in Comparison with Its Putative Prodrugs 6-Mono-Alkylamino- and 6,6-Dialkylaminoquinazoline in a Human Osteosarcoma Xenograft Model" Cells 12, no. 6: 914. https://doi.org/10.3390/cells12060914
APA StyleFacchin, C., Fraga-Timiraos, A. B., Schmitt, J., Babaa, N., Pannu, N., Aliaga, A., Larroque, A. -L., & Jean-Claude, B. J. (2023). Molecular Analysis of the Superior Efficacy of a Dual Epidermal Growth Factor Receptor (EGFR)-DNA-Targeting Combi-Molecule in Comparison with Its Putative Prodrugs 6-Mono-Alkylamino- and 6,6-Dialkylaminoquinazoline in a Human Osteosarcoma Xenograft Model. Cells, 12(6), 914. https://doi.org/10.3390/cells12060914