
Mitochondria: A Promising Convergent Target for the Treatment of Amyotrophic Lateral Sclerosis

 ,
,  , and
, and
Abstract
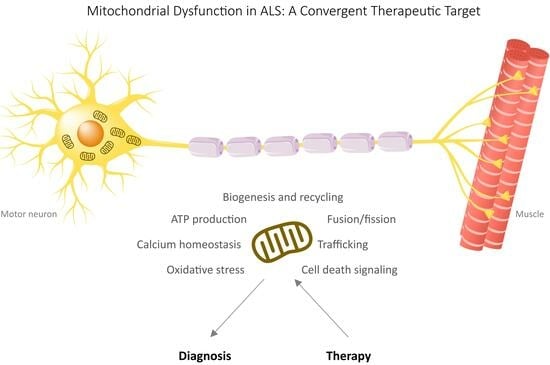
:
1. Introduction
2. Mitochondrial Dysfunctions in ALS
2.1. Alterations in Mitochondrial Respiration and ATP Production
2.2. Role of Oxidative Stress Mechanisms
2.3. Metabolic Dysregulation
2.4. Mitochondrial Dynamics and Biogenesis
2.5. Mitochondrial Trafficking in ALS
2.6. Mitochondria and Endoplasmic Reticulum Crosstalk
2.7. Autophagy and Mitophagy
2.8. Apoptotic Mechanisms
2.9. Mitochondrial Dysfunction as Cause and/or Consequence in ALS
3. Preclinical and Clinical Endeavors Targeting Mitochondria
3.1. Approaches for Direct Enhancement of Mitochondrial Function
3.1.1. Dichloroacetate
3.1.2. Ketogenic and High-Fat Diets
3.1.3. Acetyl-Carnitine
3.2. Antioxidants
3.2.1. N-acetyl-L-cysteine (NAC)
3.2.2. Edaravone
3.2.3. Melatonin
3.2.4. Mitochondria-Targeted Antioxidants
3.3. Antiapoptotic Agents
3.3.1. mPTP-Targeting Agents
3.3.2. Rasagiline
4. Conclusions
- Restoring Energy Balance: Mitochondrial dysfunction in ALS often leads to a decline in cellular energy production, which is particularly detrimental to highly energy-demanding MNs. Strategies aimed at rejuvenating mitochondrial function and ensuring an adequate supply of ATP hold promise in sustaining motor neuron health and function.
- Attenuating Oxidative Stress: Mitochondria are both sources and targets of oxidative stress in ALS. Therapies aimed at reducing mitochondrial-generated ROS and bolstering endogenous antioxidant defenses offer potential avenues for alleviating oxidative damage.
- Regulating Calcium Homeostasis: Impaired calcium homeostasis in ALS contributes to mitochondrial dysfunction and subsequent neuronal death. Interventions that restore the calcium balance within neurons and mitochondria may mitigate excitotoxicity and improve neuronal survival.
- Enhancing Mitochondrial Dynamics: Maintaining a healthy mitochondrial network through processes such as fission, fusion, and mitophagy is crucial for cellular health. Targeting these dynamic processes to ensure the timely removal of damaged mitochondria and the efficient distribution of healthy ones may have therapeutic implications.
- Unveiling Genetic Insights: Genetic mutations associated with ALS, such as those in the SOD1, C9ORF72, and FUS genes, can directly impact mitochondrial function. Understanding the precise mechanisms by which these mutations affect mitochondria could pave the way for gene-specific therapies.
Funding
Conflicts of Interest
References
- Longinetti, E.; Fang, F. Epidemiology of Amyotrophic Lateral Sclerosis: An Update of Recent Literature. Curr. Opin. Neurol. 2019, 32, 771–776. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The Role of Mitochondria in Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Valdmanis, P.N.; Rouleau, G.A. Genetics of Familial Amyotrophic Lateral Sclerosis. Neurology 2008, 70, 144–152. [Google Scholar] [CrossRef]
- Zarei, S.; Carr, K.; Reiley, L.; Diaz, K.; Guerra, O.; Altamirano, P.; Pagani, W.; Lodin, D.; Orozco, G.; Chinea, A. A Comprehensive Review of Amyotrophic Lateral Sclerosis. Surg. Neurol. Int. 2015, 6, 171. [Google Scholar] [CrossRef]
- Boylan, K. Familial Amyotrophic Lateral Sclerosis. Neurol. Clin. 2015, 33, 807–830. [Google Scholar] [CrossRef]
- Kodavati, M.; Wang, H.; Hegde, M.L. Altered Mitochondrial Dynamics in Motor Neuron Disease: An Emerging Perspective. Cells 2020, 9, 1065. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Deng, H.X.; Hentati, A.; Tainer, J.A.; Iqbal, Z.; Cayabyab, A.; Hung, W.Y.; Getzoff, E.D.; Hu, P.; Herzfeldt, B.; Roos, R.P.; et al. Amyotrophic Lateral Sclerosis and Structural Defects in Cu,Zn Superoxide Dismutase. Science 1993, 261, 1047–1051. [Google Scholar] [CrossRef]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 Mediated Pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn Superoxide Dismutase Gene Are Associated with Familial Amyotrophic Lateral Sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Synofzik, M.; Ronchi, D.; Keskin, I.; Basak, A.N.; Wilhelm, C.; Gobbi, C.; Birve, A.; Biskup, S.; Zecca, C.; Fernandez-Santiago, R.; et al. Mutant Superoxide Dismutase-1 Indistinguishable from Wild-Type Causes ALS. Hum. Mol. Genet. 2012, 21, 3568–3574. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA Processing Protein, Cause Familial Amyotrophic Lateral Sclerosis Type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef]
- Lacorte, E.; Ferrigno, L.; Leoncini, E.; Corbo, M.; Boccia, S.; Vanacore, N. Physical Activity, and Physical Activity Related to Sports, Leisure and Occupational Activity as Risk Factors for ALS: A Systematic Review. Neurosci. Biobehav. Rev. 2016, 66, 61–79. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Hardiman, O. The Epidemiology of ALS: A Conspiracy of Genes, Environment and Time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef]
- Yu, B.; Pamphlett, R. Environmental Insults: Critical Triggers for Amyotrophic Lateral Sclerosis. Transl. Neurodegener. 2017, 6, 15. [Google Scholar] [CrossRef]
- Ingre, C.; Roos, P.M.; Piehl, F.; Kamel, F.; Fang, F. Risk Factors for Amyotrophic Lateral Sclerosis. Clin. Epidemiol. 2015, 7, 181–193. [Google Scholar] [CrossRef]
- Tafuri, F.; Ronchi, D.; Magri, F.; Comi, G.P.; Corti, S. SOD1 Misplacing and Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis Pathogenesis. Front. Cell. Neurosci. 2015, 9, 336. [Google Scholar] [CrossRef]
- Bacman, S.R.; Bradley, W.G.; Moraes, C.T. Mitochondrial Involvement in Amyotrophic Lateral Sclerosis: Trigger or Target? Mol. Neurobiol. 2006, 33, 113–131. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Montezinho, L.; Mendes, C.; Firuzi, O.; Saso, L.; Oliveira, P.J.; Silva, F.S.G. Oxidative Stress in Amyotrophic Lateral Sclerosis: Pathophysiology and Opportunities for Pharmacological Intervention. Oxidative Med. Cell. Longev. 2020, 2020, 5021694. [Google Scholar] [CrossRef] [PubMed]
- Curti, D.; Malaspina, A.; Facchetti, G.; Camana, C.; Mazzini, L.; Tosca, P.; Zerbi, F.; Ceroni, M. Amyotrophic Lateral Sclerosis: Oxidative Energy Metabolism and Calcium Homeostasis in Peripheral Blood Lymphocytes. Neurology 1996, 47, 1060–1064. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, F.R.; Winkler, K.; Kuznetsov, A.V.; Bartels, C.; Vielhaber, S.; Feistner, H.; Kunz, W.S. Impairment of Mitochondrial Function in Skeletal Muscle of Patients with Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 1998, 156, 65–72. [Google Scholar] [CrossRef]
- Vielhaber, S.; Winkler, K.; Kirches, E.; Kunz, D.; Buchner, M.; Feistner, H.; Elger, C.E.; Ludolph, A.C.; Riepe, M.W.; Kunz, W.S. Visualization of Defective Mitochondrial Function in Skeletal Muscle Fibers of Patients with Sporadic Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 1999, 169, 133–139. [Google Scholar] [CrossRef]
- Nakano, Y.; Hirayama, K.; Terao, K. Hepatic Ultrastructural Changes and Liver Dysfunction in Amyotrophic Lateral Sclerosis. Arch. Neurol. 1987, 44, 103–106. [Google Scholar] [CrossRef]
- Palomo, G.M.; Manfredi, G. Exploring New Pathways of Neurodegeneration in ALS: The Role of Mitochondria Quality Control. Brain Res. 2015, 1607, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Carri, M.T. Mitochondrial Dysfunction in ALS. Prog. Neurobiol. 2012, 97, 54–66. [Google Scholar] [CrossRef]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative Diseases: Pathogenesis and Treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef]
- Vande Velde, C.; McDonald, K.K.; Boukhedimi, Y.; McAlonis-Downes, M.; Lobsiger, C.S.; Bel Hadj, S.; Zandona, A.; Julien, J.P.; Shah, S.B.; Cleveland, D.W. Misfolded SOD1 Associated with Motor Neuron Mitochondria Alters Mitochondrial Shape and Distribution Prior to Clinical Onset. PLoS ONE 2011, 6, e22031. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yamashita, T.; Tian, F.; Morimoto, N.; Ikeda, Y.; Deguchi, K.; Abe, K. Mitochondrial Fusion and Fission Proteins Expression Dynamically Change in a Murine Model of Amyotrophic Lateral Sclerosis. Curr. Neurovasc. Res. 2013, 10, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Xu, Z. Massive Mitochondrial Degeneration in Motor Neurons Triggers the Onset of Amyotrophic Lateral Sclerosis in Mice Expressing a Mutant SOD1. J. Neurosci. 1998, 18, 3241–3250. [Google Scholar] [CrossRef]
- Deng, J.; Yang, M.; Chen, Y.; Chen, X.; Liu, J.; Sun, S.; Cheng, H.; Li, Y.; Bigio, E.H.; Mesulam, M.; et al. FUS Interacts with HSP60 to Promote Mitochondrial Damage. PLoS Genet. 2015, 11, e1005357. [Google Scholar] [CrossRef]
- Stribl, C.; Samara, A.; Trumbach, D.; Peis, R.; Neumann, M.; Fuchs, H.; Gailus-Durner, V.; Hrabe de Angelis, M.; Rathkolb, B.; Wolf, E.; et al. Mitochondrial Dysfunction and Decrease in Body Weight of a Transgenic Knock-in Mouse Model for TDP-43. J. Biol. Chem. 2014, 289, 10769–10784. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS Disease-Associated Mutant TDP-43 Impairs Mitochondrial Dynamics and Function in Motor Neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef]
- Choi, S.Y.; Lopez-Gonzalez, R.; Krishnan, G.; Phillips, H.L.; Li, A.N.; Seeley, W.W.; Yao, W.D.; Almeida, S.; Gao, F.B. C9ORF72-ALS/FTD-Associated Poly(GR) Binds Atp5a1 and Compromises Mitochondrial Function in Vivo. Nat. Neurosci. 2019, 22, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H., Jr. Amyotrophic Lateral Sclerosis-Associated SOD1 Mutant Proteins Bind and Aggregate with Bcl-2 in Spinal Cord Mitochondria. Neuron 2004, 43, 19–30. [Google Scholar] [CrossRef]
- Israelson, A.; Arbel, N.; Da Cruz, S.; Ilieva, H.; Yamanaka, K.; Shoshan-Barmatz, V.; Cleveland, D.W. Misfolded Mutant SOD1 Directly Inhibits VDAC1 Conductance in a Mouse Model of Inherited ALS. Neuron 2010, 67, 575–587. [Google Scholar] [CrossRef]
- Kawamata, H.; Magrane, J.; Kunst, C.; King, M.P.; Manfredi, G. Lysyl-tRNA Synthetase is a Target for Mutant SOD1 Toxicity in Mitochondria. J. Biol. Chem. 2008, 283, 28321–28328. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; da Rocha, E.L.; et al. The Inhibition of TDP-43 Mitochondrial Localization Blocks Its Neuronal Toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.L.; Coady, T.H.; Lu, L.; Zheng, D.; Alland, I.; Tian, B.; Shneider, N.A.; Manley, J.L. ALS/FTD-Associated Protein FUS Induces Mitochondrial Dysfunction by Preferentially Sequestering Respiratory Chain Complex mRNAs. Genes Dev. 2020, 34, 785–805. [Google Scholar] [CrossRef]
- Blokhuis, A.M.; Koppers, M.; Groen, E.J.N.; van den Heuvel, D.M.A.; Dini Modigliani, S.; Anink, J.J.; Fumoto, K.; van Diggelen, F.; Snelting, A.; Sodaar, P.; et al. Comparative Interactomics Analysis of Different ALS-Associated Proteins Identifies Converging Molecular Pathways. Acta Neuropathol. 2016, 132, 175–196. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.B. Poly(GR) in C9ORF72-Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in iPSC-Derived Motor Neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Kausar, S.; Wang, F.; Cui, H. The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef]
- Wiedemann, F.R.; Manfredi, G.; Mawrin, C.; Beal, M.F.; Schon, E.A. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J. Neurochem. 2002, 80, 616–625. [Google Scholar] [CrossRef]
- Browne, S.E.; Bowling, A.C.; Baik, M.J.; Gurney, M.; Brown, R.H., Jr.; Beal, M.F. Metabolic dysfunction in familial, but not sporadic, amyotrophic lateral sclerosis. J. Neurochem. 1998, 71, 281–287. [Google Scholar] [CrossRef]
- Vielhaber, S.; Kunz, D.; Winkler, K.; Wiedemann, F.R.; Kirches, E.; Feistner, H.; Heinze, H.J.; Elger, C.E.; Schubert, W.; Kunz, W.S. Mitochondrial DNA Abnormalities in Skeletal Muscle of Patients with Sporadic Amyotrophic Lateral Sclerosis. Brain 2000, 123 Pt 7, 1339–1348. [Google Scholar] [CrossRef]
- Ghiasi, P.; Hosseinkhani, S.; Noori, A.; Nafissi, S.; Khajeh, K. Mitochondrial complex I Deficiency and ATP/ADP Ratio in Lymphocytes of Amyotrophic Lateral Sclerosis Patients. Neurol. Res. 2012, 34, 297–303. [Google Scholar] [CrossRef]
- Kirk, K.; Gennings, C.; Hupf, J.C.; Tadesse, S.; D’Aurelio, M.; Kawamata, H.; Valsecchi, F.; Mitsumoto, H.; Groups, A.P.C.S.; Manfredi, G. Bioenergetic Markers in Skin Fibroblasts of Sporadic Amyotrophic Lateral Sclerosis and Progressive Lateral Sclerosis Patients. Ann. Neurol. 2014, 76, 620–624. [Google Scholar] [CrossRef]
- Konrad, C.; Kawamata, H.; Bredvik, K.G.; Arreguin, A.J.; Cajamarca, S.A.; Hupf, J.C.; Ravits, J.M.; Miller, T.M.; Maragakis, N.J.; Hales, C.M.; et al. Fibroblast Bioenergetics to Classify Amyotrophic Lateral Sclerosis Patients. Mol. Neurodegener. 2017, 12, 76. [Google Scholar] [CrossRef] [PubMed]
- Mattiazzi, M.; D’Aurelio, M.; Gajewski, C.D.; Martushova, K.; Kiaei, M.; Beal, M.F.; Manfredi, G. Mutated Human SOD1 Causes Dysfunction of Oxidative Phosphorylation in Mitochondria of Transgenic Mice. J. Biol. Chem. 2002, 277, 29626–29633. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Higgins, C.M.; Xu, Z. Mitochondrial Electron Transport Chain Complex Dysfunction in a Transgenic Mouse Model for Amyotrophic Lateral Sclerosis. J. Neurochem. 2002, 83, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Kirkinezos, I.G.; Bacman, S.R.; Hernandez, D.; Oca-Cossio, J.; Arias, L.J.; Perez-Pinzon, M.A.; Bradley, W.G.; Moraes, C.T. Cytochrome C Association with the Inner Mitochondrial Membrane is Impaired in the CNS of G93A-SOD1 Mice. J. Neurosci. 2005, 25, 164–172. [Google Scholar] [CrossRef]
- Menzies, F.M.; Cookson, M.R.; Taylor, R.W.; Turnbull, D.M.; Chrzanowska-Lightowlers, Z.M.; Dong, L.; Figlewicz, D.A.; Shaw, P.J. Mitochondrial Dysfunction in a Cell Culture Model of Familial Amyotrophic Lateral Sclerosis. Brain 2002, 125, 1522–1533. [Google Scholar] [CrossRef] [PubMed]
- Calabria, E.; Scambi, I.; Bonafede, R.; Schiaffino, L.; Peroni, D.; Potrich, V.; Capelli, C.; Schena, F.; Mariotti, R. ASCs-Exosomes Recover Coupling Efficiency and Mitochondrial Membrane Potential in an in Vitro Model of ALS. Front. Neurosci. 2019, 13, 1070. [Google Scholar] [CrossRef] [PubMed]
- Bowling, A.C.; Schulz, J.B.; Brown, R.H., Jr.; Beal, M.F. Superoxide Dismutase Activity, Oxidative Damage, and Mitochondrial Energy Metabolism in Familial and Sporadic Amyotrophic Lateral Sclerosis. J. Neurochem. 1993, 61, 2322–2325. [Google Scholar] [CrossRef]
- Jaarsma, D.; Rognoni, F.; van Duijn, W.; Verspaget, H.W.; Haasdijk, E.D.; Holstege, J.C. CuZn Superoxide Dismutase (SOD1) Accumulates in Vacuolated Mitochondria in Transgenic Mice Expressing Amyotrophic Lateral Sclerosis-Linked SOD1 Mutations. Acta Neuropathol. 2001, 102, 293–305. [Google Scholar] [CrossRef]
- Higgins, C.M.J.; Jung, C.; Ding, H.; Xu, Z. Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J. Neurosci. 2002, 22, RC215. [Google Scholar] [CrossRef]
- Liu, J.; Lillo, C.; Jonsson, P.A.; Vande Velde, C.; Ward, C.M.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Andersen, P.M.; et al. Toxicity of Familial ALS-Linked SOD1 Mutants from Selective Recruitment to Spinal Mitochondria. Neuron 2004, 43, 5–17. [Google Scholar] [CrossRef]
- Bergemalm, D.; Jonsson, P.A.; Graffmo, K.S.; Andersen, P.M.; Brannstrom, T.; Rehnmark, A.; Marklund, S.L. Overloading of Stable and Exclusion of Unstable Human Superoxide Dismutase-1 Variants in Mitochondria of Murine Amyotrophic Lateral Sclerosis Models. J. Neurosci. 2006, 26, 4147–4154. [Google Scholar] [CrossRef]
- Deng, H.X.; Shi, Y.; Furukawa, Y.; Zhai, H.; Fu, R.; Liu, E.; Gorrie, G.H.; Khan, M.S.; Hung, W.Y.; Bigio, E.H.; et al. Conversion to the Amyotrophic Lateral Sclerosis Phenotype is Associated with Intermolecular Linked Insoluble Aggregates of SOD1 in Mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7142–7147. [Google Scholar] [CrossRef]
- Vande Velde, C.; Miller, T.M.; Cashman, N.R.; Cleveland, D.W. Selective Association of Misfolded ALS-Linked Mutant SOD1 with the Cytoplasmic Face of Mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 4022–4027. [Google Scholar] [CrossRef]
- Vijayvergiya, C.; Beal, M.F.; Buck, J.; Manfredi, G. Mutant Superoxide Dismutase 1 Forms Aggregates in the Brain Mitochondrial Matrix of Amyotrophic Lateral Sclerosis Mice. J. Neurosci. 2005, 25, 2463–2470. [Google Scholar] [CrossRef]
- Philips, T.; Rothstein, J.D. Rodent Models of Amyotrophic Lateral Sclerosis. Curr. Protoc. Pharmacol. 2015, 69, 5–67. [Google Scholar] [CrossRef]
- Ludolph, A.C.; Bendotti, C.; Blaugrund, E.; Chio, A.; Greensmith, L.; Loeffler, J.P.; Mead, R.; Niessen, H.G.; Petri, S.; Pradat, P.F.; et al. Guidelines for Preclinical Animal Research in ALS/MND: A Consensus Meeting. Amyotroph. Lateral Scler. 2010, 11, 38–45. [Google Scholar] [CrossRef]
- Kim, B.W.; Ryu, J.; Jeong, Y.E.; Kim, J.; Martin, L.J. Human Motor Neurons with SOD1-G93A Mutation Generated from CRISPR/Cas9 Gene-Edited iPSCs Develop Pathological Features of Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 2020, 14, 604171. [Google Scholar] [CrossRef]
- Lu, J.; Duan, W.; Guo, Y.; Jiang, H.; Li, Z.; Huang, J.; Hong, K.; Li, C. Mitochondrial Dysfunction in Human TDP-43 Transfected NSC34 Cell Lines and the Protective Effect of Dimethoxy Curcumin. Brain Res. Bull. 2012, 89, 185–190. [Google Scholar] [CrossRef]
- Wang, M.D.; Little, J.; Gomes, J.; Cashman, N.R.; Krewski, D. Identification of Risk Factors Associated with Onset and Progression of Amyotrophic Lateral Sclerosis Using Systematic Review and Meta-Analysis. Neurotoxicology 2016, 61, 101–130. [Google Scholar] [CrossRef]
- Warraich, S.T.; Yang, S.; Nicholson, G.A.; Blair, I.P. TDP-43: A DNA and RNA Binding Protein with Roles in Neurodegenerative Diseases. Int. J. Biochem. Cell Biol. 2010, 42, 1606–1609. [Google Scholar] [CrossRef]
- Onesto, E.; Colombrita, C.; Gumina, V.; Borghi, M.O.; Dusi, S.; Doretti, A.; Fagiolari, G.; Invernizzi, F.; Moggio, M.; Tiranti, V.; et al. Gene-Specific Mitochondria Dysfunctions in Human TARDBP and C9ORF72 Fibroblasts. Acta Neuropathol. Commun. 2016, 4, 47. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Takanashi, K. FUS Interacts with Nuclear Matrix-Associated Protein SAFB1 as Well as Matrin3 to Regulate Splicing and Ligand-Mediated Transcription. Sci. Rep. 2016, 6, 35195. [Google Scholar] [CrossRef]
- Lai, J.D.; Ichida, J.K. C9ORF72 Protein Function and Immune Dysregulation in Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2019, 713, 134523. [Google Scholar] [CrossRef] [PubMed]
- Dafinca, R.; Scaber, J.; Ababneh, N.; Lalic, T.; Weir, G.; Christian, H.; Vowles, J.; Douglas, A.G.; Fletcher-Jones, A.; Browne, C.; et al. C9orf72 Hexanucleotide Expansions Are Associated with Altered Endoplasmic Reticulum Calcium Homeostasis and Stress Granule Formation in Induced Pluripotent Stem Cell-Derived Neurons from Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Stem Cells 2016, 34, 2063–2078. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A. The Role of Mitochondria in Reactive Oxygen Species Metabolism and Signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Esra, B.; MuratUmit, S.; Cansin, S.; Serpil, E.; Omer, K. Oxidative Stress and Antioxidant Defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar]
- Barber, S.C.; Mead, R.J.; Shaw, P.J. Oxidative Stress in ALS: A Mechanism of Neurodegeneration and a Therapeutic Target. Biochim. Biophys. Acta 2006, 1762, 1051–1067. [Google Scholar] [CrossRef]
- Bodega, G.; Alique, M.; Puebla, L.; Carracedo, J.; Ramírez, R.M. Microvesicles: ROS scavengers and ROS producers. J. Extracell. Vesicles 2019, 8, 1626654. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Calingasan, N.Y.; Chen, J.; Kiaei, M.; Beal, M.F. Beta-Amyloid 42 Accumulation in the Lumbar Spinal Cord Motor Neurons of Amyotrophic Lateral Sclerosis Patients. Neurobiol. Dis. 2005, 19, 340–347. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H., Jr.; Beal, M.F. Evidence of Increased Oxidative Damage in Both Sporadic and Familial Amyotrophic Lateral Sclerosis. J. Neurochem. 1997, 69, 2064–2074. [Google Scholar] [CrossRef]
- Shaw, P.J.; Ince, P.G.; Falkous, G.; Mantle, D. Oxidative Damage to Protein in Sporadic Motor Neuron Disease Spinal Cord. Ann. Neurol. 1995, 38, 691–695. [Google Scholar] [CrossRef]
- Simpson, E.P.; Henry, Y.K.; Henkel, J.S.; Smith, R.G.; Appel, S.H. Increased Lipid Peroxidation in Sera of ALS Patients: A Potential Biomarker of Disease Burden. Neurology 2004, 62, 1758–1765. [Google Scholar] [CrossRef]
- Smith, R.G.; Henry, Y.K.; Mattson, M.P.; Appel, S.H. Presence of 4-Hydroxynonenal in Cerebrospinal Fluid of Patients with Sporadic Amyotrophic Lateral Sclerosis. Ann. Neurol. 1998, 44, 696–699. [Google Scholar] [CrossRef]
- Ihara, Y.; Nobukuni, K.; Takata, H.; Hayabara, T. Oxidative Stress and Metal Content in Blood and Cerebrospinal Fluid of Amyotrophic Lateral Sclerosis Patients with and without a Cu, Zn-Superoxide Dismutase Mutation. Neurol. Res. 2005, 27, 105–108. [Google Scholar] [CrossRef]
- Tohgi, H.; Abe, T.; Yamazaki, K.; Murata, T.; Ishizaki, E.; Isobe, C. Remarkable Increase in Cerebrospinal Fluid 3-Nitrotyrosine in Patients with Sporadic Amyotrophic Lateral Sclerosis. Ann. Neurol. 1999, 46, 129–131. [Google Scholar] [CrossRef]
- Weishaupt, J.H.; Bartels, C.; Polking, E.; Dietrich, J.; Rohde, G.; Poeggeler, B.; Mertens, N.; Sperling, S.; Bohn, M.; Huther, G.; et al. Reduced Oxidative Damage in ALS by High-Dose Enteral Melatonin Treatment. J. Pineal Res. 2006, 41, 313–323. [Google Scholar] [CrossRef]
- Mitsumoto, H.; Santella, R.M.; Liu, X.; Bogdanov, M.; Zipprich, J.; Wu, H.C.; Mahata, J.; Kilty, M.; Bednarz, K.; Bell, D.; et al. Oxidative Stress Biomarkers in Sporadic ALS. Amyotroph. Lateral Scler. 2008, 9, 177–183. [Google Scholar] [CrossRef]
- Niebrój-Dobosz, I.; Dziewulska, D.; Kwieciński, H. Oxidative Damage to Proteins in the Spinal Cord in Amyotrophic Lateral Sclerosis (ALS). Folia Neuropathol. 2004, 42, 151–156. [Google Scholar]
- Shibata, N.; Nagai, R.; Uchida, K.; Horiuchi, S.; Yamada, S.; Hirano, A.; Kawaguchi, M.; Yamamoto, T.; Sasaki, S.; Kobayashi, M. Morphological Evidence for Lipid Peroxidation and Protein Glycoxidation in Spinal Cords from Sporadic Amyotrophic Lateral Sclerosis Patients. Brain Res. 2001, 917, 97–104. [Google Scholar] [CrossRef]
- Pedersen, W.A.; Fu, W.; Keller, J.N.; Markesbery, W.R.; Appel, S.; Smith, R.G.; Kasarskis, E.; Mattson, M.P. Protein Modification by the Lipid Peroxidation Product 4-Hydroxynonenal in the Spinal Cords of Amyotrophic Lateral Sclerosis Patients. Ann. Neurol. 1998, 44, 819–824. [Google Scholar] [CrossRef]
- Beal, M.F.; Ferrante, R.J.; Browne, S.E.; Matthews, R.T.; Kowall, N.W.; Brown, R.H., Jr. Increased 3-Nitrotyrosine in Both Sporadic and Familial Amyotrophic Lateral Sclerosis. Ann. Neurol. 1997, 42, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Pan, L.H.; Watanabe, M.; Konno, H.; Kato, T.; Itoyama, Y. Upregulation of Protein-Tyrosine Nitration in the Anterior Horn Cells of Amyotrophic Lateral Sclerosis. Neurol. Res. 1997, 19, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Pan, L.H.; Watanabe, M.; Kato, T.; Itoyama, Y. Induction of Nitrotyrosine-Like Immunoreactivity in the Lower Motor Neuron of Amyotrophic Lateral Sclerosis. Neurosci. Lett. 1995, 199, 152–154. [Google Scholar] [CrossRef] [PubMed]
- Babu, G.N.; Kumar, A.; Chandra, R.; Puri, S.K.; Singh, R.L.; Kalita, J.; Misra, U.K. Oxidant-Antioxidant Imbalance in the Erythrocytes of Sporadic Amyotrophic Lateral Sclerosis Patients Correlates with the Progression of Disease. Neurochem. Int. 2008, 52, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Bozzo, F.; Mirra, A.; Carri, M.T. Oxidative Stress and Mitochondrial Damage in the Pathogenesis of ALS: New Perspectives. Neurosci. Lett. 2017, 636, 3–8. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Silva, D.F.; Segura, L.; Baldeiras, I.; Marques, R.; Rosenstock, T.; Oliveira, P.J.; Silva, F.S.G. Redox Profiles of Amyotrophic Lateral Sclerosis Lymphoblasts with or without Known SOD1 Mutations. Eur. J. Clin. Investig. 2022, 52, e13798. [Google Scholar] [CrossRef] [PubMed]
- Field, L.S.; Furukawa, Y.; O’Halloran, T.V.; Culotta, V.C. Factors Controlling the Uptake of Yeast Copper/Zinc Superoxide Dismutase into Mitochondria. J. Biol. Chem. 2003, 278, 28052–28059. [Google Scholar] [CrossRef]
- Kawamata, H.; Manfredi, G. Different Regulation of Wild-Type and Mutant Cu,Zn Superoxide Dismutase Localization in Mammalian Mitochondria. Hum. Mol. Genet. 2008, 17, 3303–3317. [Google Scholar] [CrossRef]
- Reddehase, S.; Grumbt, B.; Neupert, W.; Hell, K. The Disulfide Relay System of Mitochondria is Required for the Biogenesis of Mitochondrial CCS1 and SOD1. J. Mol. Biol. 2009, 385, 331–338. [Google Scholar] [CrossRef]
- Arciello, M.; Capo, C.R.; D’Annibale, S.; Cozzolino, M.; Ferri, A.; Carri, M.T.; Rossi, L. Copper Depletion Increases the Mitochondrial-Associated SOD1 in Neuronal Cells. Biometals 2011, 24, 269–278. [Google Scholar] [CrossRef]
- Menzies, F.M.; Ince, P.G.; Shaw, P.J. Mitochondrial Involvement in Amyotrophic Lateral Sclerosis. Neurochem. Int. 2002, 40, 543–551. [Google Scholar] [CrossRef]
- Beckman, J.S.; Estevez, A.G.; Crow, J.P.; Barbeito, L. Superoxide Dismutase and the Death of Motoneurons in ALS. Trends Neurosci. 2001, 24, S15–S20. [Google Scholar] [CrossRef]
- Estévez, A.G.; Crow, J.P.; Sampson, J.B.; Reiter, C.; Zhuang, Y.; Richardson, G.J.; Tarpey, M.M.; Barbeito, L.; Beckman, J.S. Induction of Nitric Oxide- Dependent Apoptosis in Motor Neurons by Zinc-Deficient Superoxide Dismutase. Science 1999, 286, 2498–2500. [Google Scholar] [CrossRef]
- Yim, M.B.; Kang, J.H.; Yim, H.S.; Kwak, H.S.; Chock, P.B.; Stadtman, E.R. A Gain-of-Function of an Amyotrophic Lateral Sclerosis-Associated Cu,Zn-Superoxide Dismutase Mutant: An Enhancement of Free Radical Formation Due to a Decrease in Km for Hydrogen Peroxide. Proc. Natl. Acad. Sci. USA 1996, 93, 5709–5714. [Google Scholar] [CrossRef] [PubMed]
- Liu, D. The roles of Free Radicals in Amyotrophic Lateral Sclerosis. J. Mol. Neurosci. 1996, 7, 159–167. [Google Scholar] [CrossRef]
- Beckman, J.S.; Carson, M.; Smith, C.D.; Koppenol, W.H. ALS, SOD and Peroxynitrite. Nature 1993, 364, 584. [Google Scholar] [CrossRef]
- Valentine, J.S.; Doucette, P.A.; Zittin Potter, S. Copper-Zinc Superoxide Dismutase and Amyotrophic Lateral Sclerosis. Annu. Rev. Biochem. 2005, 74, 563–593. [Google Scholar] [CrossRef] [PubMed]
- Benkler, C.; O’Neil, A.L.; Slepian, S.; Qian, F.; Weinreb, P.H.; Rubin, L.L. Aggregated SOD1 Causes Selective Death of Cultured Human Motor Neurons. Sci. Rep. 2018, 8, 16393. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Destroismaisons, L.; Peyrard, S.L.; Cadot, S.; Rouleau, G.A.; Brown, R.H., Jr.; Julien, J.P.; Arbour, N.; Vande Velde, C. Mitochondrial Damage Revealed by Immunoselection for ALS-Linked Misfolded SOD1. Hum. Mol. Genet. 2013, 22, 3947–3959. [Google Scholar] [CrossRef]
- Ferri, A.; Cozzolino, M.; Crosio, C.; Nencini, M.; Casciati, A.; Gralla, E.B.; Rotilio, G.; Valentine, J.S.; Carri, M.T. Familial ALS-Superoxide Dismutases Associate with Mitochondria and Shift Their Redox Potentials. Proc. Natl. Acad. Sci. USA 2006, 103, 13860–13865. [Google Scholar] [CrossRef] [PubMed]
- Killoy, K.M.; Harlan, B.A.; Pehar, M.; Helke, K.L.; Johnson, J.A.; Vargas, M.R. Decreased Glutathione Levels Cause Overt Motor Neuron Degeneration in hSOD1(WT) Over-Expressing Mice. Exp. Neurol. 2018, 302, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Vargas, M.R.; Johnson, D.A.; Johnson, J.A. Decreased Glutathione Accelerates Neurological Deficit and Mitochondrial Pathology in Familial ALS-Linked hSOD1(G93A) Mice Model. Neurobiol. Dis. 2011, 43, 543–551. [Google Scholar] [CrossRef]
- Pesaresi, M.G.; Amori, I.; Giorgi, C.; Ferri, A.; Fiorenzo, P.; Gabanella, F.; Salvatore, A.M.; Giorgio, M.; Pelicci, P.G.; Pinton, P.; et al. Mitochondrial Redox Signalling by p66Shc Mediates ALS-Like Disease through Rac1 Inactivation. Hum. Mol. Genet. 2011, 20, 4196–4208. [Google Scholar] [CrossRef]
- Dikalov, S. Cross Talk between Mitochondria and NADPH Oxidases. Free. Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef]
- Wu, D.C.; Re, D.B.; Nagai, M.; Ischiropoulos, H.; Przedborski, S. The Inflammatory NADPH Oxidase Enzyme Modulates Motor Neuron Degeneration in Amyotrophic Lateral Sclerosis Mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12132–12137. [Google Scholar] [CrossRef]
- Marden, J.J.; Harraz, M.M.; Williams, A.J.; Nelson, K.; Luo, M.; Paulson, H.; Engelhardt, J.F. Redox Modifier Genes in Amyotrophic Lateral Sclerosis in Mice. J. Clin. Investig. 2007, 117, 2913–2919. [Google Scholar] [CrossRef]
- Harraz, M.M.; Marden, J.J.; Zhou, W.; Zhang, Y.; Williams, A.; Sharov, V.S.; Nelson, K.; Luo, M.; Paulson, H.; Schöneich, C.; et al. SOD1 Mutations Disrupt Redox-Sensitive Rac Regulation of NADPH Oxidase in a Familial ALS Model. J. Clin. Investig. 2008, 118, 659–670. [Google Scholar] [CrossRef]
- Trumbull, K.A.; McAllister, D.; Gandelman, M.M.; Fung, W.Y.; Lew, T.; Brennan, L.; Lopez, N.; Morre, J.; Kalyanaraman, B.; Beckman, J.S. Diapocynin and Apocynin Administration Fails to Significantly Extend Survival in G93A SOD1 ALS Mice. Neurobiol. Dis. 2012, 45, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Kato, M.; Abe, Y.; Matsumura, T.; Nishino, T.; Aoki, M.; Itoyama, Y.; Asayama, K.; Awaya, A.; Hirano, A.; et al. Redox System Expression in the Motor Neurons in Amyotrophic Lateral Sclerosis (ALS): Immunohistochemical Studies on Sporadic ALS, Superoxide Dismutase 1 (SOD1)-Mutated Familial ALS, and SOD1-Mutated ALS Animal Models. Acta Neuropathol. 2005, 110, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-Regulated Genes Induced by the Chemopreventive Agent Sulforaphane by Oligonucleotide Microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar]
- Lee, J.M.; Calkins, M.J.; Chan, K.; Kan, Y.W.; Johnson, J.A. Identification of the NF-E2-Related Factor-2-Dependent Genes Conferring Protection against Oxidative Stress in Primary Cortical Astrocytes Using Oligonucleotide Microarray Analysis. J. Biol. Chem. 2003, 278, 12029–12038. [Google Scholar] [CrossRef] [PubMed]
- Petri, S.; Korner, S.; Kiaei, M. Nrf2/ARE Signaling Pathway: Key Mediator in Oxidative Stress and Potential Therapeutic Target in ALS. Neurol. Res. Int. 2012, 2012, 878030. [Google Scholar] [CrossRef] [PubMed]
- Nioi, P.; McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Identification of a Novel Nrf2-Regulated Antioxidant Response Element (ARE) in the Mouse NAD(P)H:Quinone Oxidoreductase 1 Gene: Reassessment of the ARE Consensus Sequence. Biochem. J. 2003, 374, 337–348. [Google Scholar] [CrossRef]
- Kwak, M.K.; Wakabayashi, N.; Greenlaw, J.L.; Yamamoto, M.; Kensler, T.W. Antioxidants enhance Mammalian Proteasome Expression through the Keap1-Nrf2 Signaling Pathway. Mol. Cell. Biol. 2003, 23, 8786–8794. [Google Scholar] [CrossRef] [PubMed]
- Kirby, J.; Halligan, E.; Baptista, M.J.; Allen, S.; Heath, P.R.; Holden, H.; Barber, S.C.; Loynes, C.A.; Wood-Allum, C.A.; Lunec, J.; et al. Mutant SOD1 Alters the Motor Neuronal Transcriptome: Implications for Familial ALS. Brain 2005, 128, 1686–1706. [Google Scholar] [CrossRef] [PubMed]
- Sarlette, A.; Krampfl, K.; Grothe, C.; Neuhoff, N.; Dengler, R.; Petri, S. Nuclear Erythroid 2-Related Factor 2-Antioxidative Response Element Signaling Pathway in Motor Cortex and Spinal Cord in Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2008, 67, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Kraft, A.D.; Resch, J.M.; Johnson, D.A.; Johnson, J.A. Activation of the Nrf2-ARE Pathway in Muscle and Spinal Cord during ALS-Like Pathology in Mice Expressing Mutant SOD1. Exp. Neurol. 2007, 207, 107–117. [Google Scholar] [CrossRef]
- Pehar, M.; Beeson, G.; Beeson, C.C.; Johnson, J.A.; Vargas, M.R. Mitochondria-Targeted Catalase Reverts the Neurotoxicity of hSOD1G(9)(3)A Astrocytes without Extending the Survival of ALS-Linked Mutant hSOD1 Mice. PLoS ONE 2014, 9, e103438. [Google Scholar] [CrossRef]
- Tian, Y.P.; Che, F.Y.; Su, Q.P.; Lu, Y.C.; You, C.P.; Huang, L.M.; Wang, S.G.; Wang, L.; Yu, J.X. Effects of Mutant TDP-43 on the Nrf2/ARE Pathway and Protein Expression of MafK and JDP2 in NSC-34 Cells. Genet. Mol. Res. 2017, 16, 1–13. [Google Scholar] [CrossRef]
- Moujalled, D.; Grubman, A.; Acevedo, K.; Yang, S.; Ke, Y.D.; Moujalled, D.M.; Duncan, C.; Caragounis, A.; Perera, N.D.; Turner, B.J.; et al. TDP-43 Mutations Causing Amyotrophic Lateral Sclerosis Are Associated with Altered Expression of RNA-Binding Protein hnRNP K and Affect the Nrf2 Antioxidant Pathway. Hum. Mol. Genet. 2017, 26, 1732–1746. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Li, X.; Shi, J.; Guo, Y.; Li, Z.; Li, C. Mutant TAR DNA-Binding Protein-43 Induces Oxidative Injury in Motor Neuron-Like Cell. Neuroscience 2010, 169, 1621–1629. [Google Scholar] [CrossRef] [PubMed]
- Birger, A.; Ben-Dor, I.; Ottolenghi, M.; Turetsky, T.; Gil, Y.; Sweetat, S.; Perez, L.; Belzer, V.; Casden, N.; Steiner, D.; et al. Human iPSC-Derived Astrocytes from ALS Patients with Mutated C9ORF72 Show Increased Oxidative Stress and Neurotoxicity. EBioMedicine 2019, 50, 274–289. [Google Scholar] [CrossRef] [PubMed]
- Bono, S.; Feligioni, M.; Corbo, M. Impaired Antioxidant KEAP1-NRF2 System in Amyotrophic Lateral Sclerosis: NRF2 Activation as a Potential Therapeutic Strategy. Mol. Neurodegener. 2021, 16, 71. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Pradat, P.F.; Ludolph, A.C.; Loeffler, J.P. Energy Metabolism in Amyotrophic Lateral Sclerosis. Lancet Neurol. 2011, 10, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Oudart, H.; Rene, F.; Gonzalez de Aguilar, J.L.; Loeffler, J.P. Evidence for Defective Energy Homeostasis in Amyotrophic Lateral Sclerosis: Benefit of a High-Energy Diet in a Transgenic Mouse Model. Proc. Natl. Acad. Sci. USA 2004, 101, 11159–11164. [Google Scholar] [CrossRef]
- Fergani, A.; Oudart, H.; Gonzalez De Aguilar, J.L.; Fricker, B.; Rene, F.; Hocquette, J.F.; Meininger, V.; Dupuis, L.; Loeffler, J.P. Increased Peripheral Lipid Clearance in an Animal Model of Amyotrophic Lateral Sclerosis. J. Lipid Res. 2007, 48, 1571–1580. [Google Scholar] [CrossRef]
- Chiang, P.M.; Ling, J.; Jeong, Y.H.; Price, D.L.; Aja, S.M.; Wong, P.C. Deletion of Tdp-43 Down-Regulates Tbc1d1, a Gene Linked to Obesity, and Alters Body Fat Metabolism. Proc. Natl. Acad. Sci. USA 2010, 107, 16320–16324. [Google Scholar] [CrossRef]
- Coughlan, K.S.; Halang, L.; Woods, I.; Prehn, J.H. A High-Fat Jelly Diet Restores Bioenergetic Balance and Extends Lifespan in the Presence of Motor Dysfunction and Lumbar Spinal Cord Motor Neuron Loss in TDP-43A315T Mutant C57BL6/J Mice. Dis. Models Mech. 2016, 9, 1029–1037. [Google Scholar] [CrossRef]
- Manzo, E.; Lorenzini, I.; Barrameda, D.; O’Conner, A.G.; Barrows, J.M.; Starr, A.; Kovalik, T.; Rabichow, B.E.; Lehmkuhl, E.M.; Shreiner, D.D.; et al. Glycolysis Upregulation Is Neuroprotective as a Compensatory Mechanism in ALS. eLife 2019, 8, e45114. [Google Scholar] [CrossRef]
- Schmitt, F.; Hussain, G.; Dupuis, L.; Loeffler, J.P.; Henriques, A. A Plural Role for Lipids in Motor Neuron Diseases: Energy, Signaling and Structure. Front. Cell. Neurosci. 2014, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Wong, M. Skeletal Muscle-Restricted Expression of Human SOD1 in Transgenic Mice Causes a Fatal ALS-Like Syndrome. Front. Neurol. 2020, 11, 592851. [Google Scholar] [CrossRef]
- Park, Y.; Park, J.; Kim, Y.; Baek, H.; Kim, S.H. Association between Nutritional Status and Disease Severity Using the Amyotrophic Lateral Sclerosis (ALS) Functional Rating Scale in ALS Patients. Nutrition 2015, 31, 1362–1367. [Google Scholar] [CrossRef]
- Desport, J.C.; Preux, P.M.; Truong, C.T.; Courat, L.; Vallat, J.M.; Couratier, P. Nutritional Assessment and Survival in ALS Patients. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 91–96. [Google Scholar] [CrossRef]
- Desport, J.C.; Preux, P.M.; Truong, T.C.; Vallat, J.M.; Sautereau, D.; Couratier, P. Nutritional Status Is a Prognostic Factor for Survival in ALS Patients. Neurology 1999, 53, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Paganoni, S.; Deng, J.; Jaffa, M.; Cudkowicz, M.E.; Wills, A.M. Body Mass Index, Not Dyslipidemia, Is an Independent Predictor of Survival in Amyotrophic Lateral Sclerosis. Muscle Nerve 2011, 44, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Kasarskis, E.J.; Neville, H.E. Management of ALS: Nutritional care. Neurology 1996, 47, S118–S120. [Google Scholar] [CrossRef]
- Bouteloup, C.; Desport, J.C.; Clavelou, P.; Guy, N.; Derumeaux-Burel, H.; Ferrier, A.; Couratier, P. Hypermetabolism in ALS Patients: An Early and Persistent Phenomenon. J. Neurol. 2009, 256, 1236–1242. [Google Scholar] [CrossRef]
- Kasarskis, E.J.; Berryman, S.; Vanderleest, J.G.; Schneider, A.R.; McClain, C.J. Nutritional Status of Patients with Amyotrophic Lateral Sclerosis: Relation to the Proximity of Death. Am. J. Clin. Nutr. 1996, 63, 130–137. [Google Scholar] [CrossRef]
- Desport, J.C.; Preux, P.M.; Magy, L.; Boirie, Y.; Vallat, J.M.; Beaufrere, B.; Couratier, P. Factors Correlated with Hypermetabolism in Patients with Amyotrophic Lateral Sclerosis. Am. J. Clin. Nutr. 2001, 74, 328–334. [Google Scholar] [CrossRef]
- Desport, J.C.; Torny, F.; Lacoste, M.; Preux, P.M.; Couratier, P. Hypermetabolism in ALS: Correlations with Clinical and Paraclinical Parameters. Neurodegener. Dis. 2005, 2, 202–207. [Google Scholar] [CrossRef]
- Genton, L.; Viatte, V.; Janssens, J.P.; Heritier, A.C.; Pichard, C. Nutritional State, Energy Intakes and Energy Expenditure of Amyotrophic Lateral Sclerosis (ALS) Patients. Clin. Nutr. 2011, 30, 553–559. [Google Scholar] [CrossRef]
- Gallo, V.; Wark, P.A.; Jenab, M.; Pearce, N.; Brayne, C.; Vermeulen, R.; Andersen, P.M.; Hallmans, G.; Kyrozis, A.; Vanacore, N.; et al. Prediagnostic Body Fat and Risk of Death from Amyotrophic Lateral Sclerosis: The EPIC Cohort. Neurology 2013, 80, 829–838. [Google Scholar] [CrossRef]
- O’Reilly, E.J.; Wang, H.; Weisskopf, M.G.; Fitzgerald, K.C.; Falcone, G.; McCullough, M.L.; Thun, M.; Park, Y.; Kolonel, L.N.; Ascherio, A. Premorbid Body Mass Index and Risk of Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 205–211. [Google Scholar] [CrossRef]
- Peter, R.S.; Rosenbohm, A.; Dupuis, L.; Brehme, T.; Kassubek, J.; Rothenbacher, D.; Nagel, G.; Ludolph, A.C. Life Course Body Mass Index and Risk and Prognosis of Amyotrophic Lateral Sclerosis: Results from the ALS Registry Swabia. Eur. J. Epidemiol. 2017, 32, 901–908. [Google Scholar] [CrossRef]
- Funalot, B.; Desport, J.C.; Sturtz, F.; Camu, W.; Couratier, P. High Metabolic Level in Patients with Familial Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. 2009, 10, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Vaisman, N.; Lusaus, M.; Nefussy, B.; Niv, E.; Comaneshter, D.; Hallack, R.; Drory, V.E. Do Patients with Amyotrophic Lateral Sclerosis (ALS) Have Increased Energy Needs? J. Neurol. Sci. 2009, 279, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Jesus, P.; Fayemendy, P.; Nicol, M.; Lautrette, G.; Sourisseau, H.; Preux, P.M.; Desport, J.C.; Marin, B.; Couratier, P. Hypermetabolism Is a Deleterious Prognostic Factor in Patients with Amyotrophic Lateral Sclerosis. Eur. J. Neurol. 2018, 25, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Steyn, F.J.; Ioannides, Z.A.; van Eijk, R.P.A.; Heggie, S.; Thorpe, K.A.; Ceslis, A.; Heshmat, S.; Henders, A.K.; Wray, N.R.; van den Berg, L.H.; et al. Hypermetabolism in ALS Is Associated with Greater Functional Decline and Shorter Survival. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.A.; Benedict, F.G. A Biometric Study of Human Basal Metabolism. Proc. Natl. Acad. Sci. USA 1918, 4, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.S.; Pillai, A.; Jackson, A.; Heiman-Patterson, T. Standard Equations Are Not Accurate in Assessing Resting Energy Expenditure in Patients with Amyotrophic Lateral Sclerosis. JPEN J. Parenter. Enter. Nutr. 2004, 28, 442–446. [Google Scholar] [CrossRef]
- Brito, M.D.; da Silva, G.F.G.; Tilieri, E.M.; Araujo, B.G.; Calio, M.L.; Rosenstock, T.R. Metabolic Alteration and Amyotrophic Lateral Sclerosis Outcome: A Systematic Review. Front. Neurol. 2019, 10, 1205. [Google Scholar] [CrossRef] [PubMed]
- Pharaoh, G.; Sataranatarajan, K.; Street, K.; Hill, S.; Gregston, J.; Ahn, B.; Kinter, C.; Kinter, M.; Van Remmen, H. Metabolic and Stress Response Changes Precede Disease Onset in the Spinal Cord of Mutant SOD1 ALS Mice. Front. Neurosci. 2019, 13, 487. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Fai Poon, H.; Hensley, K.; Pierce, W.M.; Klein, J.B.; Calabrese, V.; De Marco, C.; Butterfield, D.A. Proteomic Analysis of 4-Hydroxy-2-Nonenal-Modified Proteins in G93A-SOD1 Transgenic Mice—A Model of Familial Amyotrophic Lateral Sclerosis. Free. Radic. Biol. Med. 2005, 38, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Poon, H.F.; Hensley, K.; Thongboonkerd, V.; Merchant, M.L.; Lynn, B.C.; Pierce, W.M.; Klein, J.B.; Calabrese, V.; Butterfield, D.A. Redox Proteomics Analysis of Oxidatively Modified Proteins in G93A-SOD1 Transgenic Mice—A Model of Familial Amyotrophic Lateral Sclerosis. Free Radic. Biol. Med. 2005, 39, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Lukas, T.J.; Luo, W.W.; Mao, H.; Cole, N.; Siddique, T. Informatics-Assisted Protein Profiling in a Transgenic Mouse Model of Amyotrophic Lateral Sclerosis. Mol. Cell. Proteom. 2006, 5, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Massignan, T.; Casoni, F.; Basso, M.; Stefanazzi, P.; Biasini, E.; Tortarolo, M.; Salmona, M.; Gianazza, E.; Bendotti, C.; Bonetto, V. Proteomic Analysis of Spinal Cord of Presymptomatic Amyotrophic Lateral Sclerosis G93A SOD1 Mouse. Biochem. Biophys. Res. Commun. 2007, 353, 719–725. [Google Scholar] [CrossRef]
- Li, Q.; Vande Velde, C.; Israelson, A.; Xie, J.; Bailey, A.O.; Dong, M.Q.; Chun, S.J.; Roy, T.; Winer, L.; Yates, J.R.; et al. ALS-Linked Mutant Superoxide Dismutase 1 (SOD1) Alters Mitochondrial Protein Composition and Decreases Protein Import. Proc. Natl. Acad. Sci. USA 2010, 107, 21146–21151. [Google Scholar] [CrossRef]
- Valbuena, G.N.; Rizzardini, M.; Cimini, S.; Siskos, A.P.; Bendotti, C.; Cantoni, L.; Keun, H.C. Metabolomic Analysis Reveals Increased Aerobic Glycolysis and Amino Acid Deficit in a Cellular Model of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2016, 53, 2222–2240. [Google Scholar] [CrossRef]
- Miyazaki, K.; Masamoto, K.; Morimoto, N.; Kurata, T.; Mimoto, T.; Obata, T.; Kanno, I.; Abe, K. Early and Progressive Impairment of Spinal Blood Flow-Glucose Metabolism Coupling in Motor Neuron Degeneration of ALS Model Mice. J. Cereb. Blood Flow Metab. 2012, 32, 456–467. [Google Scholar] [CrossRef]
- Belanger, M.; Allaman, I.; Magistretti, P.J. Brain Energy Metabolism: Focus on Astrocyte-Neuron Metabolic Cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef]
- Sharp, P.S.; Dick, J.R.; Greensmith, L. The Effect of Peripheral Nerve Injury on Disease Progression in the SOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. Neuroscience 2005, 130, 897–910. [Google Scholar] [CrossRef]
- Palamiuc, L.; Schlagowski, A.; Ngo, S.T.; Vernay, A.; Dirrig-Grosch, S.; Henriques, A.; Boutillier, A.L.; Zoll, J.; Echaniz-Laguna, A.; Loeffler, J.P.; et al. A Metabolic Switch toward Lipid Use in Glycolytic Muscle Is an Early Pathologic Event in a Mouse Model of Amyotrophic Lateral Sclerosis. EMBO Mol. Med. 2015, 7, 526–546. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Oliveira, T.; Carvalho, M.; Sardao, V.; Ferreiro, E.; Mena, D.; Pereira, F.B.; Borges, F.; Oliveira, P.J.; Silva, F.S.G. Integrative Profiling of Amyotrophic Lateral Sclerosis Lymphoblasts Identifies Unique Metabolic and Mitochondrial Disease Fingerprints. Mol. Neurobiol. 2022, 59, 6373–6396. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.; Blazquez, C. Ketone Body Synthesis in the Brain: Possible Neuroprotective Effects. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 287–292. [Google Scholar] [CrossRef] [PubMed]
- LaManna, J.C.; Salem, N.; Puchowicz, M.; Erokwu, B.; Koppaka, S.; Flask, C.; Lee, Z. Ketones Suppress Brain Glucose Consumption. Adv. Exp. Med. Biol. 2009, 645, 301–306. [Google Scholar] [CrossRef]
- Yi, C.X.; Habegger, K.M.; Chowen, J.A.; Stern, J.; Tschop, M.H. A Role for Astrocytes in the Central Control of Metabolism. Neuroendocrinology 2011, 93, 143–149. [Google Scholar] [CrossRef]
- Le Foll, C.; Levin, B.E. Fatty Acid-Induced Astrocyte Ketone Production and the Control of Food Intake. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R1186–R1192. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, J.; Morales, L.; Barreto, G.E. Metabolic and Inflammatory Adaptation of Reactive Astrocytes: Role of PPARs. Mol. Neurobiol. 2017, 54, 2518–2538. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging Mechanisms in ALS and FTD: Disrupted RNA and Protein Homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef]
- Lawton, K.A.; Brown, M.V.; Alexander, D.; Li, Z.; Wulff, J.E.; Lawson, R.; Jaffa, M.; Milburn, M.V.; Ryals, J.A.; Bowser, R.; et al. Plasma Metabolomic Biomarker Panel to Distinguish Patients with Amyotrophic Lateral Sclerosis from Disease Mimics. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 362–370. [Google Scholar] [CrossRef]
- Lawton, K.A.; Cudkowicz, M.E.; Brown, M.V.; Alexander, D.; Caffrey, R.; Wulff, J.E.; Bowser, R.; Lawson, R.; Jaffa, M.; Milburn, M.V.; et al. Biochemical Alterations Associated with ALS. Amyotroph. Lateral Scler. 2012, 13, 110–118. [Google Scholar] [CrossRef]
- Allen, S.P.; Hall, B.; Woof, R.; Francis, L.; Gatto, N.; Shaw, A.C.; Myszczynska, M.; Hemingway, J.; Coldicott, I.; Willcock, A.; et al. C9orf72 Expansion within Astrocytes Reduces Metabolic Flexibility in Amyotrophic Lateral Sclerosis. Brain 2019, 142, 3771–3790. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Corcia, P.; Moreau, C.; Veau, S.; Fournier, C.; Vourc’h, P.; Emond, P.; Gordon, P.; Pradat, P.F.; Praline, J.; et al. 1H-NMR-Based Metabolomic Profiling of CSF in Early Amyotrophic Lateral Sclerosis. PLoS ONE 2010, 5, e13223. [Google Scholar] [CrossRef]
- Kumar, A.; Bala, L.; Kalita, J.; Misra, U.K.; Singh, R.L.; Khetrapal, C.L.; Babu, G.N. Metabolomic Analysis of Serum by (1) H NMR Spectroscopy in Amyotrophic Lateral Sclerosis. Clin. Chim. Acta 2010, 411, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Wuolikainen, A.; Andersen, P.M.; Moritz, T.; Marklund, S.L.; Antti, H. ALS Patients with Mutations in the SOD1 Gene Have an Unique Metabolomic Profile in the Cerebrospinal Fluid Compared with ALS Patients without Mutations. Mol. Genet. Metab. 2012, 105, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Wuolikainen, A.; Moritz, T.; Marklund, S.L.; Antti, H.; Andersen, P.M. Disease-Related Changes in the Cerebrospinal Fluid Metabolome in Amyotrophic Lateral Sclerosis Detected by GC/TOFMS. PLoS ONE 2011, 6, e17947. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.; Larkin, J.R.; Claridge, T.D.; Talbot, K.; Sibson, N.R.; Turner, M.R. The Longitudinal Cerebrospinal Fluid Metabolomic Profile of Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Nadal-Desbarats, L.; Pradat, P.F.; Gordon, P.H.; Madji Hounoum, B.; Patin, F.; Veyrat-Durebex, C.; Mavel, S.; Beltran, S.; Emond, P.; et al. Biomarkers in Amyotrophic Lateral Sclerosis: Combining Metabolomic and Clinical Parameters to Define Disease Progression. Eur. J. Neurol. 2016, 23, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Patin, F.; Descat, A.; Garcon, G.; Corcia, P.; Gele, P.; Lenglet, T.; Bede, P.; Meininger, V.; Devos, D.; et al. A Pharmaco-Metabolomics Approach in a Clinical Trial of ALS: Identification of Predictive Markers of Progression. PLoS ONE 2018, 13, e0198116. [Google Scholar] [CrossRef]
- Patin, F.; Baranek, T.; Vourc’h, P.; Nadal-Desbarats, L.; Goossens, J.F.; Marouillat, S.; Dessein, A.F.; Descat, A.; Hounoum, B.M.; Bruno, C.; et al. Combined Metabolomics and Transcriptomics Approaches to Assess the IL-6 Blockade as a Therapeutic of ALS: Deleterious Alteration of Lipid Metabolism. Neurotherapeutics 2016, 13, 905–917. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Balancing Mitochondrial Biogenesis and Mitophagy to Maintain Energy Metabolism Homeostasis. Cell Death Differ. 2015, 22, 1399–1401. [Google Scholar] [CrossRef]
- Nakada, K.; Inoue, K.; Ono, T.; Isobe, K.; Ogura, A.; Goto, Y.I.; Nonaka, I.; Hayashi, J.I. Inter-Mitochondrial Complementation: Mitochondria-Specific System Preventing Mice from Expression of Disease Phenotypes by Mutant mtDNA. Nat. Med. 2001, 7, 934–940. [Google Scholar] [CrossRef]
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of Fusion Results in Mitochondrial Heterogeneity and Dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar] [CrossRef]
- Khalil, B.; Lievens, J.C. Mitochondrial Quality Control in Amyotrophic Lateral Sclerosis: Towards a Common Pathway? Neural Regen. Res. 2017, 12, 1052–1061. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-Related Protein Drp1 is Required for Mitochondrial Division in Mammalian Cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef]
- Tamura, Y.; Itoh, K.; Sesaki, H. SnapShot: Mitochondrial Dynamics. Cell 2011, 145, 1158.e1. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an Essential Factor for Mitochondrial Recruitment of Drp1 During Mitochondrial Fission in Mammalian Cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [PubMed]
- James, D.I.; Parone, P.A.; Mattenberger, Y.; Martinou, J.C. Hfis1, a Novel Component of the Mammalian Mitochondrial Fission Machinery. J. Biol. Chem. 2003, 278, 36373–36379. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, New Components of the Mitochondrial Fission Machinery. EMBO Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef]
- Mears, J.A.; Lackner, L.L.; Fang, S.; Ingerman, E.; Nunnari, J.; Hinshaw, J.E. Conformational Changes in Dnm1 Support a Contractile Mechanism for Mitochondrial Fission. Nat. Struct. Mol. Biol. 2011, 18, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, N.H.; Brisch, E.; Keegan, B.R.; Bleazard, W.; Shaw, J.M. The GTPase Effector Domain Sequence of the Dnm1p GTPase Regulates Self-Assembly and Controls a Rate-Limiting Step in Mitochondrial Fission. Mol. Biol. Cell 2001, 12, 2756–2766. [Google Scholar] [CrossRef] [PubMed]
- Santel, A.; Fuller, M.T. Control of Mitochondrial Morphology by a Human Mitofusin. J. Cell Sci. 2001, 114 Pt 5, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Physiological Functions of Mitochondrial Fusion. Ann. N. Y. Acad. Sci. 2010, 1201, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 Mediate Sequential Steps in Mitochondrial Membrane Fusion. Mol. Biol. Cell 2009, 20, 3525–3532. [Google Scholar] [CrossRef]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 Requires Mitofusin 1 to Promote Mitochondrial Fusion. Proc Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; McCaffery, J.M.; Chan, D.C. Mitochondrial Fusion Protects against Neurodegeneration in the Cerebellum. Cell 2007, 130, 548–562. [Google Scholar] [CrossRef]
- Ishihara, N.; Nomura, M.; Jofuku, A.; Kato, H.; Suzuki, S.O.; Masuda, K.; Otera, H.; Nakanishi, Y.; Nonaka, I.; Goto, Y.; et al. Mitochondrial Fission Factor Drp1 is Essential for Embryonic Development and Synapse Formation in Mice. Nat. Cell Biol. 2009, 11, 958–966. [Google Scholar] [CrossRef]
- Wakabayashi, J.; Zhang, Z.; Wakabayashi, N.; Tamura, Y.; Fukaya, M.; Kensler, T.W.; Iijima, M.; Sesaki, H. The Dynamin-Related GTPase Drp1 is Required for Embryonic and Brain Development in Mice. J. Cell Biol. 2009, 186, 805–816. [Google Scholar] [CrossRef]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear Gene OPA1, Encoding a Mitochondrial Dynamin-Related Protein, is Mutated in Dominant Optic Atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- Song, W.; Song, Y.; Kincaid, B.; Bossy, B.; Bossy-Wetzel, E. Mutant SOD1G93A Triggers Mitochondrial Fragmentation in Spinal Cord Motor Neurons: Neuroprotection by SIRT3 and PGC-1alpha. Neurobiol. Dis. 2013, 51, 72–81. [Google Scholar] [CrossRef]
- Xu, Y.F.; Gendron, T.F.; Zhang, Y.J.; Lin, W.L.; D’Alton, S.; Sheng, H.; Casey, M.C.; Tong, J.; Knight, J.; Yu, X.; et al. Wild-Type Human TDP-43 Expression Causes TDP-43 Phosphorylation, Mitochondrial Aggregation, Motor Deficits, and Early Mortality in Transgenic Mice. J. Neurosci. 2010, 30, 10851–10859. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.P.; Wada, S.; Vergani, L.; Hock, M.B.; Lamon, S.; Leger, B.; Ushida, T.; Cartoni, R.; Wadley, G.D.; Hespel, P.; et al. Disruption of Skeletal Muscle Mitochondrial Network Genes and miRNAs in Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2013, 49, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Vinsant, S.; Mansfield, C.; Jimenez-Moreno, R.; Del Gaizo Moore, V.; Yoshikawa, M.; Hampton, T.G.; Prevette, D.; Caress, J.; Oppenheim, R.W.; Milligan, C. Characterization of Early Pathogenesis in the SOD1(G93A) Mouse Model of ALS: Part II, Results and Discussion. Brain Behav. 2013, 3, 431–457. [Google Scholar] [CrossRef]
- Shutt, T.; Geoffrion, M.; Milne, R.; McBride, H.M. The Intracellular Redox State Is a Core Determinant of Mitochondrial Fusion. EMBO Rep. 2012, 13, 909–915. [Google Scholar] [CrossRef]
- Kim, Y.M.; Youn, S.W.; Sudhahar, V.; Das, A.; Chandhri, R.; Cuervo Grajal, H.; Kweon, J.; Leanhart, S.; He, L.; Toth, P.T.; et al. Redox Regulation of Mitochondrial Fission Protein Drp1 by Protein Disulfide Isomerase Limits Endothelial Senescence. Cell Rep. 2018, 23, 3565–3578. [Google Scholar] [CrossRef] [PubMed]
- Tradewell, M.L.; Yu, Z.; Tibshirani, M.; Boulanger, M.C.; Durham, H.D.; Richard, S. Arginine Methylation by PRMT1 Regulates Nuclear-Cytoplasmic Localization and Toxicity of FUS/TLS Harbouring ALS-Linked Mutations. Hum. Mol. Genet. 2012, 21, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Holler, C.J.; Taylor, G.; Hudson, K.F.; Watkins, W.; Gearing, M.; Ito, D.; Murray, M.E.; Dickson, D.W.; Seyfried, N.T.; et al. FUS Is Phosphorylated by DNA-PK and Accumulates in the Cytoplasm after DNA Damage. J. Neurosci. 2014, 34, 7802–7813. [Google Scholar] [CrossRef]
- Li, P.A.; Hou, X.; Hao, S. Mitochondrial Biogenesis in Neurodegeneration. J. Neurosci. Res. 2017, 95, 2025–2029. [Google Scholar] [CrossRef]
- Panchal, K.; Tiwari, A.K. Mitochondrial Dynamics, a Key Executioner in Neurodegenerative Diseases. Mitochondrion 2019, 47, 151–173. [Google Scholar] [CrossRef]
- Bayer, H.; Lang, K.; Buck, E.; Higelin, J.; Barteczko, L.; Pasquarelli, N.; Sprissler, J.; Lucas, T.; Holzmann, K.; Demestre, M.; et al. ALS-Causing Mutations Differentially Affect PGC-1alpha Expression and Function in the Brain vs. Peripheral Tissues. Neurobiol. Dis. 2017, 97 Pt A, 36–45. [Google Scholar] [CrossRef]
- Palomo, G.M.; Granatiero, V.; Kawamata, H.; Konrad, C.; Kim, M.; Arreguin, A.J.; Zhao, D.; Milner, T.A.; Manfredi, G. Parkin Is a Disease Modifier in the Mutant SOD1 Mouse Model of ALS. EMBO Mol. Med. 2018, 10, e8888. [Google Scholar] [CrossRef]
- Thau, N.; Knippenberg, S.; Korner, S.; Rath, K.J.; Dengler, R.; Petri, S. Decreased mRNA Expression of PGC-1alpha and PGC-1alpha-Regulated Factors in the SOD1G93A ALS Mouse Model and in Human Sporadic ALS. J. Neuropathol. Exp. Neurol. 2012, 71, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Masser, D.R.; Clark, N.W.; Van Remmen, H.; Freeman, W.M. Loss of the Antioxidant Enzyme CuZnSOD (Sod1) Mimics an Age-Related Increase in Absolute Mitochondrial DNA Copy Number in the Skeletal Muscle. Age 2016, 38, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Stoccoro, A.; Mosca, L.; Carnicelli, V.; Cavallari, U.; Lunetta, C.; Marocchi, A.; Migliore, L.; Coppede, F. Mitochondrial DNA Copy Number and D-Loop Region Methylation in Carriers of Amyotrophic Lateral Sclerosis Gene Mutations. Epigenomics 2018, 10, 1431–1443. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.H.; Cai, Q. Mitochondrial Transport in Neurons: Impact on Synaptic Homeostasis and Neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef]
- Melkov, A.; Abdu, U. Regulation of Long-Distance Transport of Mitochondria along Microtubules. Cell. Mol. Life Sci. 2017, 75, 163–176. [Google Scholar] [CrossRef]
- Conde, C.; Caceres, A. Microtubule Assembly, Organization and Dynamics in Axons and Dendrites. Nat. Rev. Neurosci. 2009, 10, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kanai, Y.; Okada, Y.; Nonaka, S.; Takeda, S.; Harada, A.; Hirokawa, N. Targeted Disruption of Mouse Conventional Kinesin Heavy Chain, kif5B, Results in Abnormal Perinuclear Clustering of Mitochondria. Cell 1998, 93, 1147–1158. [Google Scholar] [CrossRef]
- Pilling, A.D.; Horiuchi, D.; Lively, C.M.; Saxton, W.M. Kinesin-1 and Dynein Are the Primary Motors for Fast Transport of Mitochondria in Drosophila Motor Axons. Mol. Biol. Cell 2006, 17, 2057–2068. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Niwa, S.; Tanaka, Y. Molecular Motors in Neurons: Transport Mechanisms and Roles in Brain Function, Development, and Disease. Neuron 2010, 68, 610–638. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Macleod, G.T.; Wellington, A.; Hu, F.; Panchumarthi, S.; Schoenfield, M.; Marin, L.; Charlton, M.P.; Atwood, H.L.; Zinsmaier, K.E. The GTPase dMiro is Required for Axonal Transport of Mitochondria to Drosophila Synapses. Neuron 2005, 47, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Stowers, R.S.; Megeath, L.J.; Gorska-Andrzejak, J.; Meinertzhagen, I.A.; Schwarz, T.L. Axonal Transport of Mitochondria to Synapses Depends on Milton, a Novel Drosophila Protein. Neuron 2002, 36, 1063–1077. [Google Scholar] [CrossRef] [PubMed]
- MacAskill, A.F.; Brickley, K.; Stephenson, F.A.; Kittler, J.T. GTPase Dependent Recruitment of Grif-1 by Miro1 Regulates Mitochondrial Trafficking in Hippocampal Neurons. Mol. Cell. Neurosci. 2009, 40, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Brickley, K.; Stephenson, F.A. Trafficking Kinesin Protein (TRAK)-Mediated Transport of Mitochondria in Axons of Hippocampal Neurons. J. Biol. Chem. 2011, 286, 18079–18092. [Google Scholar] [CrossRef]
- Cai, Q.; Gerwin, C.; Sheng, Z.H. Syntabulin-Mediated Anterograde Transport of Mitochondria along Neuronal Processes. J. Cell Biol. 2005, 170, 959–969. [Google Scholar] [CrossRef]
- Cho, K.I.; Cai, Y.; Yi, H.; Yeh, A.; Aslanukov, A.; Ferreira, P.A. Association of the Kinesin-Binding Domain of RanBP2 to KIF5B and KIF5C Determines Mitochondria Localization and Function. Traffic 2007, 8, 1722–1735. [Google Scholar] [CrossRef]
- Altmann, K.; Frank, M.; Neumann, D.; Jakobs, S.; Westermann, B. The Class V Myosin Motor Protein, Myo2, Plays a Major Role in Mitochondrial Motility in Saccharomyces Cerevisiae. J. Cell Biol. 2008, 181, 119–130. [Google Scholar] [CrossRef]
- Boldogh, I.R.; Yang, H.C.; Nowakowski, W.D.; Karmon, S.L.; Hays, L.G.; Yates, J.R., 3rd; Pon, L.A. Arp2/3 Complex and Actin Dynamics Are Required for Actin-Based Mitochondrial Motility in Yeast. Proc. Natl. Acad. Sci. USA 2001, 98, 3162–3167. [Google Scholar] [CrossRef]
- Sheng, Z.H. Mitochondrial Trafficking and Anchoring in Neurons: New Insight and Implications. J. Cell Biol. 2014, 204, 1087–1098. [Google Scholar] [CrossRef]
- Kang, J.S.; Tian, J.H.; Pan, P.Y.; Zald, P.; Li, C.; Deng, C.; Sheng, Z.H. Docking of Axonal Mitochondria by Syntaphilin Controls Their Mobility and Affects Short-Term Facilitation. Cell 2008, 132, 137–148. [Google Scholar] [CrossRef]
- Chen, Y.M.; Gerwin, C.; Sheng, Z.H. Dynein Light Chain LC8 Regulates Syntaphilin-Mediated Mitochondrial Docking in Axons. J. Neurosci. 2009, 29, 9429–9438. [Google Scholar] [CrossRef]
- De Vos, K.J.; Hafezparast, M. Neurobiology of Axonal Transport Defects in Motor Neuron Diseases: Opportunities for Translational Research? Neurobiol. Dis. 2017, 105, 283–299. [Google Scholar] [CrossRef]
- Magrane, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal Mitochondrial Transport and Morphology Are Common Pathological Denominators in SOD1 and TDP43 ALS Mouse Models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef]
- Okamoto, K.; Hirai, S.; Shoji, M.; Senoh, Y.; Yamazaki, T. Axonal Swellings in the Corticospinal Tracts in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 1990, 80, 222–226. [Google Scholar] [CrossRef]
- Sasaki, S.; Iwata, M. Ultrastructural Study of Synapses in the Anterior Horn Neurons of Patients with Amyotrophic Lateral Sclerosis. Neurosci. Lett. 1996, 204, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Moller, A.; Bauer, C.S.; Cohen, R.N.; Webster, C.P.; De Vos, K.J. Amyotrophic Lateral Sclerosis-Associated Mutant SOD1 Inhibits Anterograde Axonal Transport of Mitochondria by Reducing Miro1 Levels. Hum. Mol. Genet. 2017, 26, 4668–4679. [Google Scholar] [CrossRef] [PubMed]
- De Vos, K.J.; Chapman, A.L.; Tennant, M.E.; Manser, C.; Tudor, E.L.; Lau, K.F.; Brownlees, J.; Ackerley, S.; Shaw, P.J.; McLoughlin, D.M.; et al. Familial Amyotrophic Lateral Sclerosis-Linked SOD1 Mutants Perturb Fast Axonal Transport to Reduce Axonal Mitochondria Content. Hum. Mol. Genet. 2007, 16, 2720–2728. [Google Scholar] [CrossRef] [PubMed]
- Moloney, E.B.; de Winter, F.; Verhaagen, J. ALS as a Distal Axonopathy: Molecular Mechanisms Affecting Neuromuscular Junction Stability in the Presymptomatic Stages of the Disease. Front. Neurosci. 2014, 8, 252. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin Target Miro for Phosphorylation and Degradation to Arrest Mitochondrial Motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.N.; Ticozzi, N.; Fallini, C.; Gkazi, A.S.; Topp, S.; Kenna, K.P.; Scotter, E.L.; Kost, J.; Keagle, P.; Miller, J.W.; et al. Exome-Wide Rare Variant Analysis Identifies TUBA4A Mutations Associated with Familial ALS. Neuron 2014, 84, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Puls, I.; Jonnakuty, C.; LaMonte, B.H.; Holzbaur, E.L.; Tokito, M.; Mann, E.; Floeter, M.K.; Bidus, K.; Drayna, D.; Oh, S.J.; et al. Mutant Dynactin in Motor Neuron Disease. Nat. Genet. 2003, 33, 455–456. [Google Scholar] [CrossRef]
- Wu, C.H.; Fallini, C.; Ticozzi, N.; Keagle, P.J.; Sapp, P.C.; Piotrowska, K.; Lowe, P.; Koppers, M.; McKenna-Yasek, D.; Baron, D.M.; et al. Mutations in the Profilin 1 Gene Cause Familial Amyotrophic Lateral Sclerosis. Nature 2012, 488, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Naia, L.; Ferreira, I.L.; Ferreiro, E.; Rego, A.C. Mitochondrial Ca(2+) Handling in Huntington’s and Alzheimer’s Diseases—Role of ER-Mitochondria Crosstalk. Biochem. Biophys. Res. Commun. 2017, 483, 1069–1077. [Google Scholar] [CrossRef]
- Ji, C.; Zhang, Z.; Li, Z.; She, X.; Wang, X.; Li, B.; Xu, X.; Song, D.; Zhang, D. Mitochondria-Associated Endoplasmic Reticulum Membranes: Inextricably Linked with Autophagy Process. Oxidative Med. Cell. Longev. 2022, 2022, 7086807. [Google Scholar] [CrossRef]
- Sack, R.B.; Kline, R.L.; Spira, W.M. Oral Immunization of Rabbits with Enterotoxigenic Escherichia coli Protects against Intraintestinal Challenge. Infect. Immun. 1988, 56, 387–394. [Google Scholar] [CrossRef]
- He, Q.; Qu, M.; Shen, T.; Su, J.; Xu, Y.; Xu, C.; Barkat, M.Q.; Cai, J.; Zhu, H.; Zeng, L.H.; et al. Control of Mitochondria-Associated Endoplasmic Reticulum Membranes by Protein S-Palmitoylation: Novel Therapeutic Targets for Neurodegenerative Diseases. Ageing Res. Rev. 2023, 87, 101920. [Google Scholar] [CrossRef]
- Sala-Vila, A.; Navarro-Lerida, I.; Sanchez-Alvarez, M.; Bosch, M.; Calvo, C.; Lopez, J.A.; Calvo, E.; Ferguson, C.; Giacomello, M.; Serafini, A.; et al. Interplay between Hepatic Mitochondria-Associated Membranes, Lipid Metabolism and Caveolin-1 in Mice. Sci. Rep. 2016, 6, 27351. [Google Scholar] [CrossRef]
- Tubbs, E.; Theurey, P.; Vial, G.; Bendridi, N.; Bravard, A.; Chauvin, M.A.; Ji-Cao, J.; Zoulim, F.; Bartosch, B.; Ovize, M.; et al. Mitochondria-Associated Endoplasmic Reticulum Membrane (MAM) Integrity Is Required for Insulin Signaling and Is Implicated in Hepatic Insulin Resistance. Diabetes 2014, 63, 3279–3294. [Google Scholar] [CrossRef]
- D’Eletto, M.; Rossin, F.; Occhigrossi, L.; Farrace, M.G.; Faccenda, D.; Desai, R.; Marchi, S.; Refolo, G.; Falasca, L.; Antonioli, M.; et al. Transglutaminase Type 2 Regulates ER-Mitochondria Contact Sites by Interacting with GRP75. Cell Rep. 2018, 25, 3573–3581.e4. [Google Scholar] [CrossRef]
- Couly, S.; Yasui, Y.; Su, T.P. SIGMAR1 Confers Innate Resilience against Neurodegeneration. Int. J. Mol. Sci. 2023, 24, 7767. [Google Scholar] [CrossRef]
- Gutierrez, T.; Simmen, T. Endoplasmic Reticulum Chaperones Tweak the Mitochondrial Calcium Rheostat to Control Metabolism and Cell Death. Cell Calcium 2018, 70, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-Mitochondria Contact Sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.F.; Vizcay-Barrena, G.; Lin, W.L.; Xu, Y.F.; Lewis, J.; et al. ER-Mitochondria Associations Are Regulated by the VAPB-PTPIP51 Interaction and Are Disrupted by ALS/FTD-Associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef]
- Bernal, A.F.; Mota, N.; Pamplona, R.; Area-Gomez, E.; Portero-Otin, M. Hakuna MAM-Tata: Investigating the Role of Mitochondrial-Associated Membranes in ALS. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1869, 166716. [Google Scholar] [CrossRef]
- Stoica, R.; Paillusson, S.; Gomez-Suaga, P.; Mitchell, J.C.; Lau, D.H.; Gray, E.H.; Sancho, R.M.; Vizcay-Barrena, G.; De Vos, K.J.; Shaw, C.E.; et al. ALS/FTD-Associated FUS Activates GSK-3beta to Disrupt the VAPB-PTPIP51 Interaction and ER-Mitochondria Associations. EMBO Rep. 2016, 17, 1326–1342. [Google Scholar] [CrossRef]
- Karagas, N.E.; Gupta, R.; Rastegari, E.; Tan, K.L.; Leung, H.H.; Bellen, H.J.; Venkatachalam, K.; Wong, C.O. Loss of Activity-Induced Mitochondrial ATP Production Underlies the Synaptic Defects in a Drosophila Model of ALS. J. Neurosci. 2022, 42, 8019–8037. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Suaga, P.; Morotz, G.M.; Markovinovic, A.; Martin-Guerrero, S.M.; Preza, E.; Arias, N.; Mayl, K.; Aabdien, A.; Gesheva, V.; Nishimura, A.; et al. Disruption of ER-Mitochondria Tethering and Signalling in C9orf72-Associated Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Aging Cell 2022, 21, e13549. [Google Scholar] [CrossRef]
- Pilotto, F.; Schmitz, A.; Maharjan, N.; Diab, R.; Odriozola, A.; Tripathi, P.; Yamoah, A.; Scheidegger, O.; Oestmann, A.; Dennys, C.N.; et al. PolyGA Targets the ER Stress-Adaptive Response by Impairing GRP75 Function at the MAM in C9ORF72-ALS/FTD. Acta Neuropathol. 2022, 144, 939–966. [Google Scholar] [CrossRef]
- Jeon, Y.M.; Kwon, Y.; Lee, S.; Kim, H.J. Potential Roles of the Endoplasmic Reticulum Stress Pathway in Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2023, 15, 1047897. [Google Scholar] [CrossRef]
- Saxena, S.; Cabuy, E.; Caroni, P. A Role for Motoneuron Subtype-Selective ER Stress in Disease Manifestations of FALS Mice. Nat. Neurosci. 2009, 12, 627–636. [Google Scholar] [CrossRef]
- Ono, Y.; Tanaka, H.; Takata, M.; Nagahara, Y.; Noda, Y.; Tsuruma, K.; Shimazawa, M.; Hozumi, I.; Hara, H. SA4503, a Sigma-1 Receptor Agonist, Suppresses Motor Neuron Damage in In Vitro and In Vivo Amyotrophic Lateral Sclerosis Models. Neurosci. Lett. 2014, 559, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Das, I.; Krzyzosiak, A.; Schneider, K.; Wrabetz, L.; D’Antonio, M.; Barry, N.; Sigurdardottir, A.; Bertolotti, A. Preventing Proteostasis Diseases by Selective Inhibition of a Phosphatase Regulatory Subunit. Science 2015, 348, 239–242. [Google Scholar] [CrossRef]
- Chen, Y.; Podojil, J.R.; Kunjamma, R.B.; Jones, J.; Weiner, M.; Lin, W.; Miller, S.D.; Popko, B. Sephin1, Which Prolongs the Integrated Stress Response, Is a Promising Therapeutic for Multiple Sclerosis. Brain 2019, 142, 344–361. [Google Scholar] [CrossRef]
- Fujikake, N.; Shin, M.; Shimizu, S. Association between Autophagy and Neurodegenerative Diseases. Front. Neurosci. 2018, 12, 255. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and Function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Levine, B. Autophagic Cell Death: The Story of a Misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Song, C.Y.; Guo, J.F.; Liu, Y.; Tang, B.S. Autophagy and Its Comprehensive Impact on ALS. Int. J. Neurosci. 2012, 122, 695–703. [Google Scholar] [CrossRef]
- Park, H.; Kang, J.H.; Lee, S. Autophagy in Neurodegenerative Diseases: A Hunter for Aggregates. Int. J. Mol. Sci. 2020, 21, 3369. [Google Scholar] [CrossRef]
- King, A.; Al-Sarraj, S.; Troakes, C.; Smith, B.N.; Maekawa, S.; Iovino, M.; Spillantini, M.G.; Shaw, C.E. Mixed tau, TDP-43 and p62 Pathology in FTLD Associated with a C9ORF72 Repeat Expansion and p.Ala239Thr MAPT (tau) Variant. Acta Neuropathol. 2013, 125, 303–310. [Google Scholar] [CrossRef]
- Al-Sarraj, S.; King, A.; Troakes, C.; Smith, B.; Maekawa, S.; Bodi, I.; Rogelj, B.; Al-Chalabi, A.; Hortobagyi, T.; Shaw, C.E. p62 Positive, TDP-43 Negative, Neuronal Cytoplasmic and Intranuclear Inclusions in the Cerebellum and Hippocampus Define the Pathology of C9orf72-Linked FTLD and MND/ALS. Acta Neuropathol. 2011, 122, 691–702. [Google Scholar] [CrossRef]
- Duan, W.; Guo, M.; Yi, L.; Zhang, J.; Bi, Y.; Liu, Y.; Li, Y.; Li, Z.; Ma, Y.; Zhang, G.; et al. Deletion of Tbk1 Disrupts Autophagy and Reproduces Behavioral and Locomotor Symptoms of FTD-ALS in Mice. Aging 2019, 11, 2457–2476. [Google Scholar] [CrossRef]
- Chen, T.; Huang, B.; Shi, X.; Gao, L.; Huang, C. Mutant UBQLN2(P497H) in Motor Neurons Leads to ALS-Like Phenotypes and Defective Autophagy in Rats. Acta Neuropathol. Commun. 2018, 6, 122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Zhou, Q.M.; Chen, S.; Le, W.D. Repurposing Carbamazepine for the Treatment of Amyotrophic Lateral Sclerosis in SOD1-G93A Mouse Model. CNS Neurosci. Ther. 2018, 24, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, E.; Beltran, S.; Labrador, L.; Manque, P.; Nassif, M.; Woehlbier, U. Implications of Selective Autophagy Dysfunction for ALS Pathology. Cells 2020, 9, 381. [Google Scholar] [CrossRef]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Muller, K.; Marroquin, N.; Nordin, F.; Hubers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 Causes Familial ALS and Fronto-Temporal Dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef]
- Rea, S.L.; Majcher, V.; Searle, M.S.; Layfield, R. SQSTM1 Mutations—Bridging Paget Disease of Bone and ALS/FTLD. Exp. Cell Res. 2014, 325, 27–37. [Google Scholar] [CrossRef]
- Gal, J.; Strom, A.L.; Kwinter, D.M.; Kilty, R.; Zhang, J.; Shi, P.; Fu, W.; Wooten, M.W.; Zhu, H. Sequestosome 1/p62 Links Familial ALS Mutant SOD1 to LC3 via an Ubiquitin-Independent Mechanism. J. Neurochem. 2009, 111, 1062–1073. [Google Scholar] [CrossRef]
- Korac, J.; Schaeffer, V.; Kovacevic, I.; Clement, A.M.; Jungblut, B.; Behl, C.; Terzic, J.; Dikic, I. Ubiquitin-Independent Function of Optineurin in Autophagic Clearance of Protein Aggregates. J. Cell Sci. 2013, 126, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.C.; Li, H.Y.; Chen, G.C.; Chern, Y.; Tu, P.H. Mutations in the Ubiquitin-Binding Domain of OPTN/Optineurin Interfere with Autophagy-Mediated Degradation of Misfolded Proteins by a Dominant-Negative Mechanism. Autophagy 2015, 11, 685–700. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From Genes to Mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Wei, Y. Autophagic Induction of Amyotrophic Lateral Sclerosis-Linked Cu/Zn Superoxide Dismutase 1 G93A Mutant in NSC34 Cells. Neural Regen. Res. 2014, 9, 16–24. [Google Scholar] [CrossRef]
- Morimoto, N.; Nagai, M.; Ohta, Y.; Miyazaki, K.; Kurata, T.; Morimoto, M.; Murakami, T.; Takehisa, Y.; Ikeda, Y.; Kamiya, T.; et al. Increased Autophagy in Transgenic Mice with a G93A Mutant SOD1 Gene. Brain Res. 2007, 1167, 112–117. [Google Scholar] [CrossRef]
- An, T.; Shi, P.; Duan, W.; Zhang, S.; Yuan, P.; Li, Z.; Wu, D.; Xu, Z.; Li, C.; Guo, Y. Oxidative Stress and Autophagic Alteration in Brainstem of SOD1-G93A Mouse Model of ALS. Mol. Neurobiol. 2014, 49, 1435–1448. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Ballabio, A. TFEB Regulates Autophagy: An Integrated Coordination of Cellular Degradation and Recycling Processes. Autophagy 2011, 7, 1379–1381. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 Network Regulates Autophagy and Apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, H.; Guan, Y.; Wang, Q.; Zhou, F.; Jie, L.; Ju, J.; Pu, L.; Du, H.; Wang, X. The Altered Autophagy Mediated by TFEB in Animal and Cell Models of Amyotrophic Lateral Sclerosis. Am. J. Transl. Res. 2015, 7, 1574–1587. [Google Scholar] [PubMed]
- Zhang, F.; Strom, A.L.; Fukada, K.; Lee, S.; Hayward, L.J.; Zhu, H. Interaction between Familial Amyotrophic Lateral Sclerosis (ALS)-Linked SOD1 Mutants and the Dynein Complex. J. Biol. Chem. 2007, 282, 16691–16699. [Google Scholar] [CrossRef]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 Is Intrinsically Aggregation-Prone, and Amyotrophic Lateral Sclerosis-Linked Mutations Accelerate Aggregation and Increase Toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Diaz, Z.; Fang, X.; Hart, M.P.; Chesi, A.; Shorter, J.; Gitler, A.D. Molecular Determinants and Genetic Modifiers of Aggregation and Toxicity for the ALS Disease Protein FUS/TLS. PLoS Biol. 2011, 9, e1000614. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.H.; Jun, M.H.; Min, K.J.; Jang, D.J.; Lee, Y.S.; Kim, H.K.; Lee, J.A. Autophagy Regulates Amyotrophic Lateral Sclerosis-Linked Fused in Sarcoma-Positive Stress Granules in Neurons. Neurobiol. Aging 2014, 35, 2822–2831. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.W.; Lin, M.J.; Shen, C.K. Rapamycin Alleviates Pathogenesis of a New Drosophila Model of ALS-TDP. J. Neurogenet. 2015, 29, 59–68. [Google Scholar] [CrossRef]
- Wang, I.F.; Guo, B.S.; Liu, Y.C.; Wu, C.C.; Yang, C.H.; Tsai, K.J.; Shen, C.K. Autophagy Activators Rescue and Alleviate Pathogenesis of a Mouse Model with Proteinopathies of the TAR DNA-Binding Protein 43. Proc. Natl. Acad. Sci. USA 2012, 109, 15024–15029. [Google Scholar] [CrossRef] [PubMed]
- Almeida, S.; Gascon, E.; Tran, H.; Chou, H.J.; Gendron, T.F.; Degroot, S.; Tapper, A.R.; Sellier, C.; Charlet-Berguerand, N.; Karydas, A.; et al. Modeling Key Pathological Features of Frontotemporal Dementia with C9ORF72 Repeat Expansion in iPSC-Derived Human Neurons. Acta Neuropathol. 2013, 126, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Ugolino, J.; Ji, Y.J.; Conchina, K.; Chu, J.; Nirujogi, R.S.; Pandey, A.; Brady, N.R.; Hamacher-Brady, A.; Wang, J. Loss of C9orf72 Enhances Autophagic Activity via Deregulated mTOR and TFEB Signaling. PLoS Genet. 2016, 12, e1006443. [Google Scholar] [CrossRef]
- Amick, J.; Roczniak-Ferguson, A.; Ferguson, S.M. C9orf72 Binds SMCR8, Localizes to Lysosomes, and Regulates mTORC1 Signaling. Mol. Biol. Cell 2016, 27, 3040–3051. [Google Scholar] [CrossRef]
- Sullivan, P.M.; Zhou, X.; Robins, A.M.; Paushter, D.H.; Kim, D.; Smolka, M.B.; Hu, F. The ALS/FTLD Associated Protein C9orf72 Associates with SMCR8 and WDR41 to Regulate the Autophagy-Lysosome Pathway. Acta Neuropathol. Commun. 2016, 4, 51. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, N.; Lu, B. Mechanisms and Roles of Mitophagy in Neurodegenerative Diseases. CNS Neurosci. Ther. 2019, 25, 859–875. [Google Scholar] [CrossRef]
- Edens, B.M.; Miller, N.; Ma, Y.C. Impaired Autophagy and Defective Mitochondrial Function: Converging Paths on the Road to Motor Neuron Degeneration. Front. Cell. Neurosci. 2016, 10, 44. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The Ubiquitin Kinase PINK1 Recruits Autophagy Receptors to Induce Mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Holzbaur, E.L. Dynamic Recruitment and Activation of ALS-Associated TBK1 with Its Target Optineurin Are Required for Efficient Mitophagy. Proc. Natl. Acad. Sci. USA 2016, 113, E3349–E3358. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L. Optineurin Is an Autophagy Receptor for Damaged Mitochondria in Parkin-Mediated Mitophagy That Is Disrupted by an ALS-Linked Mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef]
- Gautam, M.; Xie, E.F.; Kocak, N.; Ozdinler, P.H. Mitoautophagy: A Unique Self-Destructive Path Mitochondria of Upper Motor Neurons with TDP-43 Pathology Take, Very Early in ALS. Front. Cell. Neurosci. 2019, 13, 489. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Bigio, E.H.; Ince, P.G.; Geser, F.; Neumann, M.; Cairns, N.J.; Kwong, L.K.; Forman, M.S.; Ravits, J.; Stewart, H.; et al. Pathological TDP-43 Distinguishes Sporadic Amyotrophic Lateral Sclerosis from Amyotrophic Lateral Sclerosis with SOD1 Mutations. Ann. Neurol. 2007, 61, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J. Neuronal Death in Amyotrophic Lateral Sclerosis Is Apoptosis: Possible Contribution of a Programmed Cell Death Mechanism. J. Neuropathol. Exp. Neurol. 1999, 58, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Ilzecka, J.; Stelmasiak, Z.; Dobosz, B. Interleukin-1beta Converting Enzyme/Caspase-1 (ICE/Caspase-1) and Soluble APO-1/Fas/CD 95 Receptor in Amyotrophic Lateral Sclerosis Patients. Acta Neurol. Scand. 2001, 103, 255–258. [Google Scholar] [CrossRef]
- Li, M.; Ona, V.O.; Guegan, C.; Chen, M.; Jackson-Lewis, V.; Andrews, L.J.; Olszewski, A.J.; Stieg, P.E.; Lee, J.P.; Przedborski, S.; et al. Functional Role of Caspase-1 and Caspase-3 in an ALS Transgenic Mouse Model. Science 2000, 288, 335–339. [Google Scholar] [CrossRef]
- Pasinelli, P.; Houseweart, M.K.; Brown, R.H., Jr.; Cleveland, D.W. Caspase-1 and -3 Are Sequentially Activated in Motor Neuron Death in Cu,Zn Superoxide Dismutase-Mediated Familial Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 2000, 97, 13901–13906. [Google Scholar] [CrossRef]
- Vukosavic, S.; Stefanis, L.; Jackson-Lewis, V.; Guegan, C.; Romero, N.; Chen, C.; Dubois-Dauphin, M.; Przedborski, S. Delaying Caspase Activation by Bcl-2: A Clue to Disease Retardation in a Transgenic Mouse Model of Amyotrophic Lateral Sclerosis. J. Neurosci. 2000, 20, 9119–9125. [Google Scholar] [CrossRef]
- Guégan, C.; Vila, M.; Rosoklija, G.; Hays, A.P.; Przedborski, S. Recruitment of the Mitochondrial-Dependent Apoptotic Pathway in Amyotrophic Lateral Sclerosis. J. Neurosci. 2001, 21, 6569–6576. [Google Scholar] [CrossRef]
- Damiano, M.; Starkov, A.A.; Petri, S.; Kipiani, K.; Kiaei, M.; Mattiazzi, M.; Flint Beal, M.; Manfredi, G. Neural Mitochondrial Ca2+ Capacity Impairment Precedes the Onset of Motor Symptoms in G93A Cu/Zn-Superoxide Dismutase Mutant Mice. J. Neurochem. 2006, 96, 1349–1361. [Google Scholar] [CrossRef]
- Silva, F.S.; Costa, C.F.; Marques, R.J.; Oliveira, P.J.; Pereira, G.C. Pharmacological Targeting of the Mitochondrial Permeability Transition Pore for Cardioprotection. In Mitochondrial Biology and Experimental Therapeutics; Oliveira, P.J., Ed.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 423–490. [Google Scholar] [CrossRef]
- Kalani, K.; Yan, S.F.; Yan, S.S. Mitochondrial Permeability Transition Pore: A Potential Drug Target for Neurodegeneration. Drug Discov. Today 2018, 23, 1983–1989. [Google Scholar] [CrossRef]
- Halestrap, A.P. What Is the Mitochondrial Permeability Transition Pore? J. Mol. Cell. Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Kinnally, K.W.; Peixoto, P.M.; Ryu, S.Y.; Dejean, L.M. Is mPTP the Gatekeeper for Necrosis, Apoptosis, or Both? Biochim. Biophys. Acta 2011, 1813, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Hurst, S.; Hoek, J.; Sheu, S.S. Mitochondrial Ca(2+) and Regulation of the Permeability Transition Pore. J. Bioenerg. Biomembr. 2017, 49, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol. Rev. 2015, 95, 1111–1155. [Google Scholar] [CrossRef]
- Bonora, M.; Morganti, C.; Morciano, G.; Pedriali, G.; Lebiedzinska-Arciszewska, M.; Aquila, G.; Giorgi, C.; Rizzo, P.; Campo, G.; Ferrari, R.; et al. Mitochondrial Permeability Transition Involves Dissociation of F1FO ATP Synthase Dimers and C-Ring Conformation. EMBO Rep. 2017, 18, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Gertz, B.; Pan, Y.; Price, A.C.; Molkentin, J.D.; Chang, Q. The Mitochondrial Permeability Transition Pore in Motor Neurons: Involvement in the Pathobiology of ALS Mice. Exp. Neurol. 2009, 218, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Parone, P.A.; Da Cruz, S.; Han, J.S.; McAlonis-Downes, M.; Vetto, A.P.; Lee, S.K.; Tseng, E.; Cleveland, D.W. Enhancing Mitochondrial Calcium Buffering Capacity Reduces Aggregation of Misfolded SOD1 and Motor Neuron Cell Death without Extending Survival in Mouse Models of Inherited Amyotrophic Lateral Sclerosis. J. Neurosci. 2013, 33, 4657–4671. [Google Scholar] [CrossRef]
- Xiao, Y.; Karam, C.; Yi, J.; Zhang, L.; Li, X.; Yoon, D.; Wang, H.; Dhakal, K.; Ramlow, P.; Yu, T.; et al. ROS-Related Mitochondrial Dysfunction in Skeletal Muscle of an ALS Mouse Model during the Disease Progression. Pharmacol. Res. 2018, 138, 25–36. [Google Scholar] [CrossRef]
- Karam, C.; Yi, J.; Xiao, Y.; Dhakal, K.; Zhang, L.; Li, X.; Manno, C.; Xu, J.; Li, K.; Cheng, H.; et al. Absence of Physiological Ca(2+) Transients Is an Initial Trigger for Mitochondrial Dysfunction in Skeletal Muscle Following Denervation. Skelet. Muscle 2017, 7, 6. [Google Scholar] [CrossRef]
- Gautam, M.; Gunay, A.; Chandel, N.S.; Ozdinler, P.H. Mitochondrial Dysregulation Occurs early in ALS Motor Cortex with TDP-43 Pathology and Suggests Maintaining NAD(+) Balance as a Therapeutic Strategy. Sci. Rep. 2022, 12, 4287. [Google Scholar] [CrossRef] [PubMed]
- So, E.; Mitchell, J.C.; Memmi, C.; Chennell, G.; Vizcay-Barrena, G.; Allison, L.; Shaw, C.E.; Vance, C. Mitochondrial Abnormalities and Disruption of the Neuromuscular Junction Precede the Clinical Phenotype and Motor Neuron Loss in hFUSWT Transgenic Mice. Hum. Mol. Genet. 2018, 27, 463–474. [Google Scholar] [CrossRef]
- Belosludtseva, N.V.; Matveeva, L.A.; Belosludtsev, K.N. Mitochondrial Dyshomeostasis as an Early Hallmark and a Therapeutic Target in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2023, 24, 16833. [Google Scholar] [CrossRef] [PubMed]
- Goyal, N.A.; Berry, J.D.; Windebank, A.; Staff, N.P.; Maragakis, N.J.; van den Berg, L.H.; Genge, A.; Miller, R.; Baloh, R.H.; Kern, R.; et al. Addressing Heterogeneity in Amyotrophic Lateral Sclerosis Clinical Trials. Muscle Nerve 2020, 62, 156–166. [Google Scholar] [CrossRef]
- Beghi, E.; Mennini, T.; Bendotti, C.; Bigini, P.; Logroscino, G.; Chio, A.; Hardiman, O.; Mitchell, D.; Swingler, R.; Traynor, B.J.; et al. The Heterogeneity of Amyotrophic Lateral Sclerosis: A Possible Explanation of Treatment Failure. Curr. Med. Chem. 2007, 14, 3185–3200. [Google Scholar] [CrossRef] [PubMed]
- Obrador, E.; Salvador-Palmer, R.; Lopez-Blanch, R.; Jihad-Jebbar, A.; Valles, S.L.; Estrela, J.M. The Link between Oxidative Stress, Redox Status, Bioenergetics and Mitochondria in the Pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 6352. [Google Scholar] [CrossRef]
- Jha, M.K.; Jeon, S.; Suk, K. Pyruvate Dehydrogenase Kinases in the Nervous System: Their Principal Functions in Neuronal-glial Metabolic Interaction and Neuro-metabolic Disorders. Curr. Neuropharmacol. 2012, 10, 393–403. [Google Scholar] [CrossRef]
- Miquel, E.; Cassina, A.; Martinez-Palma, L.; Bolatto, C.; Trias, E.; Gandelman, M.; Radi, R.; Barbeito, L.; Cassina, P. Modulation of Astrocytic Mitochondrial Function by Dichloroacetate Improves Survival and Motor Performance in Inherited Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, e34776. [Google Scholar] [CrossRef]
- Martinez-Palma, L.; Miquel, E.; Lagos-Rodriguez, V.; Barbeito, L.; Cassina, A.; Cassina, P. Mitochondrial Modulation by Dichloroacetate Reduces Toxicity of Aberrant Glial Cells and Gliosis in the SOD1G93A Rat Model of Amyotrophic Lateral Sclerosis. Neurotherapeutics 2019, 16, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Varghese, M.; Vempati, P.; Dzhun, A.; Cheng, A.; Wang, J.; Lange, D.; Bilski, A.; Faravelli, I.; Pasinetti, G.M. Caprylic Triglyceride as a Novel Therapeutic Approach to Effectively Improve the Performance and Attenuate the Symptoms Due to the Motor Neuron Loss in ALS Disease. PLoS ONE 2012, 7, e49191. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Lange, D.J.; Voustianiouk, A.; MacGrogan, D.; Ho, L.; Suh, J.; Humala, N.; Thiyagarajan, M.; Wang, J.; Pasinetti, G.M. A Ketogenic Diet as a Potential Novel Therapeutic Intervention in Amyotrophic Lateral Sclerosis. BMC Neurosci. 2006, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, W.A.; Mattson, M.P. No Benefit of Dietary Restriction on Disease Onset or Progression in Amyotrophic Lateral Sclerosis Cu/Zn-Superoxide Dismutase Mutant Mice. Brain Res. 1999, 833, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Hamadeh, M.J.; Rodriguez, M.C.; Kaczor, J.J.; Tarnopolsky, M.A. Caloric Restriction Transiently Improves Motor Performance but Hastens Clinical Onset of Disease in the Cu/Zn-Superoxide Dismutase Mutant G93A Mouse. Muscle Nerve 2005, 31, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Cutler, R.G.; Camandola, S. Energy Intake and Amyotrophic Lateral Sclerosis. Neuromol. Med. 2007, 9, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Tefera, T.W.; Borges, K. Metabolic Dysfunctions in Amyotrophic Lateral Sclerosis Pathogenesis and Potential Metabolic Treatments. Front. Neurosci. 2016, 10, 611. [Google Scholar] [CrossRef] [PubMed]
- Branco, A.F.; Ferreira, A.; Simoes, R.F.; Magalhaes-Novais, S.; Zehowski, C.; Cope, E.; Silva, A.M.; Pereira, D.; Sardao, V.A.; Cunha-Oliveira, T. Ketogenic Diets: From Cancer to Mitochondrial Diseases and Beyond. Eur. J. Clin. Investig. 2016, 46, 285–298. [Google Scholar] [CrossRef]
- Laffel, L. Ketone Bodies: A Review of Physiology, Pathophysiology and Application of Monitoring to Diabetes. Diabetes Metab. Res. Rev. 1999, 15, 412–426. [Google Scholar] [CrossRef]
- Ludolph, A.C.; Dorst, J.; Dreyhaupt, J.; Weishaupt, J.H.; Kassubek, J.; Weiland, U.; Meyer, T.; Petri, S.; Hermann, A.; Emmer, A.; et al. Effect of High-Caloric Nutrition on Survival in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2020, 87, 206–216. [Google Scholar] [CrossRef]
- Wang, S.; Yuan, T.; Yang, H.; Zhou, X.; Cao, J. Effect of Complete High-Caloric Nutrition on the Nutritional Status and Survival Rate of Amyotrophic Lateral Sclerosis Patients after Gastrostomy. Am. J. Transl. Res. 2022, 14, 7842–7851. [Google Scholar] [PubMed]
- Vance, J.E.; Campenot, R.B.; Vance, D.E. The Synthesis and Transport of Lipids for Axonal Growth and Nerve Regeneration. Biochim. Biophys. Acta 2000, 1486, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Corcia, P.; Fergani, A.; Gonzalez De Aguilar, J.L.; Bonnefont-Rousselot, D.; Bittar, R.; Seilhean, D.; Hauw, J.J.; Lacomblez, L.; Loeffler, J.P.; et al. Dyslipidemia Is a Protective Factor in Amyotrophic Lateral Sclerosis. Neurology 2008, 70, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Onofrj, M.; Ciccocioppo, F.; Varanese, S.; di Muzio, A.; Calvani, M.; Chiechio, S.; Osio, M.; Thomas, A. Acetyl-L-Carnitine: From a Biological Curiosity to a Drug for the Peripheral Nervous System and Beyond. Expert Rev. Neurother. 2013, 13, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Kira, Y.; Nishikawa, M.; Ochi, A.; Sato, E.; Inoue, M. L-Carnitine Suppresses the Onset of Neuromuscular Degeneration and Increases the Life Span of Mice with Familial Amyotrophic Lateral Sclerosis. Brain Res. 2006, 1070, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Beghi, E.; Pupillo, E.; Bonito, V.; Buzzi, P.; Caponnetto, C.; Chio, A.; Corbo, M.; Giannini, F.; Inghilleri, M.; Bella, V.L.; et al. Randomized Double-Blind Placebo-Controlled Trial Of Acetyl-L-Carnitine for ALS. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Grossini, E.; De Marchi, F.; Venkatesan, S.; Mele, A.; Ferrante, D.; Mazzini, L. Effects of Acetyl-L-Carnitine on Oxidative Stress in Amyotrophic Lateral Sclerosis Patients: Evaluation on Plasma Markers and Members of the Neurovascular Unit. Antioxidants 2023, 12, 1887. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, O.A.; Dedeoglu, A.; Klivenyi, P.; Beal, M.F.; Bush, A.I. N-Acetyl-L-Cysteine Improves Survival and Preserves Motor Performance in an Animal Model of Familial Amyotrophic Lateral Sclerosis. Neuroreport 2000, 11, 2491–2493. [Google Scholar] [CrossRef]
- Burgunder, J.M.; Varriale, A.; Lauterburg, B.H. Effect of N-Acetylcysteine on Plasma Cysteine and Glutathione Following Paracetamol Administration. Eur. J. Clin. Pharmacol. 1989, 36, 127–131. [Google Scholar] [CrossRef]
- Beretta, S.; Sala, G.; Mattavelli, L.; Ceresa, C.; Casciati, A.; Ferri, A.; Carri, M.T.; Ferrarese, C. Mitochondrial Dysfunction Due to Mutant Copper/Zinc Superoxide Dismutase Associated with Amyotrophic Lateral Sclerosis Is Reversed by N-Acetylcysteine. Neurobiol. Dis. 2003, 13, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Louwerse, E.S.; Weverling, G.J.; Bossuyt, P.M.; Meyjes, F.E.; de Jong, J.M. Randomized, Double-Blind, Controlled Trial of Acetylcysteine in Amyotrophic Lateral Sclerosis. Arch. Neurol. 1995, 52, 559–564. [Google Scholar] [CrossRef]
- Kurano, T.; Kanazawa, T.; Iioka, S.; Kondo, H.; Kosuge, Y.; Suzuki, T. Intranasal Administration of N-Acetyl-L-Cysteine Combined with Cell-Penetrating Peptide-Modified Polymer Nanomicelles as a Potential Therapeutic Approach for Amyotrophic Lateral Sclerosis. Pharmaceutics 2022, 14, 2590. [Google Scholar] [CrossRef] [PubMed]
- Bavarsad Shahripour, R.; Harrigan, M.R.; Alexandrov, A.V. N-Acetylcysteine (NAC) in Neurological Disorders: Mechanisms of Action and Therapeutic Opportunities. Brain Behav. 2014, 4, 108–122. [Google Scholar] [CrossRef]
- Giustarini, D.; Milzani, A.; Dalle-Donne, I.; Tsikas, D.; Rossi, R. N-Acetylcysteine Ethyl Ester (NACET): A Novel Lipophilic Cell-Permeable Cysteine Derivative with an Unusual Pharmacokinetic Feature and Remarkable Antioxidant Potential. Biochem. Pharmacol. 2012, 84, 1522–1533. [Google Scholar] [CrossRef] [PubMed]
- Kularatne, R.N.; Bulumulla, C.; Catchpole, T.; Takacs, A.; Christie, A.; Stefan, M.C.; Csaky, K.G. Protection of Human Retinal Pigment Epithelial Cells from Oxidative Damage Using Cysteine Prodrugs. Free Radic. Biol. Med. 2020, 152, 386–394. [Google Scholar] [CrossRef]
- Sunitha, K.; Hemshekhar, M.; Thushara, R.M.; Santhosh, M.S.; Yariswamy, M.; Kemparaju, K.; Girish, K.S. N-Acetylcysteine Amide: A Derivative to Fulfill the Promises of N-Acetylcysteine. Free Radic. Res. 2013, 47, 357–367. [Google Scholar] [CrossRef]
- Atlas, D. Emerging Therapeutic Opportunities of Novel Thiol-Amides, NAC-Amide (AD4/NACA) and Thioredoxin Mimetics (TXM-Peptides) for Neurodegenerative-Related Disorders. Free Radic. Biol. Med. 2021, 176, 120–141. [Google Scholar] [CrossRef]
- Shichinohe, H.; Kuroda, S.; Yasuda, H.; Ishikawa, T.; Iwai, M.; Horiuchi, M.; Iwasaki, Y. Neuroprotective Effects of the Free Radical Scavenger Edaravone (MCI-186) in Mice Permanent Focal Brain Ischemia. Brain Res. 2004, 1029, 200–206. [Google Scholar] [CrossRef]
- Abe, K.; Itoyama, Y.; Sobue, G.; Tsuji, S.; Aoki, M.; Doyu, M.; Hamada, C.; Kondo, K.; Yoneoka, T.; Akimoto, M.; et al. Confirmatory Double-Blind, Parallel-Group, Placebo-Controlled Study of Efficacy and Safety of Edaravone (MCI-186) in Amyotrophic Lateral Sclerosis Patients. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 610–617. [Google Scholar] [CrossRef]
- Mizuno, A.; Umemura, K.; Nakashima, M. Inhibitory Effect of MCI-186, a Free Radical Scavenger, on Cerebral Ischemia Following Rat Middle Cerebral Artery Occlusion. Gen. Pharmacol. 1998, 30, 575–578. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yuki, S.; Watanabe, T.; Mitsuka, M.; Saito, K.I.; Kogure, K. Delayed Neuronal Death Prevented by Inhibition of Increased Hydroxyl Radical Formation in a Transient Cerebral Ischemia. Brain Res. 1997, 762, 240–242. [Google Scholar] [CrossRef]
- Takayasu, Y.; Nakaki, J.; Kawasaki, T.; Koda, K.; Ago, Y.; Baba, A.; Matsuda, T. Edaravone, a Radical Scavenger, Inhibits Mitochondrial Permeability Transition Pore in Rat Brain. J. Pharmacol. Sci. 2007, 103, 434–437. [Google Scholar] [CrossRef]
- Banno, M.; Mizuno, T.; Kato, H.; Zhang, G.; Kawanokuchi, J.; Wang, J.; Kuno, R.; Jin, S.; Takeuchi, H.; Suzumura, A. The Radical Scavenger Edaravone Prevents Oxidative Neurotoxicity Induced by Peroxynitrite and Activated Microglia. Neuropharmacology 2005, 48, 283–290. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, X.; Chen, S.; Zhang, X.; Zhang, S.; Youdium, M.; Le, W. Prevention of Motor Neuron Degeneration by Novel Iron Chelators in SOD1(G93A) Transgenic Mice of Amyotrophic Lateral Sclerosis. Neurodegener. Dis. 2011, 8, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, N.; Maki, T.; Pham, L.D.; Hayakawa, K.; Seo, J.H.; Mandeville, E.T.; Mandeville, J.B.; Kim, K.W.; Lo, E.H.; Arai, K. Oxidative Stress Interferes with White Matter Renewal after Prolonged Cerebral Hypoperfusion in Mice. Stroke 2013, 44, 3516–3521. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Teng, C.H.; Wu, F.F.; Ge, L.Y.; Xiao, J.; Zhang, H.Y.; Chen, D.Q. Edaravone Attenuates Traumatic Brain Injury through Anti-Inflammatory and Anti-Oxidative Modulation. Exp. Ther. Med. 2019, 18, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Aoki, M.; Warita, H.; Mizuno, H.; Suzuki, N.; Yuki, S.; Itoyama, Y. Feasibility Study for Functional Test Battery of SOD Transgenic Rat (H46R) and Evaluation of Edaravone, a Free Radical Scavenger. Brain Res. 2011, 1382, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Wate, R.; Zhang, J.; Ohnishi, S.; Kaneko, S.; Ito, H.; Nakano, S.; Kusaka, H. Treatment with Edaravone, Initiated at Symptom Onset, Slows Motor Decline and Decreases SOD1 Deposition in ALS Mice. Exp. Neurol. 2008, 213, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Kimura, A. Investigation of the Therapeutic Effects of Edaravone, a Free Radical Scavenger, on Amyotrophic Lateral Sclerosis (Phase II Study). Amyotroph. Lateral Scler. 2006, 7, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Akimoto, M.; Palumbo, J.; Sakata, T. A Double-Blind, Parallel-Group, Placebo-Controlled, 24-Week, Exploratory Study of Edaravone (MCI-186) for the Treatment of Advanced Amyotrophic Lateral Sclerosis (ALS) (P3.191). Neurology 2016, 86. [Google Scholar] [CrossRef]
- Hardiman, O.; van den Berg, L.H. Edaravone: A New Treatment for ALS on the Horizon? Lancet Neurol. 2017, 16, 490–491. [Google Scholar] [CrossRef]
- CPHI-Online. Treeway Announces Positive Data from Two Separate Phase I Tw001 Clinical Trials. Available online: https://www.cphi-online.com/treeway-announces-positive-data-from-two-separate-news038315.html (accessed on 23 January 2024).
- Genge, A.; Pattee, G.L.; Sobue, G.; Aoki, M.; Yoshino, H.; Couratier, P.; Lunetta, C.; Petri, S.; Selness, D.; Bidani, S.; et al. Oral Edaravone Demonstrated a Favorable Safety Profile in Patients with Amyotrophic Lateral Sclerosis after 48 Weeks of Treatment. Muscle Nerve 2023, 67, 124–129. [Google Scholar] [CrossRef]
- Gao, M.; Zhu, L.; Chang, J.; Cao, T.; Song, L.; Wen, C.; Chen, Y.; Zhuo, Y.; Chen, F. Safety and Efficacy of Edaravone in Patients with Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Clin. Drug Investig. 2023, 43, 1–11. [Google Scholar] [CrossRef]
- Tan, D.X.; Reiter, R.J.; Manchester, L.C.; Yan, M.T.; El-Sawi, M.; Sainz, R.M.; Mayo, J.C.; Kohen, R.; Allegra, M.; Hardeland, R. Chemical and Physical Properties and Potential Mechanisms: Melatonin as a Broad Spectrum Antioxidant and Free Radical Scavenger. Curr. Top. Med. Chem. 2002, 2, 181–197. [Google Scholar] [CrossRef]
- Ganie, S.A.; Dar, T.A.; Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A. Melatonin: A Potential Anti-Oxidant Therapeutic Agent for Mitochondrial Dysfunctions and Related Disorders. Rejuvenation Res. 2016, 19, 21–40. [Google Scholar] [CrossRef] [PubMed]
- Pandi-Perumal, S.R.; BaHammam, A.S.; Brown, G.M.; Spence, D.W.; Bharti, V.K.; Kaur, C.; Hardeland, R.; Cardinali, D.P. Melatonin Antioxidative Defense: Therapeutical Implications for Aging and Neurodegenerative Processes. Neurotox. Res. 2013, 23, 267–300. [Google Scholar] [CrossRef] [PubMed]
- Leon, J.; Acuna-Castroviejo, D.; Escames, G.; Tan, D.X.; Reiter, R.J. Melatonin Mitigates Mitochondrial Malfunction. J. Pineal Res. 2005, 38, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cook, A.; Kim, J.; Baranov, S.V.; Jiang, J.; Smith, K.; Cormier, K.; Bennett, E.; Browser, R.P.; Day, A.L.; et al. Melatonin Inhibits the Caspase-1/Cytochrome C/Caspase-3 Cell Death Pathway, Inhibits MT1 Receptor Loss and Delays Disease Progression in a Mouse Model of Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2013, 55, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Dardiotis, E.; Panayiotou, E.; Feldman, M.L.; Hadjisavvas, A.; Malas, S.; Vonta, I.; Hadjigeorgiou, G.; Kyriakou, K.; Kyriakides, T. Intraperitoneal Melatonin Is Not Neuroprotective in the G93ASOD1 Transgenic Mouse Model of Familial ALS and May Exacerbate Neurodegeneration. Neurosci. Lett. 2013, 548, 170–175. [Google Scholar] [CrossRef]
- Murphy, M.P.; Smith, R.A. Targeting Antioxidants to Mitochondria by Conjugation to Lipophilic Cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar] [CrossRef] [PubMed]
- Finichiu, P.G.; James, A.M.; Larsen, L.; Smith, R.A.; Murphy, M.P. Mitochondrial Accumulation of a Lipophilic Cation Conjugated to an Ionisable Group Depends on Membrane Potential, pH Gradient and pK(a): Implications for the Design of Mitochondrial Probes and Therapies. J. Bioenerg. Biomembr. 2013, 45, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.; Murphy, M.P. Selective Targeting of a Redox-Active Ubiquinone to Mitochondria within Cells: Antioxidant and Antiapoptotic Properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef] [PubMed]
- Cocheme, H.M.; Kelso, G.F.; James, A.M.; Ross, M.F.; Trnka, J.; Mahendiran, T.; Asin-Cayuela, J.; Blaikie, F.H.; Manas, A.R.; Porteous, C.M.; et al. Mitochondrial Targeting of Quinones: Therapeutic Implications. Mitochondrion 2007, 7, S94–S102. [Google Scholar] [CrossRef]
- Smith, R.A.; Murphy, M.P. Animal and Human Studies with the Mitochondria-Targeted Antioxidant MitoQ. Ann. N. Y. Acad. Sci. 2010, 1201, 96–103. [Google Scholar] [CrossRef]
- Solesio, M.E.; Prime, T.A.; Logan, A.; Murphy, M.P.; Del Mar Arroyo-Jimenez, M.; Jordan, J.; Galindo, M.F. The Mitochondria-Targeted Anti-Oxidant MitoQ Reduces Aspects of Mitochondrial Fission in the 6-OHDA Cell Model of Parkinson’s Disease. Biochim. Biophys. Acta 2013, 1832, 174–182. [Google Scholar] [CrossRef]
- Ghosh, A.; Chandran, K.; Kalivendi, S.V.; Joseph, J.; Antholine, W.E.; Hillard, C.J.; Kanthasamy, A.; Kanthasamy, A.; Kalyanaraman, B. Neuroprotection by a Mitochondria-Targeted Drug in a Parkinson’s Disease Model. Free Radic. Biol. Med. 2010, 49, 1674–1684. [Google Scholar] [CrossRef]
- McManus, M.J.; Murphy, M.P.; Franklin, J.L. The Mitochondria-Targeted Antioxidant MitoQ Prevents Loss of Spatial Memory Retention and Early Neuropathology in a Transgenic Mouse Model of Alzheimer’s Disease. J. Neurosci. 2011, 31, 15703–15715. [Google Scholar] [CrossRef]
- Manczak, M.; Mao, P.; Calkins, M.J.; Cornea, A.; Reddy, A.P.; Murphy, M.P.; Szeto, H.H.; Park, B.; Reddy, P.H. Mitochondria-Targeted Antioxidants Protect against Amyloid-Beta Toxicity in Alzheimer’s Disease Neurons. J. Alzheimer’s Dis. 2010, 20 (Suppl. S2), S609–S631. [Google Scholar] [CrossRef]
- Snow, B.J.; Rolfe, F.L.; Lockhart, M.M.; Frampton, C.M.; O’Sullivan, J.D.; Fung, V.; Smith, R.A.; Murphy, M.P.; Taylor, K.M.; Protect Study, G. A Double-Blind, Placebo-Controlled Study to Assess the Mitochondria-Targeted Antioxidant MitoQ as a Disease-Modifying Therapy in Parkinson’s Disease. Mov. Disord. 2010, 25, 1670–1674. [Google Scholar] [CrossRef]
- Cassina, P.; Cassina, A.; Pehar, M.; Castellanos, R.; Gandelman, M.; de Leon, A.; Robinson, K.M.; Mason, R.P.; Beckman, J.S.; Barbeito, L.; et al. Mitochondrial Dysfunction in SOD1G93A-Bearing Astrocytes Promotes Motor Neuron Degeneration: Prevention by Mitochondrial-Targeted Antioxidants. J. Neurosci. 2008, 28, 4115–4122. [Google Scholar] [CrossRef]
- Miquel, E.; Cassina, A.; Martinez-Palma, L.; Souza, J.M.; Bolatto, C.; Rodriguez-Bottero, S.; Logan, A.; Smith, R.A.; Murphy, M.P.; Barbeito, L.; et al. Neuroprotective Effects of the Mitochondria-Targeted Antioxidant MitoQ in a Model of Inherited Amyotrophic Lateral Sclerosis. Free Radic. Biol. Med. 2014, 70, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Gane, E.J.; Weilert, F.; Orr, D.W.; Keogh, G.F.; Gibson, M.; Lockhart, M.M.; Frampton, C.M.; Taylor, K.M.; Smith, R.A.; Murphy, M.P. The Mitochondria-Targeted Anti-Oxidant Mitoquinone Decreases Liver Damage in a Phase II Study of Hepatitis C Patients. Liver Int. 2010, 30, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef] [PubMed]
- Pehar, M.; Vargas, M.R.; Robinson, K.M.; Cassina, P.; Diaz-Amarilla, P.J.; Hagen, T.M.; Radi, R.; Barbeito, L.; Beckman, J.S. Mitochondrial Superoxide Production and Nuclear Factor Erythroid 2-Related Factor 2 Activation in p75 Neurotrophin Receptor-Induced Motor Neuron Apoptosis. J. Neurosci. 2007, 27, 7777–7785. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.L.; Yi, F.; Pan, H.Z.; Duan, S.L.; Ding, Z.C.; Yuan, G.H.; Qu, J.; Zhang, H.C.; Liu, G.H. Progress and Prospects in Stem Cell Therapy. Acta Pharmacol. Sin. 2013, 34, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Ohnuki, M.; Takahashi, K. Present and Future Challenges of Induced Pluripotent Stem Cells. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140367. [Google Scholar] [CrossRef] [PubMed]
- Keep, M.; Elmer, E.; Fong, K.S.; Csiszar, K. Intrathecal Cyclosporin Prolongs Survival of Late-Stage ALS Mice. Brain Res. 2001, 894, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, J.; Fong, K.S.; Hansson, M.J.; Elmer, E.; Csiszar, K.; Keep, M.F. Life Span Extension and Reduced Neuronal Death after Weekly Intraventricular Cyclosporin Injections in the G93A Transgenic Mouse Model of Amyotrophic Lateral Sclerosis. J. Neurosurg. 2004, 101, 128–137. [Google Scholar] [CrossRef]
- Kirkinezos, I.G.; Hernandez, D.; Bradley, W.G.; Moraes, C.T. An ALS Mouse Model with a Permeable Blood-Brain Barrier Benefits from Systemic Cyclosporine A Treatment. J. Neurochem. 2004, 88, 821–826. [Google Scholar] [CrossRef]
- Martin, L.J. The Mitochondrial Permeability Transition Pore: A Molecular Target for Amyotrophic Lateral Sclerosis Therapy. Biochim. Biophys. Acta 2010, 1802, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J. Olesoxime, a Cholesterol-Like Neuroprotectant for the Potential Treatment of Amyotrophic Lateral Sclerosis. IDrugs 2010, 13, 568–580. [Google Scholar]
- Bordet, T.; Buisson, B.; Michaud, M.; Drouot, C.; Galea, P.; Delaage, P.; Akentieva, N.P.; Evers, A.S.; Covey, D.F.; Ostuni, M.A.; et al. Identification and Characterization of Cholest-4-En-3-One, Oxime (TRO19622), a Novel Drug Candidate for Amyotrophic Lateral Sclerosis. J. Pharmacol. Exp. Ther. 2007, 322, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Sunyach, C.; Michaud, M.; Arnoux, T.; Bernard-Marissal, N.; Aebischer, J.; Latyszenok, V.; Gouarne, C.; Raoul, C.; Pruss, R.M.; Bordet, T.; et al. Olesoxime Delays Muscle Denervation, Astrogliosis, Microglial Activation and Motoneuron Death in an ALS Mouse Model. Neuropharmacology 2012, 62, 2346–2352. [Google Scholar] [CrossRef] [PubMed]
- Lenglet, T.; Lacomblez, L.; Abitbol, J.L.; Ludolph, A.; Mora, J.S.; Robberecht, W.; Shaw, P.J.; Pruss, R.M.; Cuvier, V.; Meininger, V.; et al. A Phase II–III Trial of Olesoxime in Subjects with Amyotrophic Lateral Sclerosis. Eur. J. Neurol. 2014, 21, 529–536. [Google Scholar] [CrossRef]
- Scott, S.; Kranz, J.E.; Cole, J.; Lincecum, J.M.; Thompson, K.; Kelly, N.; Bostrom, A.; Theodoss, J.; Al-Nakhala, B.M.; Vieira, F.G.; et al. Design, Power, and Interpretation of Studies in the Standard Murine Model of ALS. Amyotroph. Lateral Scler. 2008, 9, 4–15. [Google Scholar] [CrossRef]
- Yang, Y.M.; Gupta, S.K.; Kim, K.J.; Powers, B.E.; Cerqueira, A.; Wainger, B.J.; Ngo, H.D.; Rosowski, K.A.; Schein, P.A.; Ackeifi, C.A.; et al. A Small Molecule Screen in Stem-Cell-Derived Motor Neurons Identifies a Kinase Inhibitor as a Candidate Therapeutic for ALS. Cell Stem Cell 2013, 12, 713–726. [Google Scholar] [CrossRef]
- Martin, L.J.; Fancelli, D.; Wong, M.; Niedzwiecki, M.; Ballarini, M.; Plyte, S.; Chang, Q. GNX-4728, a Novel Small Molecule Drug Inhibitor of Mitochondrial Permeability Transition, Is Therapeutic in a Mouse Model of Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 2014, 8, 433. [Google Scholar] [CrossRef]
- Parkinson Study, G. A Controlled Trial of Rasagiline in Early Parkinson Disease: The Tempo Study. Arch. Neurol. 2002, 59, 1937–1943. [Google Scholar] [CrossRef]
- Olanow, C.W.; Rascol, O.; Hauser, R.; Feigin, P.D.; Jankovic, J.; Lang, A.; Langston, W.; Melamed, E.; Poewe, W.; Stocchi, F.; et al. A Double-Blind, Delayed-Start Trial of Rasagiline in Parkinson’s Disease. N. Engl. J. Med. 2009, 361, 1268–1278. [Google Scholar] [CrossRef]
- Youdim, M.B.; Weinstock, M. Molecular Basis of Neuroprotective Activities of Rasagiline and the Anti-Alzheimer Drug TV3326 [(N-propargyl-(3R)aminoindan-5-YL)-ethyl methyl carbamate]. Cell. Mol. Neurobiol. 2001, 21, 555–573. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, W.; Akao, Y.; Youdim, M.B.; Davis, B.A.; Naoi, M. Transfection-Enforced Bcl-2 Overexpression and an Anti-Parkinson Drug, Rasagiline, Prevent Nuclear Accumulation of Glyceraldehyde-3-Phosphate Dehydrogenase Induced by an Endogenous Dopaminergic Neurotoxin, N-methyl(R)salsolinol. J. Neurochem. 2001, 78, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, W.; Youdim, M.B.; Naoi, M. Antiapoptotic Properties of Rasagiline, N-Propargylamine-1(R)-Aminoindan, and Its Optical (S)-isomer, TV1022. Ann. N. Y. Acad. Sci. 2001, 939, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Amit, T.; Falach-Yogev, M.; Bar Am, O.; Maruyama, W.; Naoi, M. The Essentiality of Bcl-2, PKC and Proteasome-Ubiquitin Complex Activations in the Neuroprotective-Antiapoptotic Action of the Anti-Parkinson Drug, Rasagiline. Biochem. Pharmacol. 2003, 66, 1635–1641. [Google Scholar] [CrossRef] [PubMed]
- Waibel, S.; Reuter, A.; Malessa, S.; Blaugrund, E.; Ludolph, A.C. Rasagiline Alone and in Combination with Riluzole Prolongs Survival in an ALS Mouse Model. J. Neurol. 2004, 251, 1080–1084. [Google Scholar] [CrossRef]
- Macchi, Z.; Wang, Y.; Moore, D.; Katz, J.; Saperstein, D.; Walk, D.; Simpson, E.; Genge, A.; Bertorini, T.; Fernandes, J.A.; et al. A Multi-Center Screening Trial of Rasagiline in Patients with Amyotrophic Lateral Sclerosis: Possible Mitochondrial Biomarker Target Engagement. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 345–352. [Google Scholar] [CrossRef]
- Statland, J.M.; Moore, D.; Wang, Y.; Walsh, M.; Mozaffar, T.; Elman, L.; Nations, S.P.; Mitsumoto, H.; Fernandes, J.A.; Saperstein, D.; et al. Rasagiline for Amyotrophic Lateral Sclerosis: A Randomized, Controlled Trial. Muscle Nerve 2019, 59, 201–207. [Google Scholar] [CrossRef]
- Ludolph, A.C.; Schuster, J.; Dorst, J.; Dupuis, L.; Dreyhaupt, J.; Weishaupt, J.H.; Kassubek, J.; Weiland, U.; Petri, S.; Meyer, T.; et al. Safety and Efficacy of Rasagiline as an Add-on Therapy to Riluzole in Patients with Amyotrophic Lateral Sclerosis: A Randomised, Double-Blind, Parallel-Group, Placebo-Controlled, Phase 2 Trial. Lancet Neurol. 2018, 17, 681–688. [Google Scholar] [CrossRef]




{kind=link}
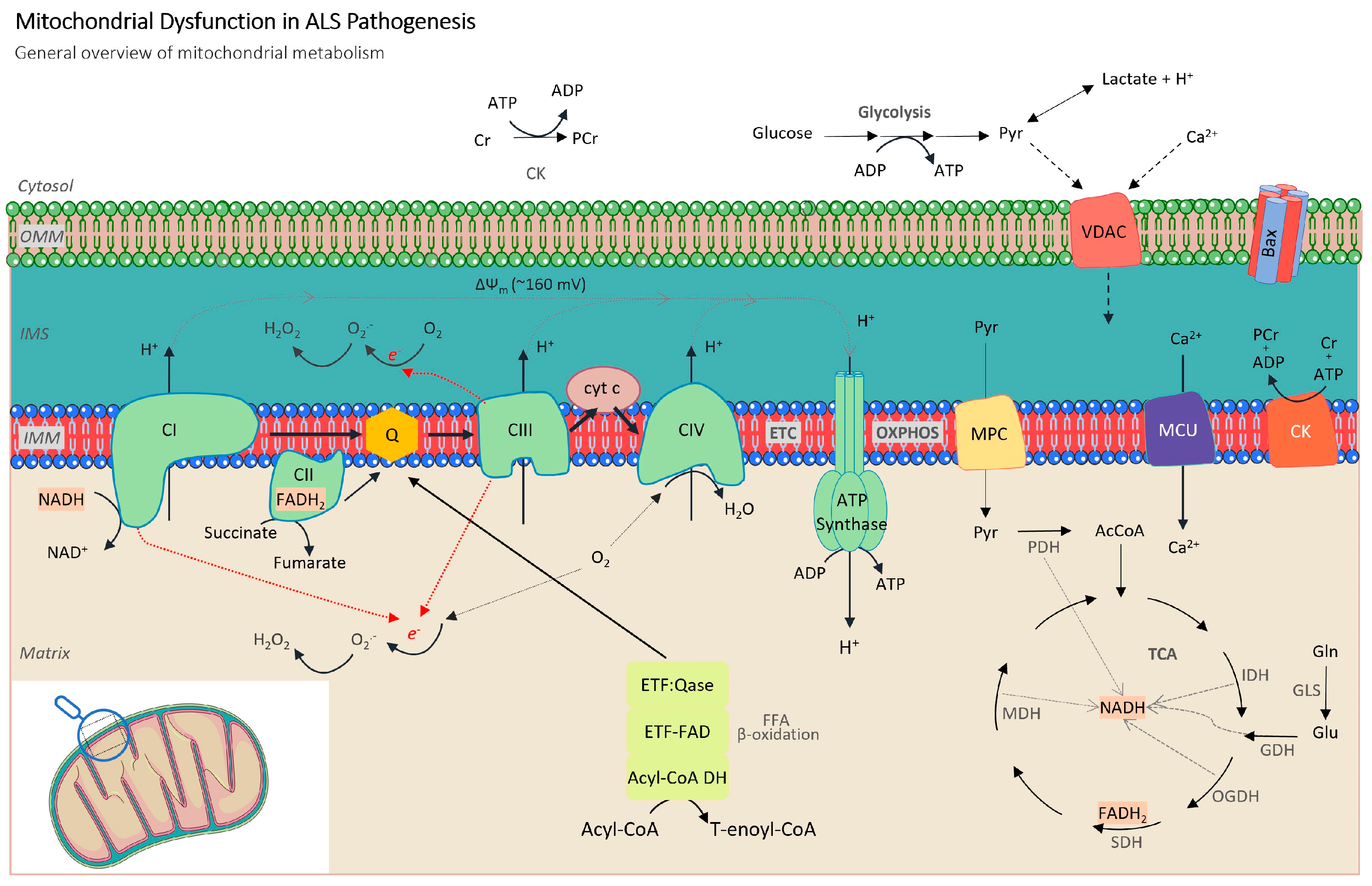{kind=link}
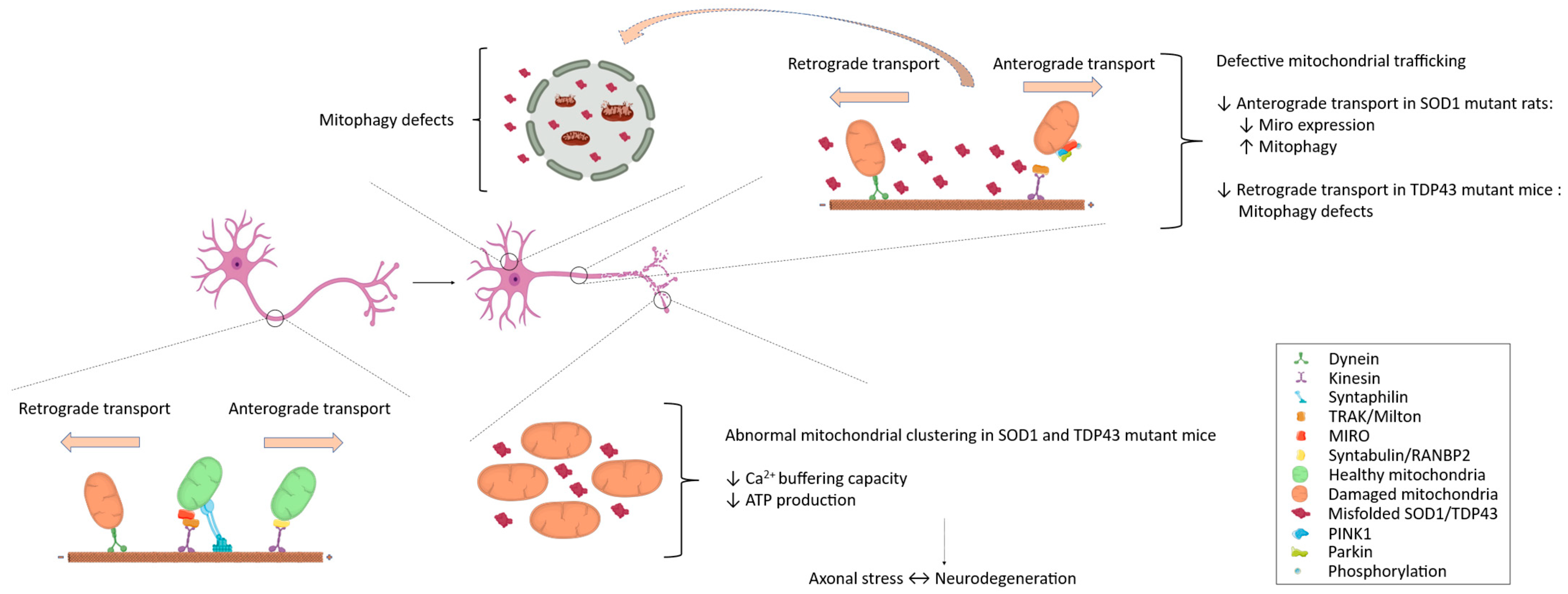{kind=link}
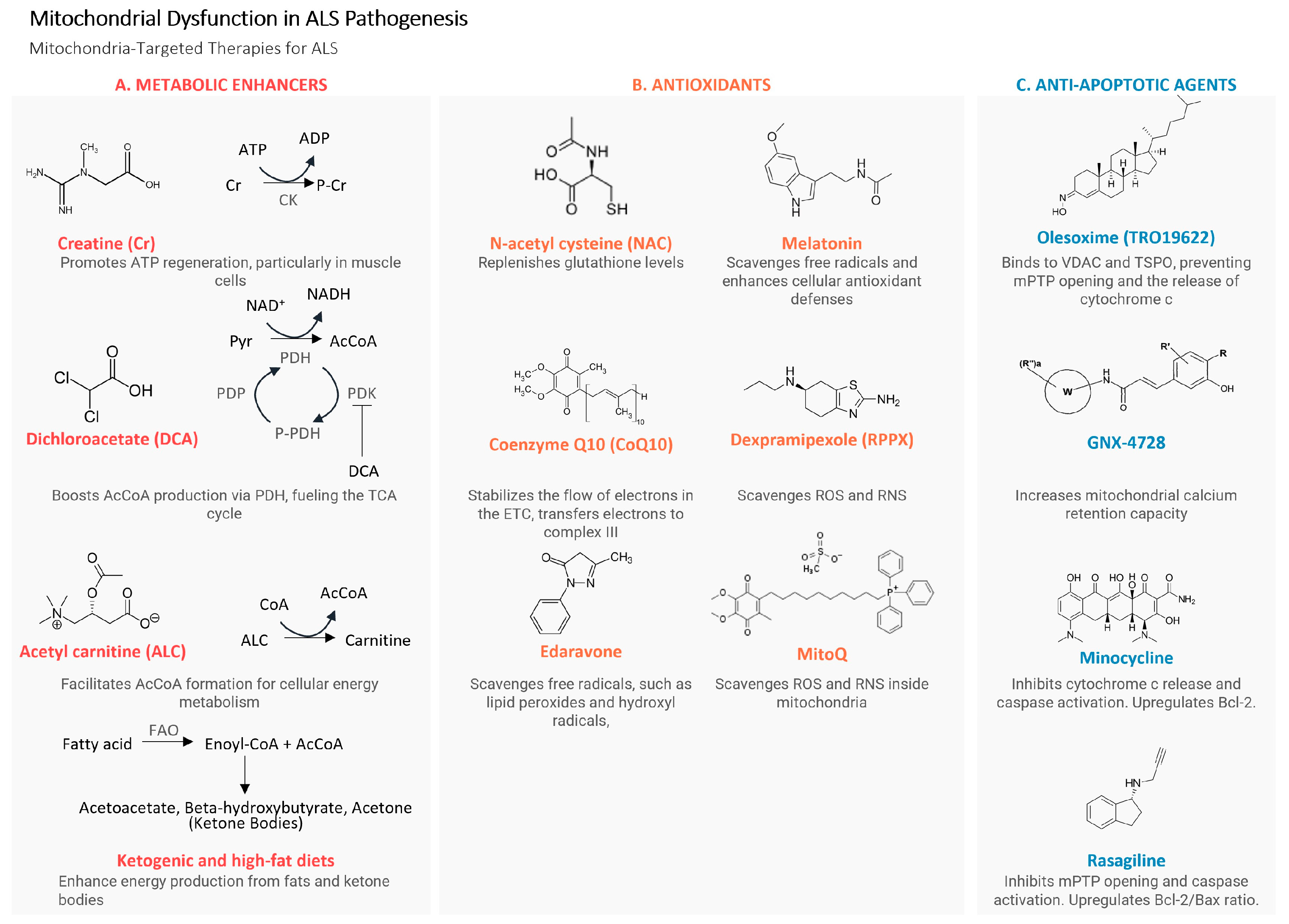{kind=link}
| Clinical Trial ID | Study Name | Condition | Phase/Type | Sponsor | Starting Date | End Date | Number of Patients |
|---|---|---|---|---|---|---|---|
| NCT01257581 | Phase 2 Selection Trial of High Dosage Creatine and Two Dosages of Tamoxifen in Amyotrophic Lateral Sclerosis (ALS) | Amyotrophic Lateral Sclerosis (ALS) | Phase 2 | Nazem Atassi, Massachusetts General Hospital | 2011-03 | 2013-02 | 60 |
| NCT02306590 | Efficacy, Safety and Tolerability of High Lipid and Calorie Supplementation in Amyotrophic Lateral Sclerosis | Amyotrophic Lateral Sclerosis (ALS) | Prospective, multicenter, randomized, stratified, parallel-group, double-blind trial | Albert Christian Ludolph, Prof., University of Ulm | 2015-02 | 2018-09 | 207 |
| NCT04172792 | Safety and Tolerability of Fat-rich vs. Carbohydrate-rich High-caloric Food Supplements in Patients with Amyotrophic Lateral Sclerosis (ALS) | Amyotrophic Lateral Sclerosis (ALS) | Phase 1 | Albert Christian Ludolph, Prof., University of Ulm | 2019-11 | 2021-04 | 64 |
| NCT00243932 | Clinical Trial of High Dose CoQ10 in ALS | Amyotrophic Lateral Sclerosis (ALS) | Phase 2 | Columbia University | 2005-04 | 2008-03 | 185 |
| NCT01492686 | Efficacy and Safety Study of MCI-186 for Treatment of Patients with Amyotrophic Lateral Sclerosis (ALS) 2 | Amyotrophic Lateral Sclerosis (ALS) | Phase 3 | Mitsubishi Tanabe Pharma Corporation | 2011-12 | 2014-10 | 137 |
| NCT04165824 | Safety Study of Oral Edaravone Administered in Subjects with ALS | Amyotrophic Lateral Sclerosis (ALS) | Phase 3 | Mitsubishi Tanabe Pharma America Inc. | 2019-11 | 2021-10 | 185 |
| NCT01281189 | A Randomized, Double-Blind, Placebo-Controlled, Multi-Center Study of the Safety and Efficacy of Dexpramipexole in Subjects with Amyotrophic Lateral Sclerosis | Amyotrophic Lateral Sclerosis (ALS) | Phase 3 | Knopp Biosciences | 2011-03 | 2012-11 | 942 |
| NCT00329056 | A Double-Blind, Prospective, Randomized Comparison of 2 Doses of MitoQ and Placebo for the Treatment of Patients with Parkinson’s Disease | Parkinson’s Disease (PD) | Double-Blind, Prospective, Randomized Comparison | Antipodean Pharmaceuticals, Inc. | 2006-05 | 2007-11 | 128 |
| NCT00047723 | Minocycline to Treat Amyotrophic Lateral Sclerosis | Amyotrophic Lateral Sclerosis (ALS) | Phase 3 | National Institute of Neurological Disorders and Stroke (NINDS) | 2003-01 | 2007-01 | 400 |
| NCT01232738 | A Multi-Center Controlled Screening Trial of Safety and Efficacy of Rasagiline in Subjects with Amyotrophic Lateral Sclerosis (ALS) | Amyotrophic Lateral Sclerosis (ALS) | Phase 2 | Yunxia Wang, MD, University of Kansas Medical Center | 2011-12 | 2013-05 | 36 |
| NCT01786603 | Rasagiline in Subjects with Amyotrophic Lateral Sclerosis (ALS) | Amyotrophic Lateral Sclerosis (ALS) | Phase 2 | Richard Barohn, MD, University of Kansas Medical Center | 2013-11 | 2016-07 | 80 |
| NCT01879241 | Efficacy, Safety, and Tolerability Study of 1 mg Rasagiline in Patients with Amyotrophic Lateral Sclerosis (ALS) Receiving Standard Therapy (Riluzole)-An AMG Trial with a Market Authorized Substance | Amyotrophic Lateral Sclerosis (ALS) | Phase 2 | Albert Christian Ludolph, Prof., University of Ulm | 2013-06 | 2016-08 | 252 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cunha-Oliveira, T.; Montezinho, L.; Simões, R.F.; Carvalho, M.; Ferreiro, E.; Silva, F.S.G. Mitochondria: A Promising Convergent Target for the Treatment of Amyotrophic Lateral Sclerosis. Cells 2024, 13, 248. https://doi.org/10.3390/cells13030248
Cunha-Oliveira T, Montezinho L, Simões RF, Carvalho M, Ferreiro E, Silva FSG. Mitochondria: A Promising Convergent Target for the Treatment of Amyotrophic Lateral Sclerosis. Cells. 2024; 13(3):248. https://doi.org/10.3390/cells13030248
Chicago/Turabian StyleCunha-Oliveira, Teresa, Liliana Montezinho, Rui F. Simões, Marcelo Carvalho, Elisabete Ferreiro, and Filomena S. G. Silva. 2024. "Mitochondria: A Promising Convergent Target for the Treatment of Amyotrophic Lateral Sclerosis" Cells 13, no. 3: 248. https://doi.org/10.3390/cells13030248
APA StyleCunha-Oliveira, T., Montezinho, L., Simões, R. F., Carvalho, M., Ferreiro, E., & Silva, F. S. G. (2024). Mitochondria: A Promising Convergent Target for the Treatment of Amyotrophic Lateral Sclerosis. Cells, 13(3), 248. https://doi.org/10.3390/cells13030248