
Suspension TRAPping Filter (sTRAP) Sample Preparation for Quantitative Proteomics in the Low µg Input Range Using a Plasmid DNA Micro-Spin Column: Analysis of the Hippocampus from the 5xFAD Alzheimer’s Disease Mouse Model

 , , ,
, , ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Sample Homogenization
2.3. sTRAP Protocol for Sample Preparation
2.4. Sample Preparation Protocols for Commercial sTRAP Column and In-Gel Digestion
2.5. LC-MS analysis
2.6. Data Analysis
3. Results
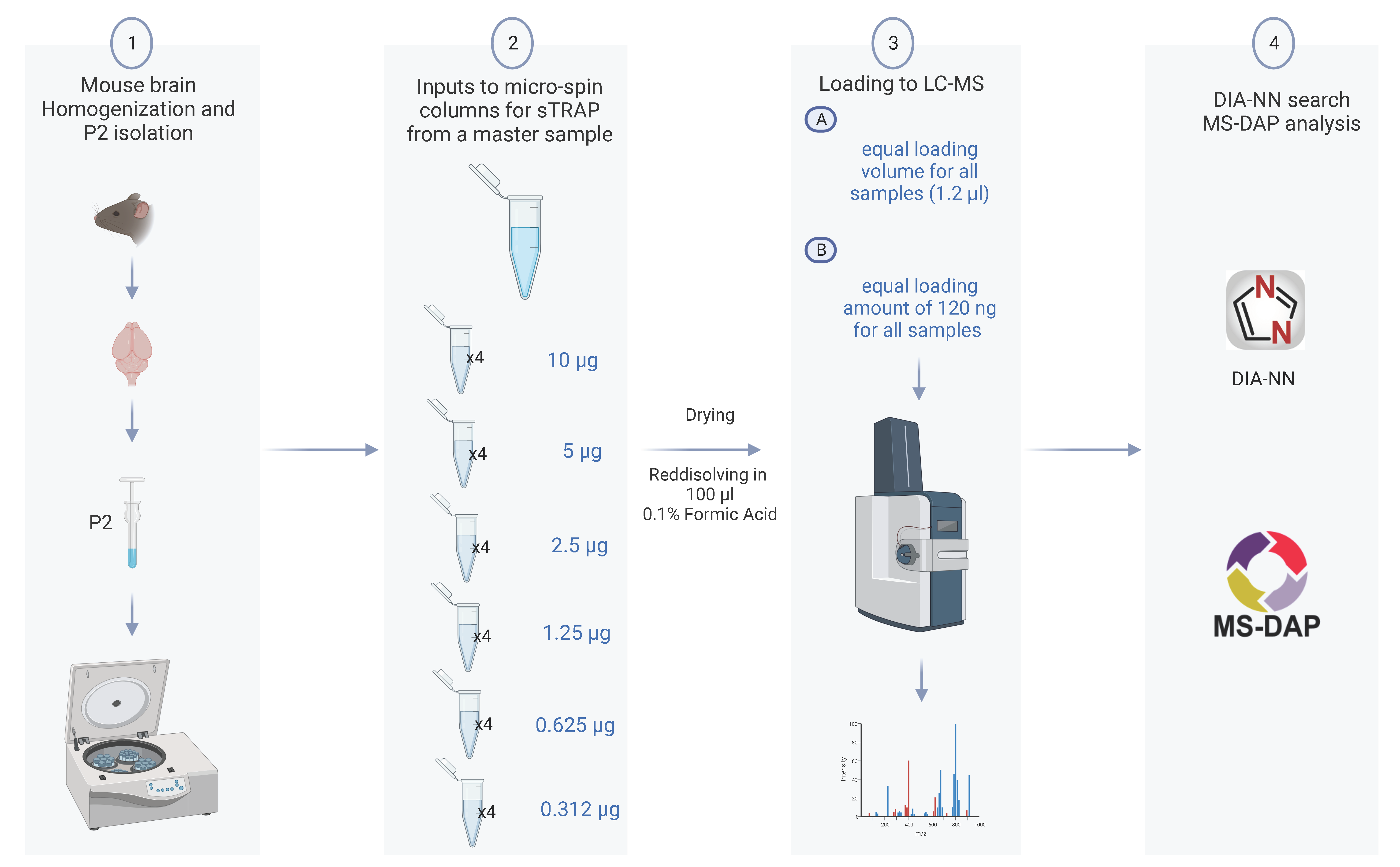
3.1. Plasmid DNA Micro-Spin Column Provides Excellent Filter for sTRAP Sample Preparation
3.2. Plasmid DNA Micro-Spin Column for sTRAP Protocol Has Low CoV—Implications from the LC-MS Study
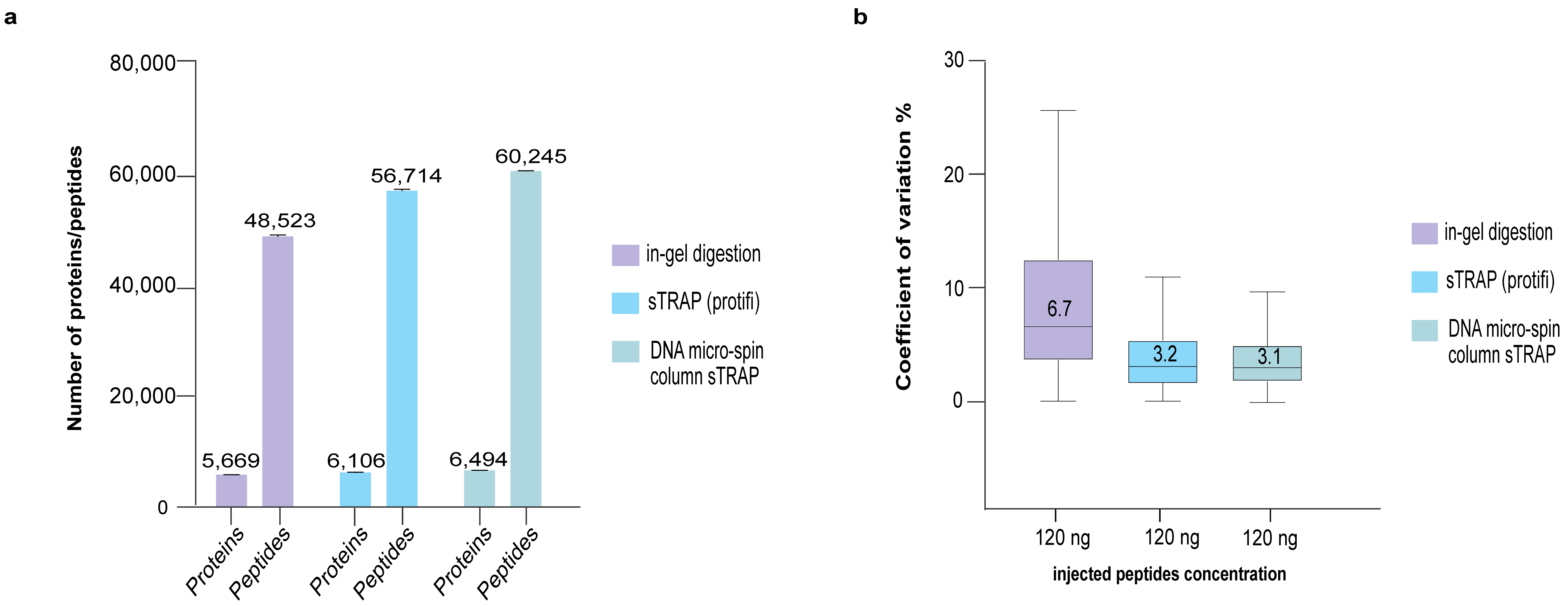
3.3. Comparison of the Sample Preparation Protocol Using Plasmid DNA Micro-Spin Column, the Commercial S-TRAP Column, and In-Gel Digestion Reveals the Favorable Properties of Plasmid DNA Micro-Spin Column for Quantitative Proteomics
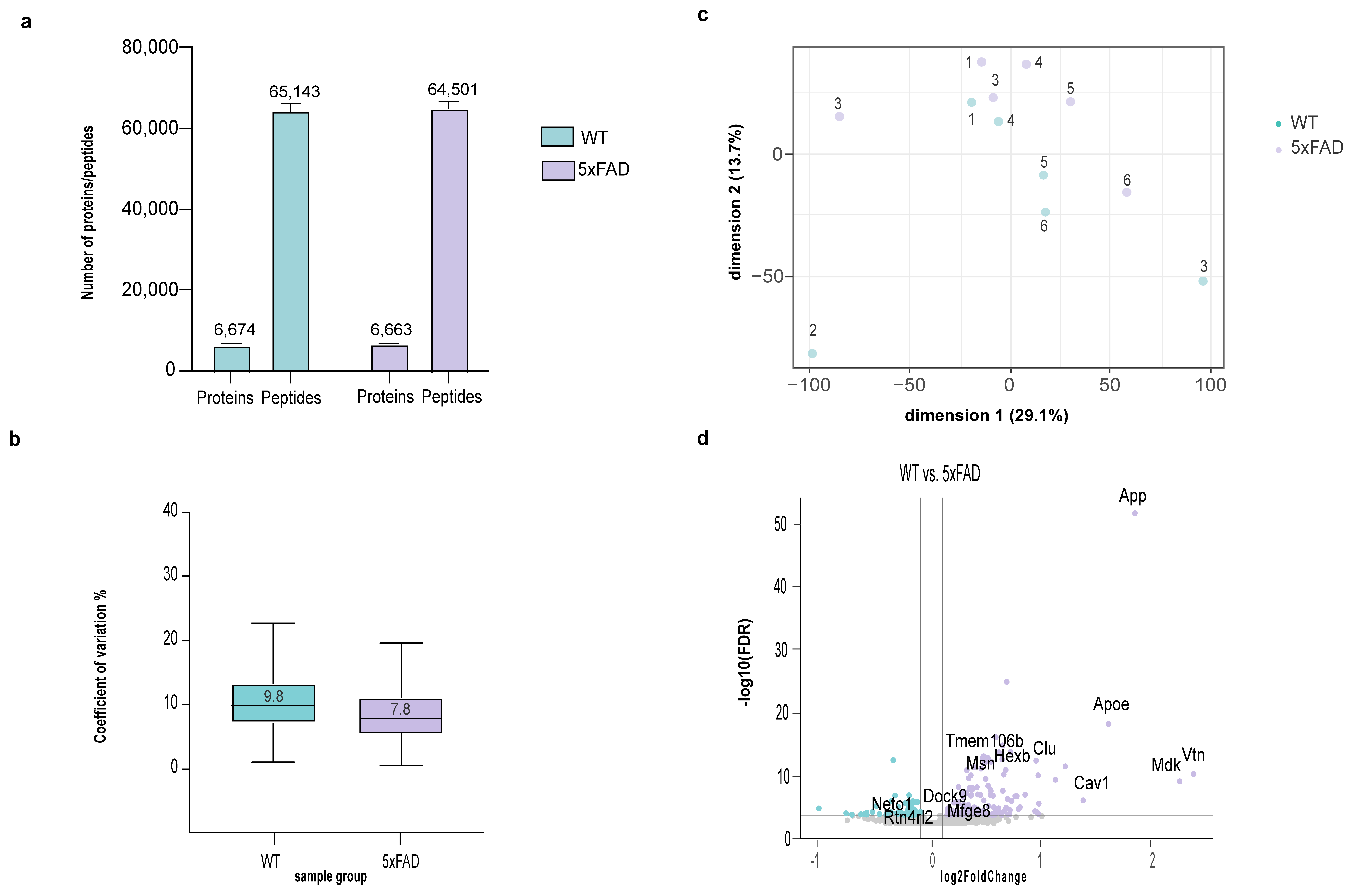
3.4. Proteomics Analysis of Hippocampi from 5xFAD and Wildtype Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van der Spek, S.J.F.; Koopmans, F.; Paliukhovich, I.; Ramsden, S.L.; Harvey, K.; Harvey, R.J.; Smit, A.B.; Li, K.W. Glycine Receptor Complex Analysis Using Immunoprecipitation-Blue Native Gel Electrophoresis-Mass Spectrometry. Proteomics 2020, 20, e1900403. [Google Scholar] [CrossRef] [PubMed]
- Li, K.W.; Chen, N.; Klemmer, P.; Koopmans, F.; Karupothula, R.; Smit, A.B. Identifying true protein complex constituents in interaction proteomics: The example of the DMXL2 protein complex. Proteomics 2012, 12, 2428–2432. [Google Scholar]
- Gonzalez-Lozano, M.A.; Wortel, J.; van der Loo, R.J.; van Weering, J.R.T.; Smit, A.B.; Li, K.W. Reduced mGluR5 Activity Modulates Mitochondrial Function. Cells 2021, 10, 1375. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Hughes, C.S.; Foehr, S.; Garfield, D.A.; Furlong, E.E.; Steinmetz, L.M.; Krijgsveld, J. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol. 2014, 10, 757. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Lozano, M.A.; Koopmans, F.; Paliukhovich, I.; Smit, A.B.; Li, K.W. A Fast and Economical Sample Preparation Protocol for Interaction Proteomics Analysis. Proteomics 2019, 19, e1900027. [Google Scholar] [CrossRef]
- Lin, Y.H.; Eguez, R.V.; Torralba, M.G.; Singh, H.; Golusinski, P.; Golusinski, W.; Masternak, M.; Nelson, K.E.; Freire, M.; Yu, Y. Self-Assembled STrap for Global Proteomics and Salivary Biomarker Discovery. J. Proteome Res. 2019, 18, 1907–1915. [Google Scholar] [CrossRef]
- Zougman, A.; Selby, P.J.; Banks, R.E. Suspension trapping (STrap) sample preparation method for bottom-up proteomics analysis. Proteomics 2014, 14, 1000–1006. [Google Scholar] [CrossRef]
- Crowell, A.M.; MacLellan, D.L.; Doucette, A.A. A two-stage spin cartridge for integrated protein precipitation, digestion and SDS removal in a comparative bottom-up proteomics workflow. J. Proteome 2015, 118, 140–150. [Google Scholar] [CrossRef]
- Varnavides, G.; Madern, M.; Anrather, D.; Hartl, N.; Reiter, W.; Hartl, M. In Search of a Universal Method: A Comparative Survey of Bottom-Up Proteomics Sample Preparation Methods. J. Proteome Res. 2022, 21, 2397–2411. [Google Scholar] [CrossRef]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef] [PubMed]
- HaileMariam, M.; Eguez, R.V.; Singh, H.; Bekele, S.; Ameni, G.; Pieper, R.; Yu, Y. S-Trap, an Ultrafast Sample-Preparation Approach for Shotgun Proteomics. J. Proteome Res. 2018, 17, 2917–2924. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S.; Vila, M.; Jackson-Lewis, V. Neurodegeneration: What is it and where are we? J. Clin. Investig. 2003, 111, 3–10. [Google Scholar] [CrossRef] [PubMed]
- McKean, N.E.; Handley, R.R.; Snell, R.G. A Review of the Current Mammalian Models of Alzheimer’s Disease and Challenges That Need to Be Overcome. Int. J. Mol. Sci. 2021, 22, 13168. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Oblak, A.L.; Lin, P.B.; Kotredes, K.P.; Pandey, R.S.; Garceau, D.; Williams, H.M.; Uyar, A.; O’Rourke, R.; O’Rourke, S.; Ingraham, C.; et al. Comprehensive Evaluation of the 5XFAD Mouse Model for Preclinical Testing Applications: A MODEL-AD Study. Front. Aging Neurosci. 2021, 13, 713726. [Google Scholar] [CrossRef]
- Meier, F.; Brunner, A.D.; Frank, M.; Ha, A.; Bludau, I.; Voytik, E.; Kaspar-Schoenefeld, S.; Lubeck, M.; Raether, O.; Bache, N.; et al. diaPASEF: Parallel accumulation-serial fragmentation combined with data-independent acquisition. Nat. Methods 2020, 17, 1229–1236. [Google Scholar] [CrossRef]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef]
- Koopmans, F.; Li, K.W.; Klaassen, R.V.; Smit, A.B. MS-DAP Platform for Downstream Data Analysis of Label-Free Proteomics Uncovers Optimal Workflows in Benchmark Data Sets and Increased Sensitivity in Analysis of Alzheimer’s Biomarker Data. J. Proteome Res. 2022, 22, 374–386. [Google Scholar] [CrossRef]
- Koopmans, F.; Ho, J.T.C.; Smit, A.B.; Li, K.W. Comparative Analyses of Data Independent Acquisition Mass Spectrometric Approaches: DIA, WiSIM-DIA, and Untargeted DIA. Proteomics 2018, 18, 1700304. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids. Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids. Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]
- Golde, T.E. Alzheimer’s disease—The journey of a healthy brain into organ failure. Mol. Neurodegener 2022, 17, 18. [Google Scholar] [CrossRef]
- Duong, V.A.; Park, J.M.; Lee, H. A review of suspension trapping digestion method in bottom-up proteomics. J. Sep. Sci. 2022, 45, 3150–3168. [Google Scholar] [CrossRef]
- van der Spek, S.J.F.; Gonzalez-Lozano, M.A.; Koopmans, F.; Miedema, S.S.M.; Paliukhovich, I.; Smit, A.B.; Li, K.W. Age-Dependent Hippocampal Proteomics in the APP/PS1 Alzheimer Mouse Model: A Comparative Analysis with Classical SWATH/DIA and directDIA Approaches. Cells 2021, 10, 1588. [Google Scholar] [CrossRef]
- Kim, D.K.; Han, D.; Park, J.; Choi, H.; Park, J.C.; Cha, M.Y.; Woo, J.; Byun, M.S.; Lee, D.Y.; Kim, Y.; et al. Deep proteome profiling of the hippocampus in the 5XFAD mouse model reveals biological process alterations and a novel biomarker of Alzheimer’s disease. Exp. Mol. Med. 2019, 51, 1–17. [Google Scholar] [CrossRef]
- Bai, B.; Wang, X.; Li, Y.; Chen, P.C.; Yu, K.; Dey, K.K.; Yarbro, J.M.; Han, X.; Lutz, B.M.; Rao, S.; et al. Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron 2020, 106, 700. [Google Scholar] [CrossRef]
- Blasco Tavares Pereira Lopes, F.; Schlatzer, D.; Wang, R.; Li, X.; Feng, E.; Koyuturk, M.; Qi, X.; Chance, M.R. Temporal and Sex-Linked Protein Expression Dynamics in a Familial Model of Alzheimer’s Disease. Mol. Cell Proteom. 2022, 21, 100280. [Google Scholar] [CrossRef]
- Li, K.W.; Ganz, A.B.; Smit, A.B. Proteomics of neurodegenerative diseases: Analysis of human post-mortem brain. J. Neurochem. 2019, 151, 435–445. [Google Scholar] [CrossRef]
- Schweighauser, M.; Arseni, D.; Bacioglu, M.; Huang, M.; Lovestam, S.; Shi, Y.; Yang, Y.; Zhang, W.; Kotecha, A.; Garringer, H.J.; et al. Age-dependent formation of TMEM106B amyloid filaments in human brains. Nature 2022, 605, 310–314. [Google Scholar] [CrossRef]
- Chang, A.; Xiang, X.; Wang, J.; Lee, C.; Arakhamia, T.; Simjanoska, M.; Wang, C.; Carlomagno, Y.; Zhang, G.; Dhingra, S.; et al. Homotypic fibrillization of TMEM106B across diverse neurodegenerative diseases. Cell 2022, 185, 1346–1355.e15. [Google Scholar] [CrossRef]
- Jiang, Y.X.; Cao, Q.; Sawaya, M.R.; Abskharon, R.; Ge, P.; DeTure, M.; Dickson, D.W.; Fu, J.Y.; Ogorzalek Loo, R.R.; Loo, J.A.; et al. Amyloid fibrils in FTLD-TDP are composed of TMEM106B and not TDP-43. Nature 2022, 605, 304–309. [Google Scholar] [CrossRef]
- Mao, F.; Robinson, J.L.; Unger, T.; Posavi, M.; Amado, D.A.; Elman, L.; Grossman, M.; Wolk, D.A.; Lee, E.B.; Van Deerlin, V.M.; et al. TMEM106B modifies TDP-43 pathology in human ALS brain and cell-based models of TDP-43 proteinopathy. Acta. Neuropathol. 2021, 142, 629–642. [Google Scholar] [CrossRef]
- Drummond, E.; Kavanagh, T.; Pires, G.; Marta-Ariza, M.; Kanshin, E.; Nayak, S.; Faustin, A.; Berdah, V.; Ueberheide, B.; Wisniewski, T. The amyloid plaque proteome in early onset Alzheimer’s disease and Down syndrome. Acta. Neuropathol Commun 2022, 10, 53. [Google Scholar] [CrossRef]
- Rayaprolu, S.; Gao, T.; Xiao, H.; Ramesha, S.; Weinstock, L.D.; Shah, J.; Duong, D.M.; Dammer, E.B.; Webster, J.A., Jr.; Lah, J.J.; et al. Flow-cytometric microglial sorting coupled with quantitative proteomics identifies moesin as a highly-abundant microglial protein with relevance to Alzheimer’s disease. Mol. Neurodegener 2020, 15, 28. [Google Scholar] [CrossRef]
- Gupta, A.; Sharma, A.; Kumar, A.; Goyal, R. Alteration in memory cognition due to activation of caveolin-1 and oxidative damage in a model of dementia of Alzheimer’s type. Indian J. Pharm. 2019, 51, 173–180. [Google Scholar]
- Wang, S.; Ichinomiya, T.; Terada, Y.; Wang, D.; Patel, H.H.; Head, B.P. Synapsin-Promoted Caveolin-1 Overexpression Maintains Mitochondrial Morphology and Function in PSAPP Alzheimer’s Disease Mice. Cells 2021, 10, 2487. [Google Scholar] [CrossRef]
- Bonds, J.A.; Shetti, A.; Bheri, A.; Chen, Z.; Disouky, A.; Tai, L.; Mao, M.; Head, B.P.; Bonini, M.G.; Haus, J.M.; et al. Depletion of Caveolin-1 in Type 2 Diabetes Model Induces Alzheimer’s Disease Pathology Precursors. J. Neurosci. 2019, 39, 8576–8583. [Google Scholar] [CrossRef]
- Shin, T.M.; Isas, J.M.; Hsieh, C.L.; Kayed, R.; Glabe, C.G.; Langen, R.; Chen, J. Formation of soluble amyloid oligomers and amyloid fibrils by the multifunctional protein vitronectin. Mol. Neurodegener 2008, 3, 16. [Google Scholar] [CrossRef]
- Ruzha, Y.; Ni, J.; Quan, Z.; Li, H.; Qing, H. Role of Vitronectin and Its Receptors in Neuronal Function and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 12387. [Google Scholar] [CrossRef]
- Shin, K.; Kent, J.E.; Singh, C.; Fujimoto, L.M.; Yu, J.; Tian, Y.; Im, W.; Marassi, F.M. Calcium and hydroxyapatite binding site of human vitronectin provides insights to abnormal deposit formation. Proc. Natl. Acad. Sci. USA 2020, 117, 18504–18510. [Google Scholar] [CrossRef]
- Boddaert, J.; Kinugawa, K.; Lambert, J.C.; Boukhtouche, F.; Zoll, J.; Merval, R.; Blanc-Brude, O.; Mann, D.; Berr, C.; Vilar, J.; et al. Evidence of a role for lactadherin in Alzheimer’s disease. Am. J. Pathol. 2007, 170, 921–929. [Google Scholar] [CrossRef]
- Wagner, J.; Degenhardt, K.; Veit, M.; Louros, N.; Konstantoulea, K.; Skodras, A.; Wild, K.; Liu, P.; Obermuller, U.; Bansal, V.; et al. Medin co-aggregates with vascular amyloid-beta in Alzheimer’s disease. Nature 2022, 612, 123–131. [Google Scholar] [CrossRef]
- Marazuela, P.; Sole, M.; Bonaterra-Pastra, A.; Pizarro, J.; Camacho, J.; Martinez-Saez, E.; Kuiperij, H.B.; Verbeek, M.M.; de Kort, A.M.; Schreuder, F.; et al. MFG-E8 (LACTADHERIN): A novel marker associated with cerebral amyloid angiopathy. Acta Neuropathol. Commun. 2021, 9, 154. [Google Scholar] [CrossRef]
- Rong, Z.; Cheng, B.; Zhong, L.; Ye, X.; Li, X.; Jia, L.; Li, Y.; Shue, F.; Wang, N.; Cheng, Y.; et al. Activation of FAK/Rac1/Cdc42-GTPase signaling ameliorates impaired microglial migration response to Abeta(42) in triggering receptor expressed on myeloid cells 2 loss-of-function murine models. FASEB J. 2020, 34, 10984–10997. [Google Scholar] [CrossRef]
- Cousins, S.L.; Innocent, N.; Stephenson, F.A. Neto1 associates with the NMDA receptor/amyloid precursor protein complex. J. Neurochem. 2013, 126, 554–564. [Google Scholar] [CrossRef]
- Wyeth, M.S.; Pelkey, K.A.; Yuan, X.; Vargish, G.; Johnston, A.D.; Hunt, S.; Fang, C.; Abebe, D.; Mahadevan, V.; Fisahn, A.; et al. Neto Auxiliary Subunits Regulate Interneuron Somatodendritic and Presynaptic Kainate Receptors to Control Network Inhibition. Cell Rep. 2017, 20, 2156–2168. [Google Scholar] [CrossRef]
- Zhao, Y.; Sivaji, S.; Chiang, M.C.; Ali, H.; Zukowski, M.; Ali, S.; Kennedy, B.; Sklyar, A.; Cheng, A.; Guo, Z.; et al. Amyloid Beta Peptides Block New Synapse Assembly by Nogo Receptor-Mediated Inhibition of T-Type Calcium Channels. Neuron 2017, 96, 355–372.e6. [Google Scholar] [CrossRef]
- Gong, B.; Chen, F.; Pan, Y.; Arrieta-Cruz, I.; Yoshida, Y.; Haroutunian, V.; Pasinetti, G.M. SCFFbx2-E3-ligase-mediated degradation of BACE1 attenuates Alzheimer’s disease amyloidosis and improves synaptic function. Aging Cell 2010, 9, 1018–1031. [Google Scholar] [CrossRef]
- Kern, F.; Sarg, B.; Stasyk, T.; Hess, D.; Lindner, H. The Nogo receptor 2 is a novel substrate of Fbs1. Biochem. Biophys. Res. Commun. 2012, 417, 977–981. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thanou, E.; Koopmans, F.; Pita-Illobre, D.; Klaassen, R.V.; Özer, B.; Charalampopoulos, I.; Smit, A.B.; Li, K.W. Suspension TRAPping Filter (sTRAP) Sample Preparation for Quantitative Proteomics in the Low µg Input Range Using a Plasmid DNA Micro-Spin Column: Analysis of the Hippocampus from the 5xFAD Alzheimer’s Disease Mouse Model. Cells 2023, 12, 1242. https://doi.org/10.3390/cells12091242
Thanou E, Koopmans F, Pita-Illobre D, Klaassen RV, Özer B, Charalampopoulos I, Smit AB, Li KW. Suspension TRAPping Filter (sTRAP) Sample Preparation for Quantitative Proteomics in the Low µg Input Range Using a Plasmid DNA Micro-Spin Column: Analysis of the Hippocampus from the 5xFAD Alzheimer’s Disease Mouse Model. Cells. 2023; 12(9):1242. https://doi.org/10.3390/cells12091242
Chicago/Turabian StyleThanou, Evangelia, Frank Koopmans, Débora Pita-Illobre, Remco V. Klaassen, Berna Özer, Ioannis Charalampopoulos, August B. Smit, and Ka Wan Li. 2023. "Suspension TRAPping Filter (sTRAP) Sample Preparation for Quantitative Proteomics in the Low µg Input Range Using a Plasmid DNA Micro-Spin Column: Analysis of the Hippocampus from the 5xFAD Alzheimer’s Disease Mouse Model" Cells 12, no. 9: 1242. https://doi.org/10.3390/cells12091242
APA StyleThanou, E., Koopmans, F., Pita-Illobre, D., Klaassen, R. V., Özer, B., Charalampopoulos, I., Smit, A. B., & Li, K. W. (2023). Suspension TRAPping Filter (sTRAP) Sample Preparation for Quantitative Proteomics in the Low µg Input Range Using a Plasmid DNA Micro-Spin Column: Analysis of the Hippocampus from the 5xFAD Alzheimer’s Disease Mouse Model. Cells, 12(9), 1242. https://doi.org/10.3390/cells12091242