
Liver X Receptor Activation Attenuates Oxysterol-Induced Inflammatory Responses in Fetoplacental Endothelial Cells

 , , , , and
, , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Isolation and Culture of Primary Human Fetoplacental Endothelial Cells
2.3. RNA Isolation, Reverse Transcription and Real-Time Quantitative PCR (RT-qPCR)
2.4. Immunoblotting
2.5. Human Cytokine Multiplex Assay
2.6. Flow Cytometry
2.7. LDH Cytotoxicity Assay
2.8. Statistical Analysis
3. Results
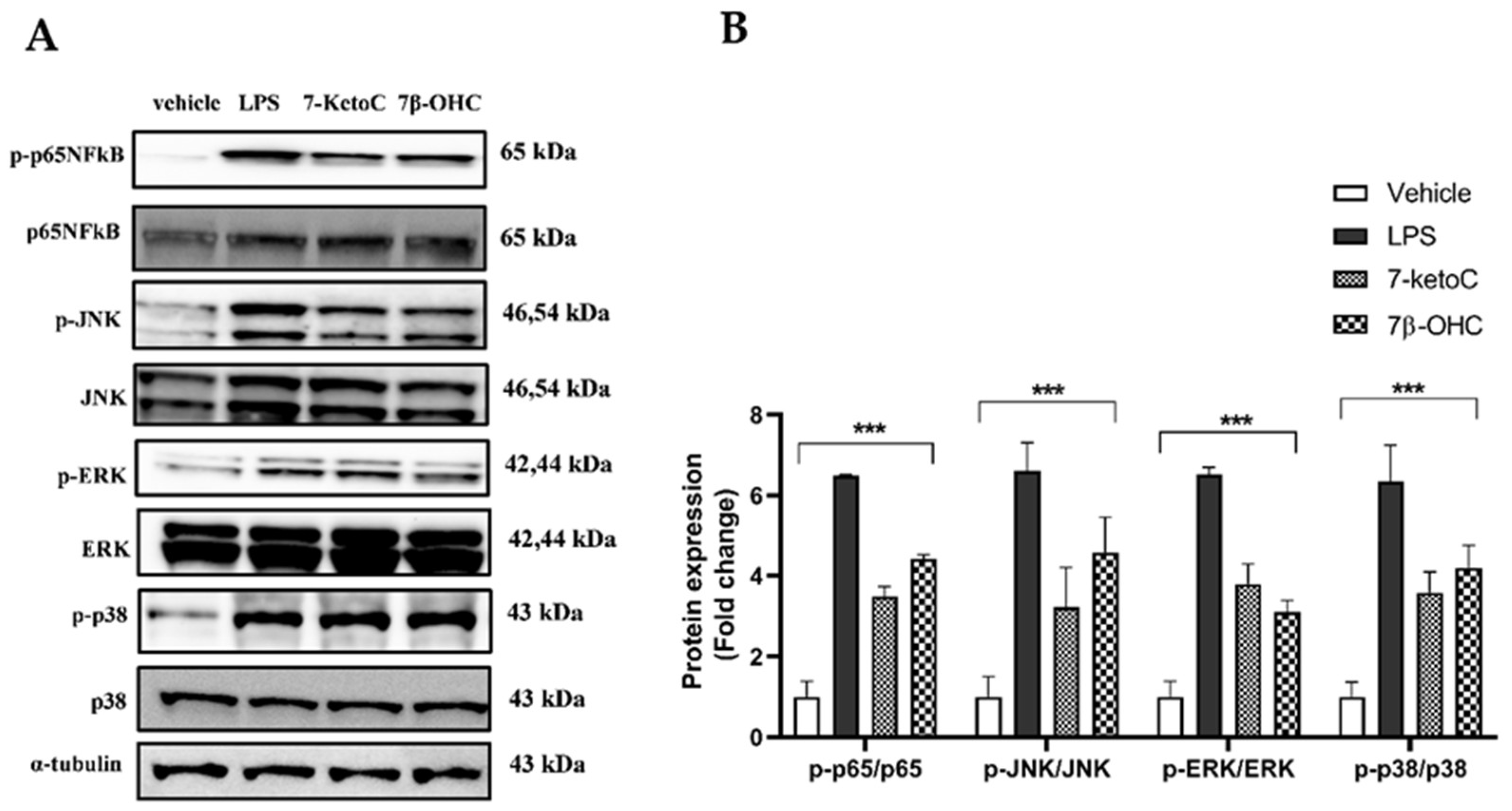
3.1. 7-KetoC and 7β-OHC Activate Pro-Inflammatory TLR-4 Signaling in fpEC
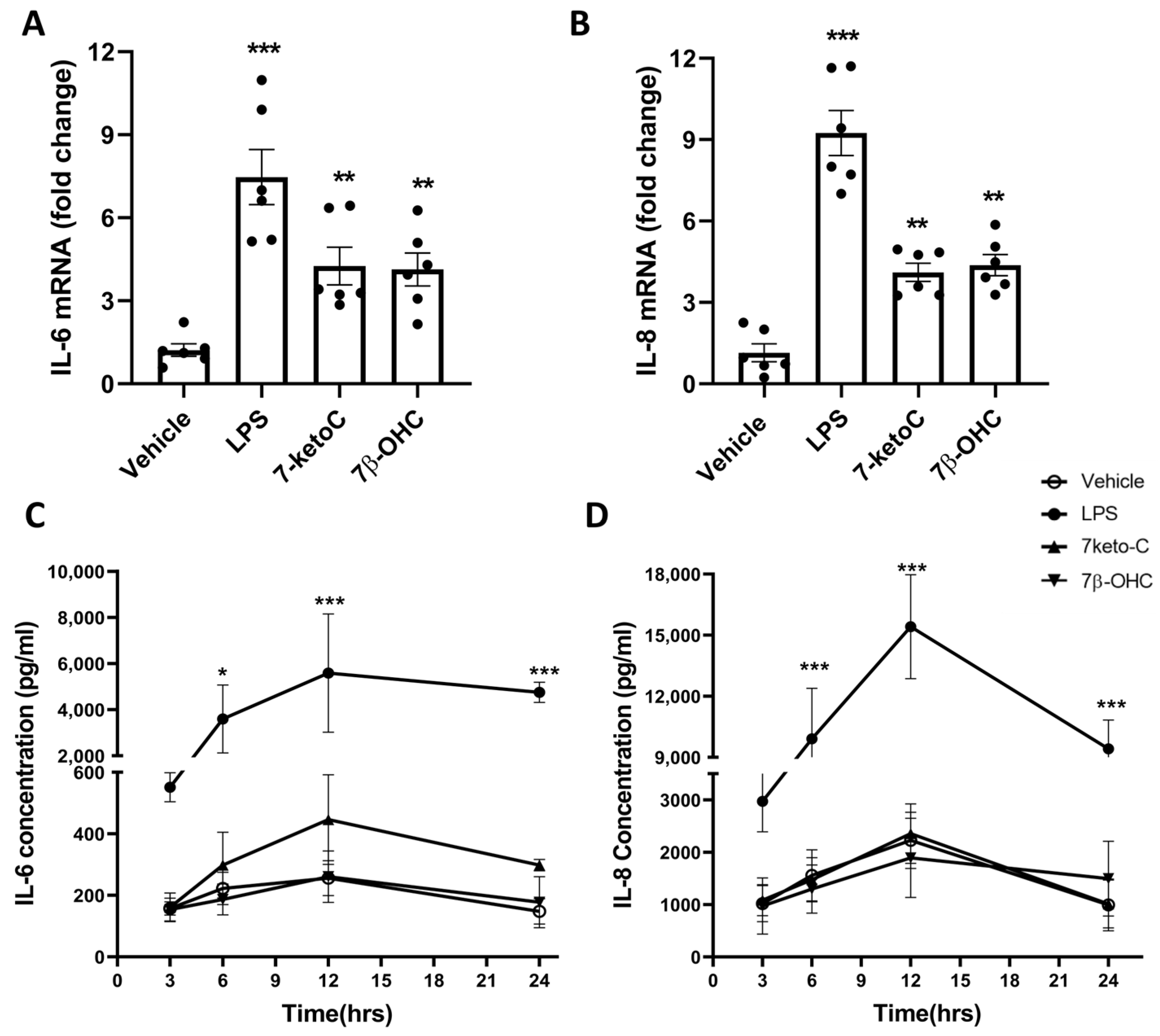
3.2. 7-KetoC and 7β-OHC Induce Transcription of Pro-Inflammatory Cytokines but Lead to Unchanged Release
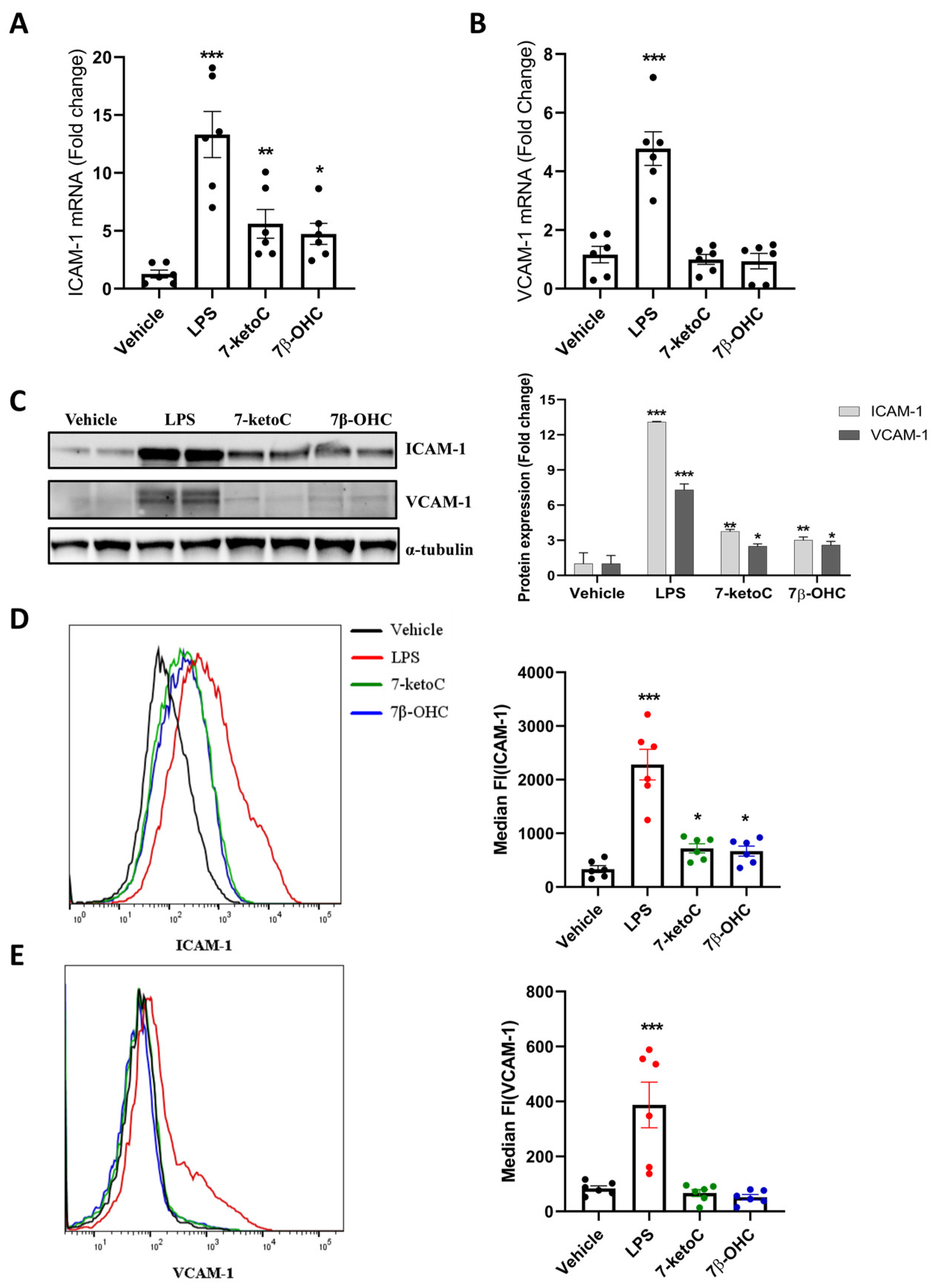
3.3. ICAM-1 Expression Is Elevated in fpEC in Response to 7-KetoC and 7β-OHC
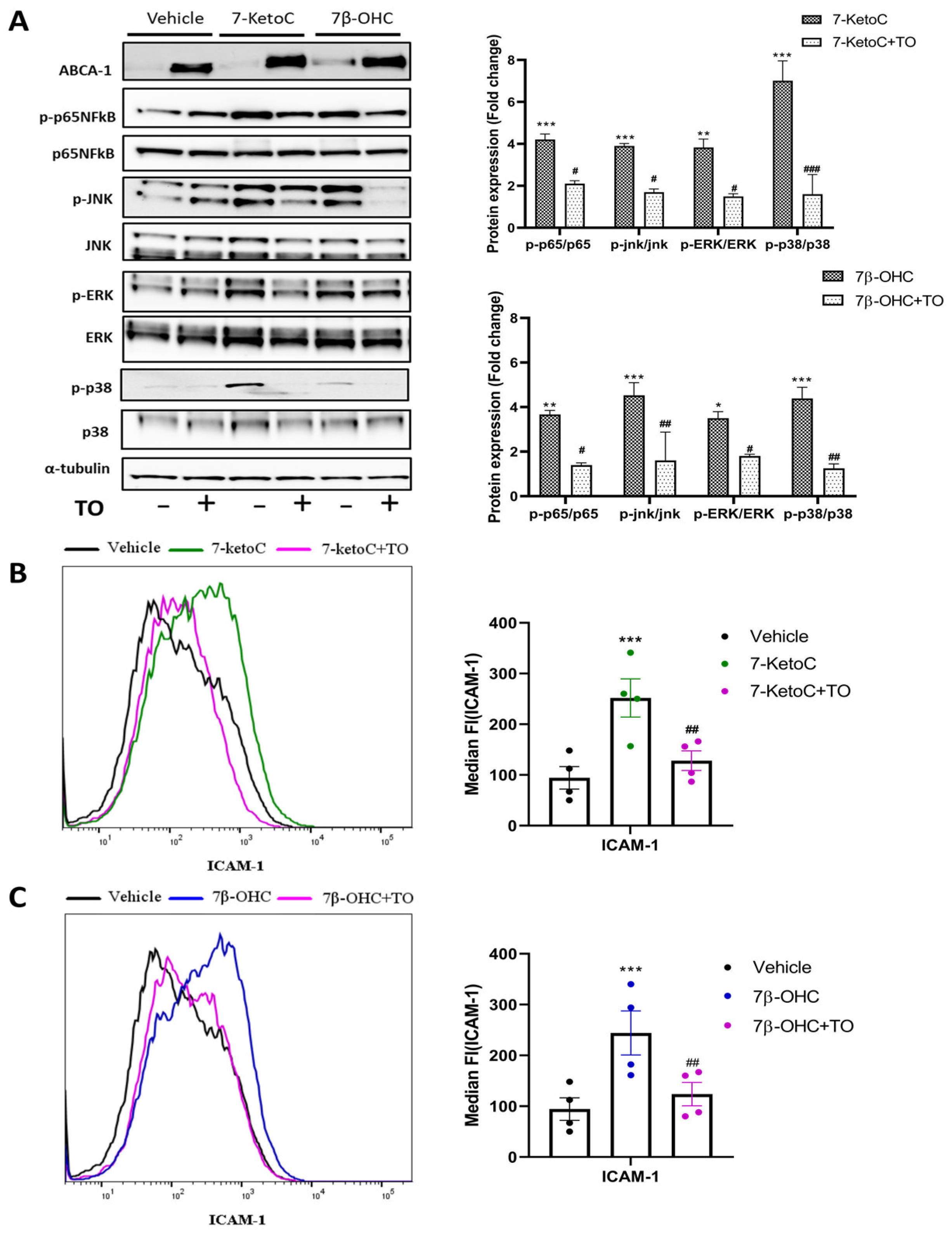
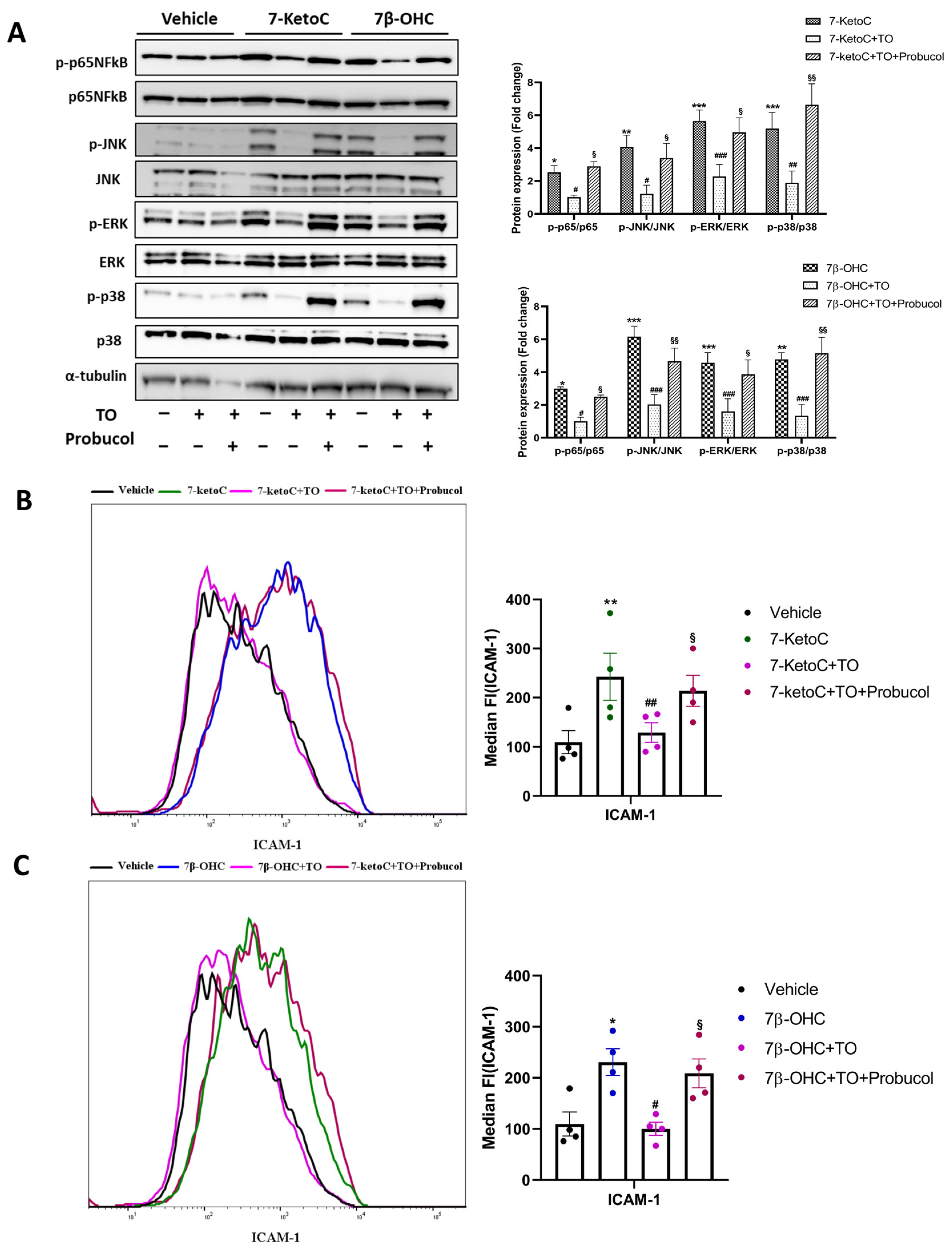
3.4. LXR Activation by Synthetic Agonist T0901317 Inhibits Inflammatory Signaling
3.5. ABCA-1 Induction Is Crucial for LXR-Mediated Repression of Inflammatory Signaling
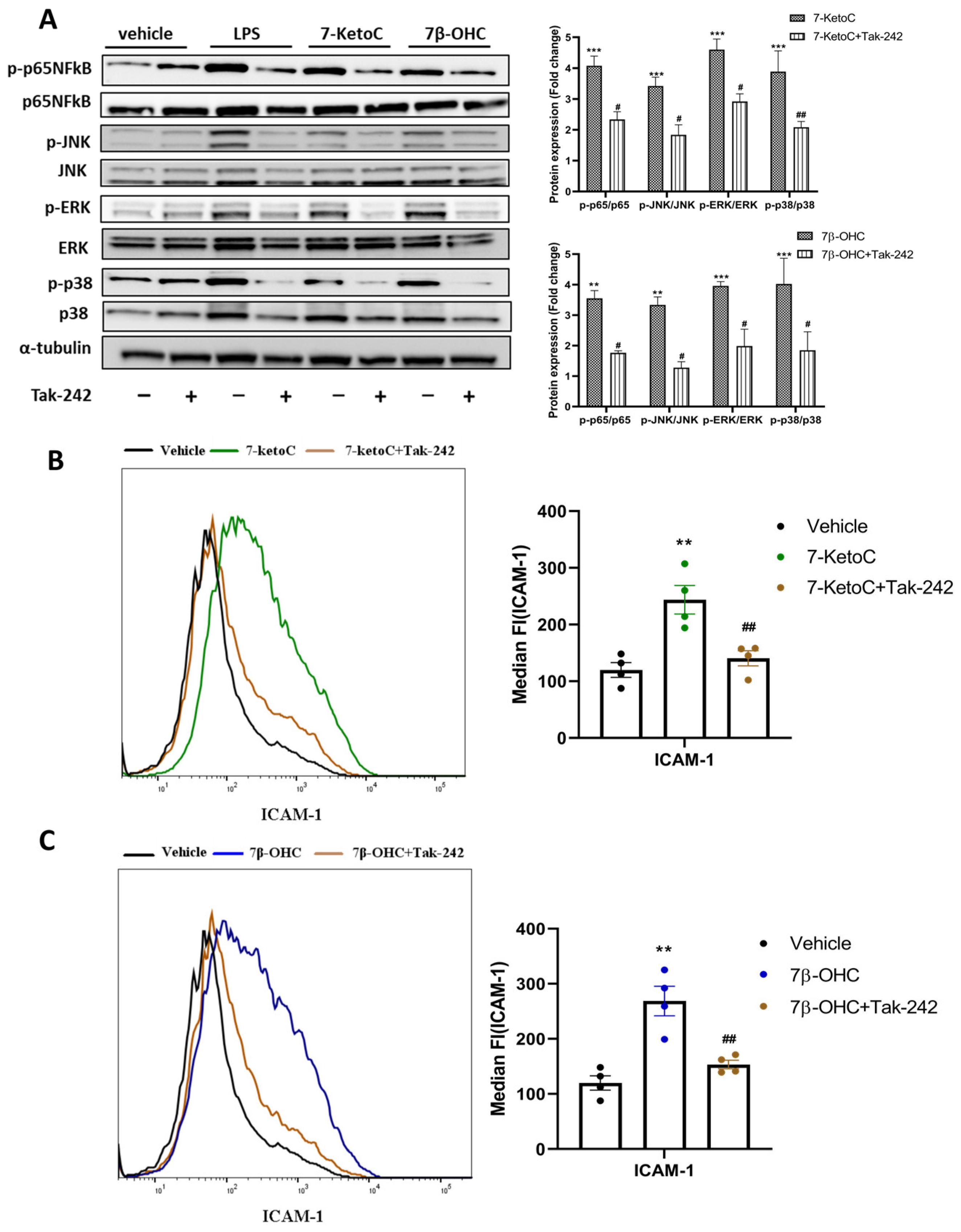
3.6. 7-KetoC and 7β-OHC Exert Inflammatory Responses in fpEC via TLR-4 Dependent Mechanisms
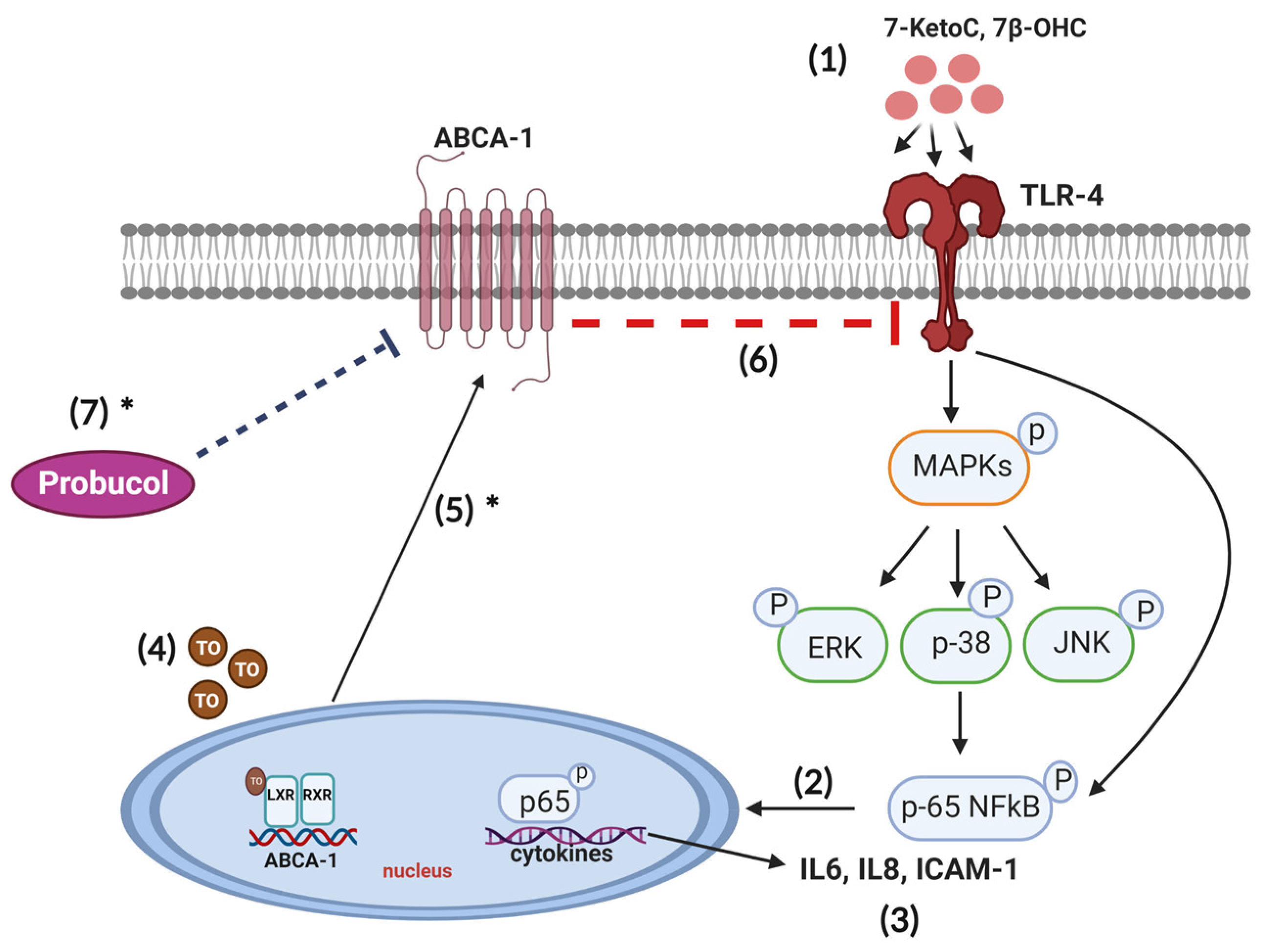
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cotechini, T.; Graham, C.H. Aberrant Maternal Inflammation as a Cause of Pregnancy Complications: A Potential Therapeutic Target? Placenta 2015, 36, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Mor, G.; Cardenas, I.; Abrahams, V.; Guller, S. Inflammation and Pregnancy: The Role of the Immune System at the Implantation Site. Ann. N. Y. Acad. Sci. 2011, 1221, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Pantham, P.; Aye, I.L.M.H.; Powell, T.L. Inflammation in Maternal Obesity and Gestational Diabetes Mellitus. Placenta 2015, 36, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Abell, S.K.; de Courten, B.; Boyle, J.A.; Teede, H.J.; Baker, P.N. Inflammatory and Other Biomarkers: Role in Pathophysiology and Prediction of Gestational Diabetes Mellitus. Int. J. Mol. Sci. 2015, 16, 13442–13473. [Google Scholar] [CrossRef]
- Sudharshana Murthy, K.A.; Bhandiwada, A.; Chandan, S.L.; Gowda, S.L.; Sindhusree, G. Evaluation of Oxidative Stress and Proinflammatory Cytokines in Gestational Diabetes Mellitus and Their Correlation with Pregnancy Outcome. Indian J. Endocrinol. Metab. 2018, 22, 79–84. [Google Scholar] [CrossRef]
- Bowen, J.M.; Chamley, L.; Mitchell, M.D.; Keelan, J.A. Cytokines of the Placenta and Extra-Placental Membranes: Biosynthesis, Secretion and Roles Establishment of Pregnancy in Women. Placenta 2002, 23, 239–256. [Google Scholar] [CrossRef]
- Radaelli, T.; Varastehpour, A.; Catalano, P.; Hauguel-de Mouzon, S. Gestational Diabetes Induces Placental Genes for Chronic Stress and Inflammatory Pathways. Diabetes 2003, 52, 2951–2958. [Google Scholar] [CrossRef]
- Kaaja, R.; Rönnemaa, T. Gestational Diabetes: Pathogenesis and Consequences to Mother and Offspring. Rev. Diabet. Stud. 2008, 5, 194–202. [Google Scholar] [CrossRef]
- Bełtowski, J. Liver X Receptors (LXR) as Therapeutic Targets in Dyslipidemia. Cardiovasc. Ther. 2008, 26, 297–316. [Google Scholar] [CrossRef]
- Yvan-Charvet, L.; Wang, N.; Tall, A.R. ATVB In Focus HDL Structure, Function, Therapeutics and Imaging Role of HDL, ABCA1, and ABCG1 Transporters in Cholesterol Efflux and Immune Responses. Arter. Thromb. Vasc. Biol. 2010, 30, 139–143. [Google Scholar] [CrossRef]
- Spyridon, M.; Moraes, L.A.; Jones, C.I.; Sage, T.; Sasikumar, P.; Bucci, G.; Gibbins, J.M. LXR as a Novel Antithrombotic Target. Blood 2011, 117, 5751–5761. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Hong, C.; Rong, X.; Zhu, X.; Tarling, E.J.; Hedde, P.N.; Gratton, E.; Parks, J.; Tontonoz, P. LXRs Link Metabolism to Inflammation through Abca1-Dependent Regulation of Membrane Composition and TLR Signaling. Elife 2015, 4, e08009. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.G.; Doran, A.C.; Fotakis, P.; Westerterp, M.; Antonson, P.; Jiang, H.; Jiang, X.C.; Gustafsson, J.Å.; Tabas, I.; Tall, A.R. LXR Suppresses Inflammatory Gene Expression and Neutrophil Migration through Cis-Repression and Cholesterol Efflux. Cell Rep. 2018, 25, 3774–3785.e4. [Google Scholar] [CrossRef] [PubMed]
- Ghisletti, S.; Huang, W.; Ogawa, S.; Pascual, G.; Lin, M.E.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. Parallel SUMOylation-Dependent Pathways Mediate Gene- and Signal-Specific Transrepression by LXRs and PPARγ. Mol. Cell 2007, 25, 57–70. [Google Scholar] [CrossRef]
- Simons, K.; Toomre, D. Lipid Rafts and Signal Transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef]
- Tajalli-Nezhad, S.; Karimian, M.; Beyer, C.; Atlasi, M.A.; Azami Tameh, A. The Regulatory Role of Toll-like Receptors after Ischemic Stroke: Neurosteroids as TLR Modulators with the Focus on TLR2/4. Cell. Mol. Life Sci. 2019, 76, 523–537. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Hayden, M.S.; Ghosh, S. Crosstalk in NF-ΚB Signaling Pathways. Nat. Immunol. 2011, 12, 695–708. [Google Scholar] [CrossRef]
- Saha, R.N.; Jana, M.; Pahan, K. MAPK P38 Regulates Transcriptional Activity of NF-ΚB in Primary Human Astrocytes via Acetylation of P651. J. Immunol. 2007, 179, 7101–7109. [Google Scholar] [CrossRef]
- Fessler, M.B.; Parks, J.S. Intracellular Lipid Flux and Membrane Microdomains as Organizing Principles in Inflammatory Cell Signaling. J. Immunol. 2011, 187, 1529–1535. [Google Scholar] [CrossRef]
- Aye, I.L.M.H.; Waddell, B.J.; Mark, P.J.; Keelan, J.A. Oxysterols Exert Proinflammatory Effects in Placental Trophoblasts via TLR4-Dependent, Cholesterol-Sensitive Activation of NF-ΚB. Mol. Hum. Reprod. 2012, 18, 341–353. [Google Scholar] [CrossRef]
- Björkhem, I.; Diczfalusy, U. Oxysterols. Arter. Thromb. Vasc. Biol. 2002, 22, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.; Jessup, W. Oxysterols: Sources, Cellular Storage and Metabolism, and New Insights into Their Roles in Cholesterol Homeostasis. Mol. Asp. Med. 2009, 30, 111–122. [Google Scholar] [CrossRef] [PubMed]
- De Freitas, F.A.; Levy, D.; Reichert, C.O.; Cunha-Neto, E.; Kalil, J.; Bydlowski, S.P. Effects of Oxysterols on Immune Cells and Related Diseases. Cells 2022, 11, 1251. [Google Scholar] [CrossRef] [PubMed]
- Mutemberezi, V.; Buisseret, B.; Masquelier, J.; Guillemot-Legris, O.; Alhouayek, M.; Muccioli, G.G. Oxysterol Levels and Metabolism in the Course of Neuroinflammation: Insights from in Vitro and in Vivo Models. J. Neuroinflammation 2018, 15, 1–16. [Google Scholar] [CrossRef]
- Lappas, M.; Hiden, U.; Desoye, G.; Froehlich, J.; Hauguel-de Mouzon, S.; Jawerbaum, A. The Role of Oxidative Stress in the Pathophysiology of Gestational Diabetes Mellitus. Antioxid. Redox Signal. 2011, 15, 3061–3100. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Jhun, B.S.; Yoon, Y. High-Glucose Stimulation Increases Reactive Oxygen Species Production Through the Calcium and Mitogen-Activated Protein Kinase-Mediated Activation of Mitochondrial Fission. Antioxid. Redox Signal. 2010, 14, 425–437. [Google Scholar] [CrossRef]
- Sun, Y.; Kopp, S.; Strutz, J.; Gali, C.C.; Zandl-Lang, M.; Fanaee-Danesh, E.; Kirsch, A.; Cvitic, S.; Frank, S.; Saffery, R.; et al. Gestational Diabetes Mellitus Modulates Cholesterol Homeostasis in Human Fetoplacental Endothelium. Biochim. Et. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2018, 1863, 968–979. [Google Scholar] [CrossRef]
- Challier, J.C.; Basu, S.; Bintein, T.; Minium, J.; Hotmire, K.; Catalano, P.M.; Hauguel-de Mouzon, S. Obesity in Pregnancy Stimulates Macrophage Accumulation and Inflammation in the Placenta. Placenta 2008, 29, 274–281. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving Functions of Endothelial Cells in Inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Stefulj, J.; Panzenboeck, U.; Becker, T.; Hirschmugl, B.; Schweinzer, C.; Lang, I.; Marsche, G.; Sadjak, A.; Lang, U.; Desoye, G.; et al. Human Endothelial Cells of the Placental Barrier Efficiently Deliver Cholesterol to the Fetal Circulation via ABCA1 and ABCG1. Circ. Res. 2009, 104, 600–608. [Google Scholar] [CrossRef]
- Strahlhofer-Augsten, M.; Schliefsteiner, C.; Cvitic, S.; George, M.; Lang-Olip, I.; Hirschmugl, B.; Marsche, G.; Lang, U.; Novakovic, B.; Saffery, R.; et al. The Distinct Role of the HDL Receptor SR-BI in Cholesterol Homeostasis of Human Placental Arterial and Venous Endothelial Cells. Int. J. Mol. Sci. 2022, 23, 5364. [Google Scholar] [CrossRef] [PubMed]
- Lang, I.; Schweizer, A.; Hiden, U.; Ghaffari-Tabrizi, N.; Hagendorfer, G.; Bilban, M.; Pabst, M.A.; Korgun, E.T.; Dohr, G.; Desoye, G. Human Fetal Placental Endothelial Cells Have a Mature Arterial and a Juvenile Venous Phenotype with Adipogenic and Osteogenic Differentiation Potential. Differentiation 2008, 76, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Scholler, M.; Wadsack, C.; Metso, J.; Manavalan, A.P.C.; Sreckovic, I.; Schweinzer, C.; Hiden, U.; Jauhiainen, M.; Desoye, G.; Panzenboeck, U. Phospholipid Transfer Protein Is Differentially Expressed in Human Arterial and Venous Placental Endothelial Cells and Enhances Cholesterol Efflux to Fetal HDL. J. Clin. Endocrinol. Metab. 2012, 97, 2466–2474. [Google Scholar] [CrossRef]
- Bah, S.Y.; Dickinson, P.; Forster, T.; Kampmann, B.; Ghazal, P. Immune Oxysterols: Role in Mycobacterial Infection and Inflammation. J. Steroid Biochem. Mol. Biol. 2017, 169, 152–163. [Google Scholar] [CrossRef]
- Steinhoff, G.; Haverich, A. Cell-Cell and Cell-Matrix Adhesion Molecules in Human Heart and Lung Transplants. Mol. Cell Biochem. 1995, 147, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Harjunpää, H.; Llort Asens, M.; Guenther, C.; Fagerholm, S.C. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front. Immunol. 2019, 10, 1078. [Google Scholar] [CrossRef] [PubMed]
- Schulman, I.G. Liver X Receptors Link Lipid Metabolism and Inflammation. FEBS. Lett. 2017, 591, 2978–2991. [Google Scholar] [CrossRef]
- Fessler, M.B. The Challenges and Promise of Targeting the Liver X Receptors for Treatment of Inflammatory Disease. Pharmacol. Ther. 2018, 181, 1–12. [Google Scholar] [CrossRef]
- Guillemot-Legris, O.; Mutemberezi, V.; Muccioli, G.G. Oxysterols in Metabolic Syndrome: From Bystander Molecules to Bioactive Lipids. Trends Mol. Med. 2016, 22, 594–614. [Google Scholar] [CrossRef]
- Arakawa, R.; Tsujita, M.; Iwamoto, N.; Ito-Ohsumi, C.; Lu, R.; Wu, C.A.; Shimizu, K.; Aotsuka, T.; Kanazawa, H.; Abe-Dohmae, S.; et al. Pharmacological Inhibition of ABCA1 Degradation Increases HDL Biogenesis and Exhibits Antiatherogenesis. J. Lipid Res. 2009, 50, 2299–2305. [Google Scholar] [CrossRef]
- Yamashita, S.; Matsuzawa, Y. Where Are We with Probucol: A New Life for an Old Drug? Atherosclerosis 2009, 207, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Favari, E.; Zanotti, I.; Zimetti, F.; Ronda, N.; Bernini, F.; Rothblat, G.H. Probucol Inhibits ABCA1-Mediated Cellular Lipid Efflux. Arter. Thromb. Vasc. Biol. 2004, 24, 2345–2350. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.A.; Tsujita, M.; Hayashi, M.; Yokoyama, S. Probucol Inactivates ABCA1 in the Plasma Membrane with Respect to Its Mediation of Apolipoprotein Binding and High Density Lipoprotein Assembly and to Its Proteolytic Degradation. J. Biol. Chem. 2004, 279, 30168–30174. [Google Scholar] [CrossRef]
- Matsunaga, N.; Tsuchimori, N.; Matsumoto, T.; Ii, M. TAK-242 (Resatorvid), a Small-Molecule Inhibitor of Toll-like Receptor (TLR) 4 Signaling, Binds Selectively to TLR4 and Interferes with Interactions between TLR4 and Its Adaptor Molecules. Mol. Pharm. 2011, 79, 34–41. [Google Scholar] [CrossRef]
- Aye, I.L.M.H.; Waddell, B.J.; Mark, P.J.; Keelan, J.A. Placental ABCA1 and ABCG1 Transporters Efflux Cholesterol and Protect Trophoblasts from Oxysterol Induced Toxicity. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2010, 1801, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Bodzek, P.; Janoszka, B.; Wielkoszynski, T.; Bodzek, D.; Sieron, A. Concentration of Chosen Oxycholesterols in Plasma of Pregnant Women with Pregnancy-Induced Hypertension. Biomed. Chromatogr. 2002, 16, 13–18. [Google Scholar] [CrossRef]
- Morello, F.; Saglio, E.; Noghero, A.; Schiavone, D.; Williams, T.A.; Verhovez, A.; Bussolino, F.; Veglio, F.; Mulatero, P. LXR-Activating Oxysterols Induce the Expression of Inflammatory Markers in Endothelial Cells through LXR-Independent Mechanisms. Atherosclerosis 2009, 207, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Aye, I.L.M.H.; Waddell, B.J.; Mark, P.J.; Keelan, J.A. Oxysterols Inhibit Differentiation and Fusion of Term Primary Trophoblasts by Activating Liver X Receptors. Placenta 2011, 32, 183–191. [Google Scholar] [CrossRef]
- Kadl, A.; Leitinger, N. The Role of Endothelial Cells in the Resolution of Acute Inflammation. Antioxid Redox Signal. 2005, 7, 1744–1754. [Google Scholar] [CrossRef]
- Desoye, G.; Hauguel-De Mouzon, S. The Human Placenta in Gestational Diabetes Mellitus: The Insulin and Cytokine Network. Diabetes Care 2007, 30, S120–S126. [Google Scholar] [CrossRef]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, Inflammation and Innate Immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Endo, K.; Oyama, T.; Saiki, A.; Ban, N.; Ohira, M.; Koide, N.; Murano, T.; Watanabe, H.; Nishii, M.; Miura, M.; et al. Determination of Serum 7-Ketocholesterol Concentrations and Their Relationships with Coronary Multiple Risks in Diabetes Mellitus. Diabetes Res. Clin. Pract. 2008, 80, 63–68. [Google Scholar] [CrossRef]
- Dzeletovic, S.; Breuer, O.; Lund, E.; Diczfalusy, U. Determination of Cholesterol Oxidation Products in Human Plasma by Isotope Dilution-Mass Spectrometry. Anal. Biochem. 1995, 225, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Cruset, S.; Carpenter, K.; Guardiola, F.; Stein, B.; Mitchinson, M. Oxysterol Profiles of Normal Human Arteries, Fatty Streaks and Advanced Lesions. Free. Radic. Res. 2001, 35, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Iuliano, L.; Micheletta, F.; Natoli, S.; Ginanni Corradini, S.; Iappelli, M.; Elisei, W.; Giovannelli, L.; Violi, F.; Diczfalusy, U. Measurement of Oxysterols and α-Tocopherol in Plasma and Tissue Samples as Indices of Oxidant Stress Status. Anal. Biochem. 2003, 312, 217–223. [Google Scholar] [CrossRef]
- Staff, A.C.; Dechend, R.; Pijnenborg, R. Learning From the Placenta. Hypertens. 2010, 56, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Pitz Jacobsen, D.; Fjeldstad, H.E.; Johnsen, G.M.; Fosheim, I.K.; Moe, K.; Alnæs-Katjavivi, P.; Dechend, R.; Sugulle, M.; Staff, A.C. Acute Atherosis Lesions at the Fetal-Maternal Border: Current Knowledge and Implications for Maternal Cardiovascular Health. Front. Immunol. 2021, 12, 791606. [Google Scholar] [CrossRef]
- López Morales, C.M.; Brito Zurita, O.R.; González Heredia, R.; Cruz López, M.; Méndez Padrón, A.; Matute Briseño, J.A. Placental Atherosclerosis and Markers of Endothelial Dysfunction in Infants Born to Mothers with Gestational Diabetes. Med. Clínica 2016, 147, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Vejux, A.; Abed-Vieillard, D.; Hajji, K.; Zarrouk, A.; Mackrill, J.J.; Ghosh, S.; Nury, T.; Yammine, A.; Zaibi, M.; Mihoubi, W.; et al. 7-Ketocholesterol and 7β-Hydroxycholesterol: In Vitro and Animal Models Used to Characterize Their Activities and to Identify Molecules Preventing Their Toxicity. Biochem. Pharm. 2020, 173, 113648. [Google Scholar] [CrossRef]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 Signal Transduction Pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Krishnaswamy, G.; Kelley, J.; Yerra, L.; Smith, J.K.; Chi, D.S. Human Endothelium as a Source of Multifunctional Cytokines: Molecular Regulation and Possible Role in Human Disease. J. Interferon. Cytokine. Res. 1999, 19, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Xing, K.; Murthy, S.; Liles, W.C.; Singh, J.M. Clinical Utility of Biomarkers of Endothelial Activation in Sepsis-a Systematic Review. Crit. Care 2012, 16, R7. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Choi, W.M.; Bae, W.H.; Anker, J.; Davis, A.A.; Agte, S.; Iams, W.T.; Cruz, M.; Matsangou, M.; Giles, F.J. Overexpression of Adhesion Molecules and Barrier Molecules Is Associated with Differential Infiltration of Immune Cells in Non-Small Cell Lung Cancer. Sci. Rep. 2018, 8, 1023. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Pérez, F.I.; Hiden, U.; Gauster, M.; Lang, I.; Konya, V.; Heinemann, A.; Lögl, J.; Saffery, R.; Desoye, G.; Cvitic, S. Post-Transcriptional down Regulation of ICAM-1 in Feto-Placental Endothelium in GDM. Cell Adh. Migr. 2016, 10, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Atègbo, J.M.; Grissa, O.; Yessoufou, A.; Hichami, A.; Dramane, K.L.; Moutairou, K.; Miled, A.; Grissa, A.; Jerbi, M.; Tabka, Z.; et al. Modulation of Adipokines and Cytokines in Gestational Diabetes and Macrosomia. J. Clin. Endocrinol. Metab. 2006, 91, 4137–4143. [Google Scholar] [CrossRef] [PubMed]
- Shih, J.H.; Tsai, Y.F.; Li, I.H.; Chen, M.H.; Huang, Y.S. Hp-S1 Ganglioside Suppresses Proinflammatory Responses by Inhibiting MyD88-Dependent NF-ΚB and JNK/P38 MAPK Pathways in Lipopolysaccharide-Stimulated Microglial Cells. Mar. Drugs 2020, 18, 496. [Google Scholar] [CrossRef]
- Ripmeester, E.G.J.; Timur, U.T.; Caron, M.M.J.; Welting, T.J.M. Recent Insights into the Contribution of the Changing Hypertrophic Chondrocyte Phenotype in the Development and Progression of Osteoarthritis. Front. Bioeng. Biotechnol. 2018, 6, 18. [Google Scholar] [CrossRef]
- Westerterp, M.; Murphy, A.J.; Wang, M.; Pagler, T.A.; Vengrenyuk, Y.; Kappus, M.S.; Gorman, D.J.; Nagareddy, P.R.; Zhu, X.; Abramowicz, S.; et al. Deficiency of ATP-Binding Cassette Transporters A1 and G1 in Macrophages Increases Inflammation and Accelerates Atherosclerosis in Mice. Circ. Res. 2013, 112, 1456–1465. [Google Scholar] [CrossRef]
- Juhl, A.D.; Wüstner, D. Pathways and Mechanisms of Cellular Cholesterol Efflux—Insight From Imaging. Front. Cell Dev. Biol. 2022, 10, 362. [Google Scholar] [CrossRef]
- Tam, S.-P.; Mok, L.; Chimini, G.; Vasa, M.; Deeley, R.G. ABCA1 Mediates High-Affinity Uptake of 25-Hydroxycholesterol by Membrane Vesicles and Rapid Efflux of Oxysterol by Intact Cells. Am. J. Physiol. Cell Physiol. 2006, 291, 490–502. [Google Scholar] [CrossRef]
- Terasaka, N.; Wang, N.; Yvan-Charvet, L.; Tall, A.R. High-Density Lipoprotein Protects Macrophages from Oxidized Low-Density Lipoprotein-Induced Apoptosis by Promoting Efflux of 7-Ketocholesterol via ABCG1. Proc. Natl. Acad. Sci. USA 2007, 104, 15093–15098. [Google Scholar] [CrossRef] [PubMed]
- Houck, K.A.; Borchert, K.M.; Hepler, C.D.; Thomas, J.S.; Bramlett, K.S.; Michael, L.F.; Burris, T.P. T0901317 Is a Dual LXR/FXR Agonist. Mol. Genet. Metab. 2004, 83, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Shaik, F.B.; Prasad, D.V.R.; Narala, V.R. Role of Farnesoid X Receptor in Inflammation and Resolution. Inflamm. Res. 2015, 64, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Rossin, D.; Barbosa-Pereira, L.; Iaia, N.; Testa, G.; Sottero, B.; Poli, G.; Zeppa, G.; Biasi, F. A Dietary Mixture of Oxysterols Induces in Vitro Intestinal Inflammation through TLR2/4 Activation: The Protective Effect of Cocoa Bean Shells. Antioxidants 2019, 8, 151. [Google Scholar] [CrossRef] [PubMed]
- Parusel, R.; Steimle, A.; Lange, A.; Schäfer, A.; Maerz, J.K.; Bender, A.; Frick, J.-S. An Important Question: Which LPS Do You Use? Virulence 2017, 8, 1890–1893. [Google Scholar] [CrossRef]
- Choi, S.-H.; Kim, J.; Gonen, A.; Viriyakosol, S.; Miller, Y.I. MD-2 Binds Cholesterol. Biochem. Biophys Res. Commun. 2016, 470, 877–880. [Google Scholar] [CrossRef]
- Choi, S.-H.; Yin, H.; Ravandi, A.; Armando, A.; Dumlao, D.; Kim, J.; Almazan, F.; Taylor, A.M.; McNamara, C.A.; Tsimikas, S.; et al. Polyoxygenated Cholesterol Ester Hydroperoxide Activates TLR4 and SYK Dependent Signaling in Macrophages. PLoS ONE 2013, 8, e83145. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Company | Forward/Reverse Primer |
|---|---|---|
| HPRT-1 | Thermo Fisher Scientific | F: GACCAGTCAACAGGGGACAT R: CTGCATTGTTTTGCCAGTGT |
| IL-6 | Thermo Fisher Scientific | F: CCACACAGACAGCCACTCAC R: TGCCTCTTTGCTGCTTTCAC |
| IL-8 | Thermo Fisher Scientific | F: GACCACACTGCGCCAACAC R: CTTCTCCACAACCCTCTGCAC |
| ICAM-1 | Thermo Fisher Scientific | F: ATGCCCAGACATCTGTGTCC R: GGGGTCTCTATGCCCAACAA |
| VCAM-1 | Thermo Fisher Scientific | F: GGGAAGATGGTCGTGATCCTT R: TCTGGGGTGGTCTCGATTTTA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
George, M.; Lang, M.; Gali, C.C.; Babalola, J.A.; Tam-Amersdorfer, C.; Stracke, A.; Strobl, H.; Zimmermann, R.; Panzenboeck, U.; Wadsack, C. Liver X Receptor Activation Attenuates Oxysterol-Induced Inflammatory Responses in Fetoplacental Endothelial Cells. Cells 2023, 12, 1186. https://doi.org/10.3390/cells12081186
George M, Lang M, Gali CC, Babalola JA, Tam-Amersdorfer C, Stracke A, Strobl H, Zimmermann R, Panzenboeck U, Wadsack C. Liver X Receptor Activation Attenuates Oxysterol-Induced Inflammatory Responses in Fetoplacental Endothelial Cells. Cells. 2023; 12(8):1186. https://doi.org/10.3390/cells12081186
Chicago/Turabian StyleGeorge, Meekha, Magdalena Lang, Chaitanya Chakravarthi Gali, Joshua Adekunle Babalola, Carmen Tam-Amersdorfer, Anika Stracke, Herbert Strobl, Robert Zimmermann, Ute Panzenboeck, and Christian Wadsack. 2023. "Liver X Receptor Activation Attenuates Oxysterol-Induced Inflammatory Responses in Fetoplacental Endothelial Cells" Cells 12, no. 8: 1186. https://doi.org/10.3390/cells12081186
APA StyleGeorge, M., Lang, M., Gali, C. C., Babalola, J. A., Tam-Amersdorfer, C., Stracke, A., Strobl, H., Zimmermann, R., Panzenboeck, U., & Wadsack, C. (2023). Liver X Receptor Activation Attenuates Oxysterol-Induced Inflammatory Responses in Fetoplacental Endothelial Cells. Cells, 12(8), 1186. https://doi.org/10.3390/cells12081186