
Dual Targeting of Glioblastoma Cells with Bispecific Killer Cell Engagers Directed to EGFR and ErbB2 (HER2) Facilitates Effective Elimination by NKG2D-CAR-Engineered NK Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Culture Conditions
2.2. NK-92 Cells Expressing an NKG2D-Based CAR and RD-IL15
2.3. Expression and Activity of RD-IL15
2.4. Production of Bispecific NKAB Antibodies
2.5. Generation of GL261 Glioblastoma Cells Expressing EGFR and ErbB2
2.6. Cytotoxicity Assays
2.7. Time-Lapse Microscopy
2.8. Statistical Analysis
3. Results
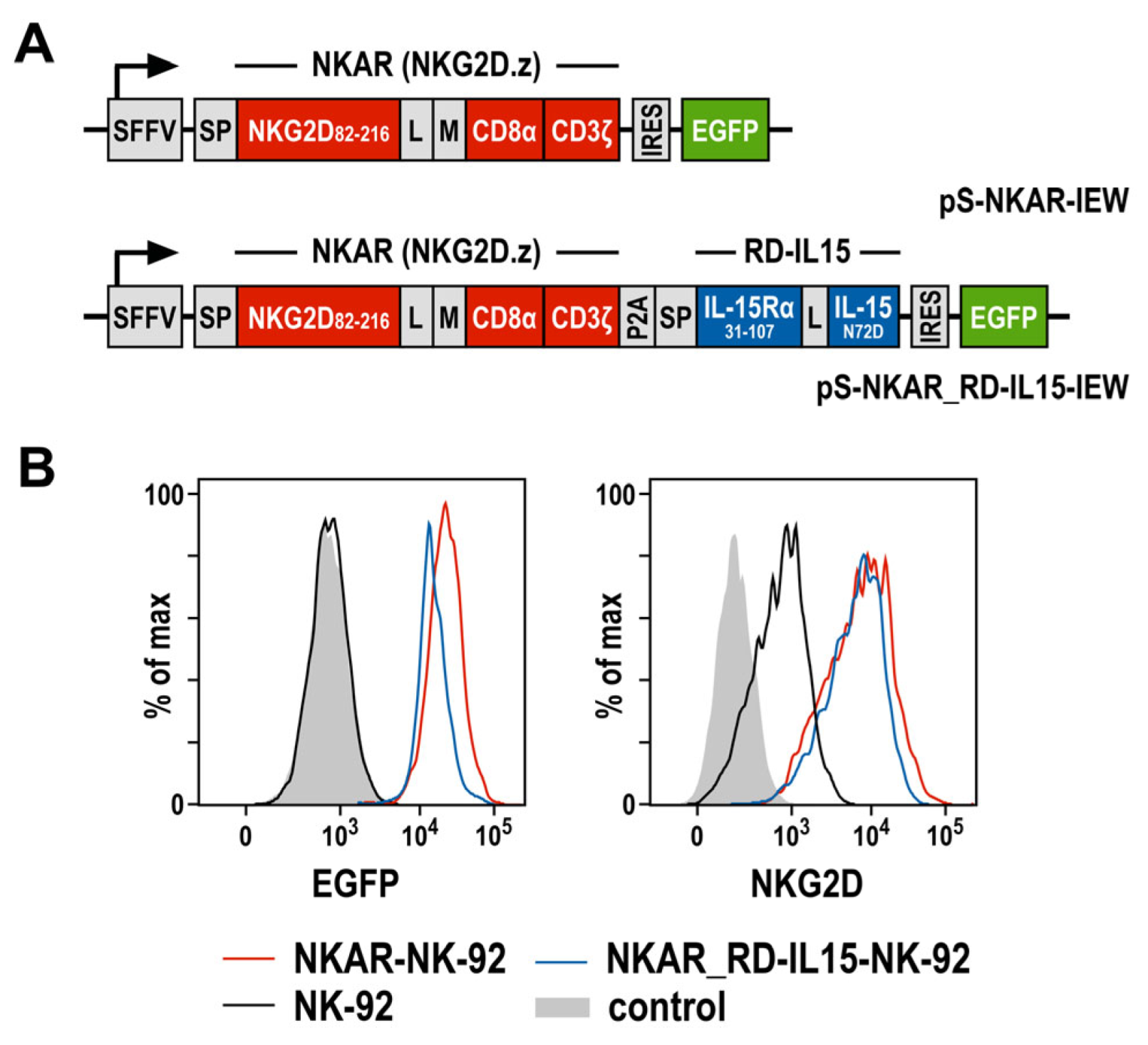
3.1. Generation of CAR-NK Cells Co-Expressing an NKG2D-Based CAR and an IL-15 Superagonist
3.2. Effect of RD-IL15 on NK Cell Activation and Proliferation
3.3. Generation of a Bispecific Killer Cell Engager Binding to NKG2D and Epidermal Growth Factor Receptor
3.4. Tumor-Specific Redirection of NKAR-Expressing NK-92 and Peripheral Blood-Derived NK Cells by NKAB-EGFR Antibody
3.5. Activity of NKAB-EGFR and NKAB-ErbB2 against Tumor Cells with Heterogeneous Target Antigen Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lanier, L.L. NKG2D receptor and its ligands in host defense. Cancer Immunol. Res. 2015, 3, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Stojanovic, A.; Correia, M.P.; Cerwenka, A. The NKG2D/NKG2DL axis in the crosstalk between lymphoid and myeloid cells in health and disease. Front. Immunol. 2018, 9, 827. [Google Scholar] [CrossRef] [PubMed]
- Lerner, E.C.; Woroniecka, K.I.; D’Anniballe, V.M.; Wilkinson, D.S.; Mohan, A.A.; Lorrey, S.J.; Waibl-Polania, J.; Wachsmuth, L.P.; Miggelbrink, A.M.; Jackson, J.D.; et al. CD8(+) T cells maintain killing of MHC-I-negative tumor cells through the NKG2D-NKG2DL axis. Nat. Cancer 2023, 4, 1258–1272. [Google Scholar] [CrossRef] [PubMed]
- Cerwenka, A.; Lanier, L.L. NKG2D ligands: Unconventional MHC class I-like molecules exploited by viruses and cancer. Tissue Antigens 2003, 61, 335–343. [Google Scholar] [CrossRef]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef]
- Tan, G.; Spillane, K.M.; Maher, J. The role and regulation of the NKG2D/NKG2D ligand system in cancer. Biology 2023, 12, 1079. [Google Scholar] [CrossRef]
- Demoulin, B.; Cook, W.J.; Murad, J.; Graber, D.J.; Sentman, M.L.; Lonez, C.; Gilham, D.E.; Sentman, C.L.; Agaugue, S. Exploiting natural killer group 2D receptors for CAR T-cell therapy. Future Oncol. 2017, 13, 1593–1605. [Google Scholar] [CrossRef]
- Curio, S.; Jonsson, G.; Marinović, S. A summary of current NKG2D-based CAR clinical trials. Immunother. Adv. 2021, 1, ltab018. [Google Scholar] [CrossRef]
- Sallman, D.A.; Brayer, J.; Sagatys, E.M.; Lonez, C.; Breman, E.; Agaugué, S.; Verma, B.; Gilham, D.E.; Lehmann, F.F.; Davila, M.L. NKG2D-based chimeric antigen receptor therapy induced remission in a relapsed/refractory acute myeloid leukemia patient. Haematologica 2018, 103, e424–e426. [Google Scholar] [CrossRef]
- Baumeister, S.H.; Murad, J.; Werner, L.; Daley, H.; Trebeden-Negre, H.; Gicobi, J.K.; Schmucker, A.; Reder, J.; Sentman, C.L.; Gilham, D.E.; et al. Phase I trial of autologous CAR T cells targeting NKG2D ligands in patients with AML/MDS and multiple myeloma. Cancer Immunol. Res. 2019, 7, 100–112. [Google Scholar] [CrossRef]
- Sallman, D.A.; Kerre, T.; Havelange, V.; Poiré, X.; Lewalle, P.; Wang, E.S.; Brayer, J.B.; Davila, M.L.; Moors, I.; Machiels, J.P.; et al. CYAD-01, an autologous NKG2D-based CAR T-cell therapy, in relapsed or refractory acute myeloid leukaemia and myelodysplastic syndromes or multiple myeloma (THINK): Haematological cohorts of the dose escalation segment of a phase 1 trial. Lancet Haematol. 2023, 10, e191–e202. [Google Scholar] [CrossRef]
- Paczulla, A.M.; Rothfelder, K.; Raffel, S.; Konantz, M.; Steinbacher, J.; Wang, H.; Tandler, C.; Mbarga, M.; Schaefer, T.; Falcone, M.; et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature 2019, 572, 254–259. [Google Scholar] [CrossRef]
- Ibáñez-Navarro, M.; Fernández, A.; Escudero, A.; Esteso, G.; Campos-Silva, C.; Navarro-Aguadero, M.; Leivas, A.; Caracuel, B.R.; Rodríguez-Antolín, C.; Ortiz, A.; et al. NKG2D-CAR memory T cells target pediatric T-cell acute lymphoblastic leukemia in vitro and in vivo but fail to eliminate leukemia initiating cells. Front. Immunol. 2023, 14, 1187665. [Google Scholar] [CrossRef]
- Salih, H.R.; Rammensee, H.G.; Steinle, A. Cutting edge: Down-regulation of MICA on human tumors by proteolytic shedding. J. Immunol. 2002, 169, 4098–4102. [Google Scholar] [CrossRef] [PubMed]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef]
- Song, H.; Kim, J.; Cosman, D.; Choi, I. Soluble ULBP suppresses natural killer cell activity via down-regulating NKG2D expression. Cell Immunol. 2006, 239, 22–30. [Google Scholar] [CrossRef]
- Kloess, S.; Huenecke, S.; Piechulek, D.; Esser, R.; Koch, J.; Brehm, C.; Soerensen, J.; Gardlowski, T.; Brinkmann, A.; Bader, P.; et al. IL-2-activated haploidentical NK cells restore NKG2D-mediated NK-cell cytotoxicity in neuroblastoma patients by scavenging of plasma MICA. Eur. J. Immunol. 2010, 40, 3255–3267. [Google Scholar] [CrossRef]
- Chitadze, G.; Lettau, M.; Bhat, J.; Wesch, D.; Steinle, A.; Fürst, D.; Mytilineos, J.; Kalthoff, H.; Janssen, O.; Oberg, H.H.; et al. Shedding of endogenous MHC class I-related chain molecules A and B from different human tumor entities: Heterogeneous involvement of the “a disintegrin and metalloproteases” 10 and 17. Int. J. Cancer 2013, 133, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Kellner, C.; Gramatzki, M.; Peipp, M. Promoting natural killer cell functions by recombinant immunoligands mimicking an induced self phenotype. Oncoimmunology 2013, 2, e24481. [Google Scholar] [CrossRef] [PubMed]
- Peipp, M.; Klausz, K.; Boje, A.S.; Zeller, T.; Zielonka, S.; Kellner, C. Immunotherapeutic targeting of activating natural killer cell receptors and their ligands in cancer. Clin. Exp. Immunol. 2022, 209, 22–32. [Google Scholar] [CrossRef]
- von Strandmann, E.P.; Hansen, H.P.; Reiners, K.S.; Schnell, R.; Borchmann, P.; Merkert, S.; Simhadri, V.R.; Draube, A.; Reiser, M.; Purr, I.; et al. A novel bispecific protein (ULBP2-BB4) targeting the NKG2D receptor on natural killer (NK) cells and CD138 activates NK cells and has potent antitumor activity against human multiple myeloma in vitro and in vivo. Blood 2006, 107, 1955–1962. [Google Scholar] [CrossRef]
- Rothe, A.; Jachimowicz, R.D.; Borchmann, S.; Madlener, M.; Keßler, J.; Reiners, K.S.; Sauer, M.; Hansen, H.P.; Ullrich, R.T.; Chatterjee, S.; et al. The bispecific immunoligand ULBP2-aCEA redirects natural killer cells to tumor cells and reveals potent anti-tumor activity against colon carcinoma. Int. J. Cancer 2014, 134, 2829–2840. [Google Scholar] [CrossRef]
- Kellner, C.; Günther, A.; Humpe, A.; Repp, R.; Klausz, K.; Derer, S.; Valerius, T.; Ritgen, M.; Brüggemann, M.; van de Winkel, J.G.; et al. Enhancing natural killer cell-mediated lysis of lymphoma cells by combining therapeutic antibodies with CD20-specific immunoligands engaging NKG2D or NKp30. Oncoimmunology 2016, 5, e1058459. [Google Scholar] [CrossRef]
- Pan, M.; Wang, F.; Nan, L.; Yang, S.; Qi, J.; Xie, J.; Shao, S.; Zou, H.; Wang, M.; Sun, F.; et al. αVEGFR2-MICA fusion antibodies enhance immunotherapy effect and synergize with PD-1 blockade. Cancer Immunol. Immunother. 2023, 72, 969–984. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.K.; Kang, S.; Youssef, Y.; Glankler, E.N.; Barrett, E.R.; Carter, A.M.; Ahmed, E.H.; Prasad, A.; Chen, L.; Zhang, J.; et al. A CS1-NKG2D bispecific antibody collectively activates cytolytic immune cells against multiple myeloma. Cancer Immunol. Res. 2018, 6, 776–787. [Google Scholar] [CrossRef]
- Raynaud, A.; Desrumeaux, K.; Vidard, L.; Termine, E.; Baty, D.; Chames, P.; Vigne, E.; Kerfelec, B. Anti-NKG2D single domain-based antibodies for the modulation of anti-tumor immune response. Oncoimmunology 2020, 10, 1854529. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Röder, J.; Scherer, A.; Bodden, M.; Pfeifer Serrahima, J.; Bhatti, A.; Waldmann, A.; Müller, N.; Oberoi, P.; Wels, W.S. Bispecific antibody-mediated redirection of NKG2D-CAR natural killer cells facilitates dual targeting and enhances antitumor activity. J. Immunother. Cancer 2021, 9, e002980. [Google Scholar] [CrossRef] [PubMed]
- Lutz, S.; Klausz, K.; Albici, A.M.; Ebinger, L.; Sellmer, L.; Teipel, H.; Frenzel, A.; Langner, A.; Winterberg, D.; Krohn, S.; et al. Novel NKG2D-directed bispecific antibodies enhance antibody-mediated killing of malignant B cells by NK cells and T cells. Front. Immunol. 2023, 14, 1227572. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Marcus, W.D.; Xu, W.; Lee, H.-I.; Han, K.; Egan, J.O.; Yovandich, J.L.; Rhode, P.R.; Wong, H.C. Novel human interleukin-15 agonists. J. Immunol. 2009, 183, 3598–3607. [Google Scholar] [CrossRef] [PubMed]
- Müller, D. Targeted cancer immunotherapy: Mimicking physiological trans-presentation of IL-15. Oncoimmunology 2012, 1, 1213–1214. [Google Scholar] [CrossRef]
- Bodden, M.; Häcker, A.; Röder, J.; Kiefer, A.; Zhang, C.; Bhatti, A.; Pfeifer Serrahima, J.; Ullrich, E.; Kühnel, I.; Wels, W.S. Co-expression of an IL-15 superagonist facilitates self-enrichment of GD(2)-targeted CAR-NK cells and mediates potent cell killing in the absence of IL-2. Cancers 2023, 15, 4310. [Google Scholar] [CrossRef]
- Akbasak, A.; Oldfield, E.H.; Saris, S.C. Expression and modulation of major histocompatibility antigens on murine primary brain tumor in vitro. J. Neurosurg. 1991, 75, 922–929. [Google Scholar] [CrossRef]
- Gong, J.H.; Maki, G.; Klingemann, H.G. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 1994, 8, 652–658. [Google Scholar]
- Demaison, C.; Parsley, K.; Brouns, G.; Scherr, M.; Battmer, K.; Kinnon, C.; Grez, M.; Thrasher, A.J. High-level transduction and gene expression in hematopoietic repopulating cells using a human immunodeficiency virus type 1-based lentiviral vector containing an internal spleen focus forming virus promoter. Hum. Gene Ther. 2002, 13, 803–813. [Google Scholar] [CrossRef]
- Schönfeld, K.; Sahm, C.; Zhang, C.; Naundorf, S.; Brendel, C.; Odendahl, M.; Nowakowska, P.; Bönig, H.; Köhl, U.; Kloess, S.; et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol. Ther. 2015, 23, 330–338. [Google Scholar] [CrossRef]
- Kwong, K.Y.; Baskar, S.; Zhang, H.; Mackall, C.L.; Rader, C. Generation, affinity maturation, and characterization of a human anti-human NKG2D monoclonal antibody with dual antagonistic and agonistic activity. J. Mol. Biol. 2008, 384, 1143–1156. [Google Scholar] [CrossRef] [PubMed]
- Genßler, S.; Burger, M.C.; Zhang, C.; Oelsner, S.; Mildenberger, I.; Wagner, M.; Steinbach, J.P.; Wels, W.S. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology 2016, 5, e1119354. [Google Scholar] [CrossRef]
- Pfeifer Serrahima, J.; Zhang, C.; Oberoi, P.; Bodden, M.; Röder, J.; Arndt, C.; Feldmann, A.; Kiefer, A.; Prüfer, M.; Kühnel, I.; et al. Multivalent adaptor proteins specifically target NK cells carrying a universal chimeric antigen receptor to ErbB2 (HER2)-expressing cancers. Cancer Immunol. Immunother. 2023, 72, 2905–2918. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Goebeler, M.E.; Bargou, R.C. T cell-engaging therapies-BiTEs and beyond. Nat. Rev. Clin. Oncol. 2020, 17, 418–434. [Google Scholar] [CrossRef] [PubMed]
- Phung, S.K.; Miller, J.S.; Felices, M. Bi-specific and tri-specific NK cell engagers: The new avenue of targeted NK cell immunotherapy. Mol. Diagn. Ther. 2021, 25, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.; Virone-Oddos, A.; Beninga, J.; Rossi, B.; Nicolazzi, C.; Amara, C.; Blanchard-Alvarez, A.; Gourdin, N.; Courta, J.; Basset, A.; et al. Control of acute myeloid leukemia by a trifunctional NKp46-CD16a-NK cell engager targeting CD123. Nat. Biotechnol. 2023, 41, 1296–1306. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Rivas, D.; Lee, S.H.; Thomas, J.S.; Kim, Y.J.; Cervantes, A.; Saavedra, O.; Shim, B.Y.; Kohlhas, L.; Hintzen, G.; et al. Leveraging innate immunity with AFM24, a novel CD16A and epidermal growth factor receptor (EGFR) bispecific innate cell engager: Interim results for the non-small cell lung cancer (NSCLC) cohort. J. Clin. Oncol. 2023, 41, 1. [Google Scholar] [CrossRef]
- Maskalenko, N.A.; Zhigarev, D.; Campbell, K.S. Harnessing natural killer cells for cancer immunotherapy: Dispatching the first responders. Nat. Rev. Drug Discov. 2022, 21, 559–577. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Xiao, L.; Cen, D.; Gan, H.; Sun, Y.; Huang, N.; Xiong, H.; Jin, Q.; Su, L.; Liu, X.; Wang, K.; et al. Adoptive transfer of NKG2D CAR mRNA-engineered natural killer cells in colorectal cancer patients. Mol. Ther. 2019, 27, 1114–1125. [Google Scholar] [CrossRef]
- Klingemann, H. The NK-92 cell line-30 years later: Its impact on natural killer cell research and treatment of cancer. Cytotherapy 2023, 25, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018, 8, 1083–1089. [Google Scholar]
- Burger, M.C.; Forster, M.T.; Romanski, A.; Straßheimer, F.; Macas, J.; Zeiner, P.S.; Steidl, E.; Herkt, S.; Weber, K.J.; Schupp, J.; et al. Intracranial injection of natural killer cells engineered with a HER2-targeted chimeric antigen receptor in patients with recurrent glioblastoma. Neuro Oncol. 2023, 25, 2058–2071. [Google Scholar] [CrossRef]
- Zhang, C.; Oberoi, P.; Oelsner, S.; Waldmann, A.; Lindner, A.; Tonn, T.; Wels, W.S. Chimeric antigen receptor-engineered NK-92 cells: An off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front. Immunol. 2017, 8, 533. [Google Scholar] [CrossRef]
- Tam, Y.K.; Maki, G.; Miyagawa, B.; Hennemann, B.; Tonn, T.; Klingemann, H.G. Characterization of genetically altered, interleukin 2-independent natural killer cell lines suitable for adoptive cellular immunotherapy. Hum. Gene Ther. 1999, 10, 1359–1373. [Google Scholar] [CrossRef] [PubMed]
- Sahm, C.; Schönfeld, K.; Wels, W.S. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol. Immunother. 2012, 61, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Felices, M.; Lenvik, A.J.; McElmurry, R.; Chu, S.; Hinderlie, P.; Bendzick, L.; Geller, M.A.; Tolar, J.; Blazar, B.R.; Miller, J.S. Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI Insight 2018, 3, e96219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, J.; Niu, J.; Zhang, J.; Tian, Z. Interleukin-15 improves cytotoxicity of natural killer cells via up-regulating NKG2D and cytotoxic effector molecule expression as well as STAT1 and ERK1/2 phosphorylation. Cytokine 2008, 42, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Yang, H.; Xiong, H.; Luo, K.Q. NK cell exhaustion in the tumor microenvironment. Front. Immunol. 2023, 14, 1303605. [Google Scholar] [CrossRef]
- Wagner, J.; Wickman, E.; DeRenzo, C.; Gottschalk, S. CAR T cell therapy for solid tumors: Bright future or dark reality? Mol. Ther. 2020, 28, 2320–2339. [Google Scholar] [CrossRef]
- Crucitta, S.; Cucchiara, F.; Mathijssen, R.; Mateo, J.; Jager, A.; Joosse, A.; Passaro, A.; Attili, I.; Petrini, I.; van Schaik, R.; et al. Treatment-driven tumour heterogeneity and drug resistance: Lessons from solid tumours. Cancer Treat. Rev. 2022, 104, 102340. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiefer, A.; Prüfer, M.; Röder, J.; Pfeifer Serrahima, J.; Bodden, M.; Kühnel, I.; Oberoi, P.; Wels, W.S. Dual Targeting of Glioblastoma Cells with Bispecific Killer Cell Engagers Directed to EGFR and ErbB2 (HER2) Facilitates Effective Elimination by NKG2D-CAR-Engineered NK Cells. Cells 2024, 13, 246. https://doi.org/10.3390/cells13030246
Kiefer A, Prüfer M, Röder J, Pfeifer Serrahima J, Bodden M, Kühnel I, Oberoi P, Wels WS. Dual Targeting of Glioblastoma Cells with Bispecific Killer Cell Engagers Directed to EGFR and ErbB2 (HER2) Facilitates Effective Elimination by NKG2D-CAR-Engineered NK Cells. Cells. 2024; 13(3):246. https://doi.org/10.3390/cells13030246
Chicago/Turabian StyleKiefer, Anne, Maren Prüfer, Jasmin Röder, Jordi Pfeifer Serrahima, Malena Bodden, Ines Kühnel, Pranav Oberoi, and Winfried S. Wels. 2024. "Dual Targeting of Glioblastoma Cells with Bispecific Killer Cell Engagers Directed to EGFR and ErbB2 (HER2) Facilitates Effective Elimination by NKG2D-CAR-Engineered NK Cells" Cells 13, no. 3: 246. https://doi.org/10.3390/cells13030246
APA StyleKiefer, A., Prüfer, M., Röder, J., Pfeifer Serrahima, J., Bodden, M., Kühnel, I., Oberoi, P., & Wels, W. S. (2024). Dual Targeting of Glioblastoma Cells with Bispecific Killer Cell Engagers Directed to EGFR and ErbB2 (HER2) Facilitates Effective Elimination by NKG2D-CAR-Engineered NK Cells. Cells, 13(3), 246. https://doi.org/10.3390/cells13030246