.png)
A Review on the Role and Function of Cinnabarinic Acid, a “Forgotten” Metabolite of the Kynurenine Pathway


Abstract
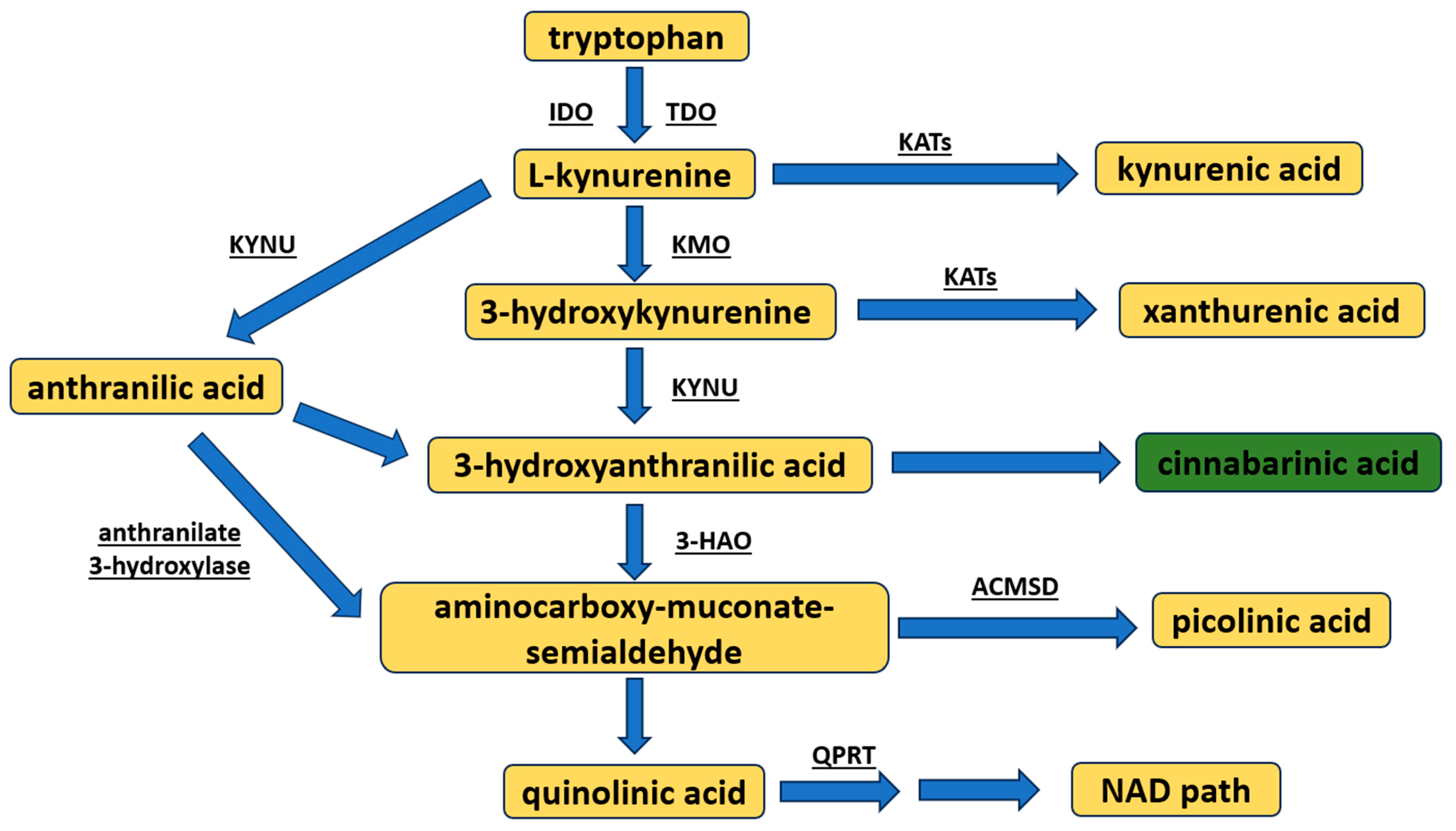
:1. Introduction
2. Chemistry and Molecular Targets of Cinnabarinic Acid
2.1. Cinnabarinic Acid Chemistry
2.2. Cinnabarinic Acid Synthesis
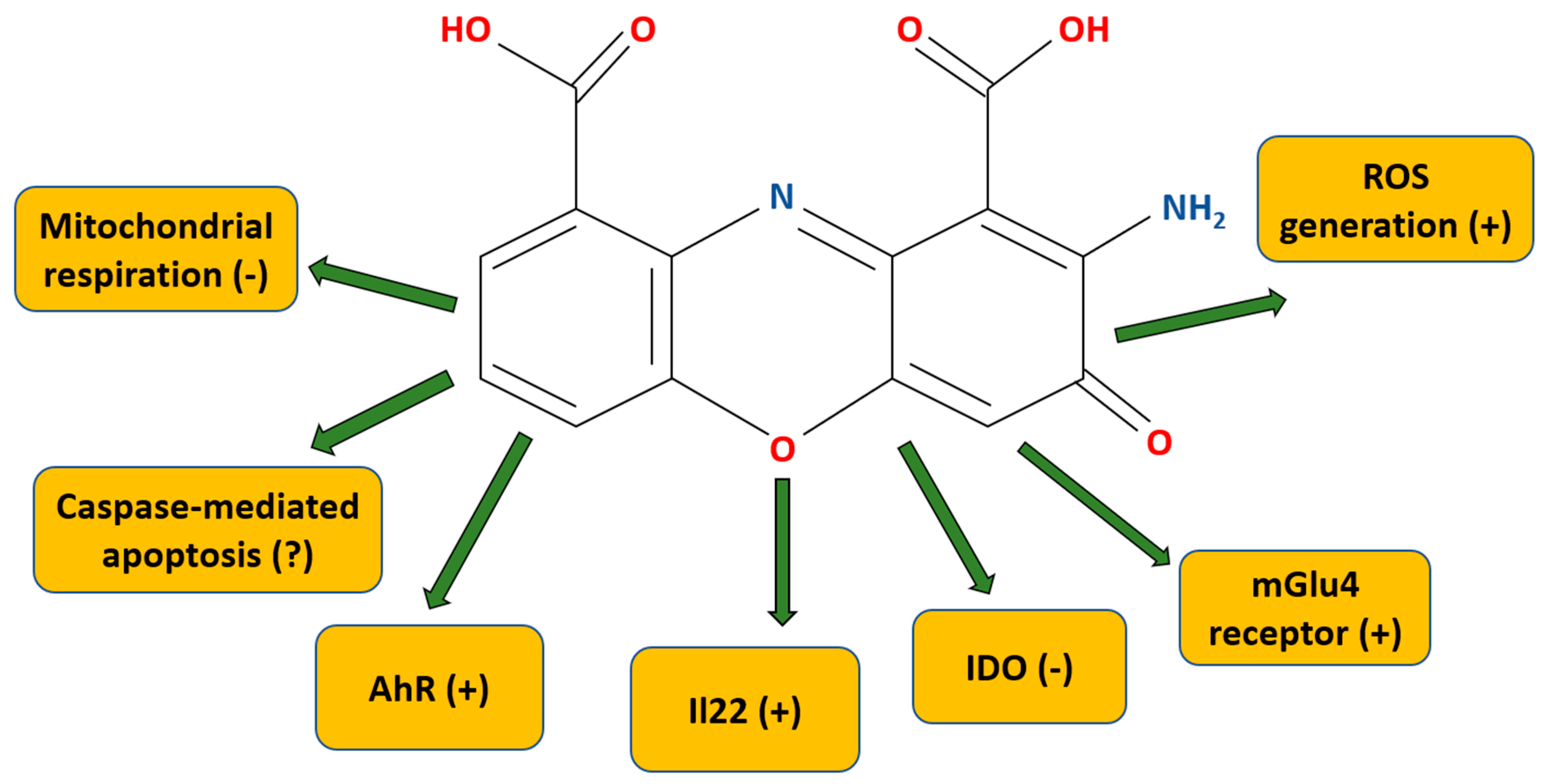
2.3. Receptors and Molecular Mechanisms of Action

{kind=link}
{kind=link}
| Molecular Target/Mechanism | CA Activity (Summary of Data Found in References) | References |
|---|---|---|
| IDO inhibition | IC50 ≈ 2 μM | [77] |
| Ki value at 326 nM IC50 was equal to 0.46 μM | [76] | |
| mGlu4 receptor orthosteric agonist | 100 μM CA increases [3H]Ins5 formation by ca. 35% (it is 5× less efficacious than the full mGlu4 agonist ACPT-I); CA binds within the glutamate-binding pocket. | [74] |
| AhR agonist; production of IL22 | CA increases (1 μM) the production of IL22 in human and mouse CD4+ T cells through AhR (the blocking of this receptor prevents the IL22 increase). | [78] |
| Inhibition of mitochondrial respiration | Complete inhibition at 5 μM; 0.5 μM of CA leads to 50% inhibition of state III respiration. | [84] |
| CA is at least 20× more efficient at inhibition than 3-hydroxyanthranilic acid. | [82] | |
| ROS generation | CA brings about the rapid induction of ROS generation (ca. 15 min, with return to the control level after 4 h). | [85] |
| Apoptosis | Induction: CA holds at least 10× higher apoptosis-inducing properties when compared with 3-hydroxyanthranilic acid. The caspase-3 activity is upregulated in the thymocytes within 6 h after simulation with 30 μM of CA. | [85] |
| Antiapoptotic properties: CA alleviates caspase-3 or caspase-3/7 upregulation in ethanol-treated hepatocytes/liver lysates. No direct effect of CA itself is indicated. | [86,87] |
3. In Vivo Studies
| Species | Group | Content of CA in Tissues/Body Fluids [Value in pM Recalculated for Comparison between Studies] | Comment(s) (If Applicable) | Method for Quantification | References |
|---|---|---|---|---|---|
| Human | 23 adult individuals with schizophrenia (16 males and 7 females) and 26 non-schizophrenic patients |
|
| UPLC-MS/MS | [88] |
| |||||
| Human | 23 female, adult patients with schizophrenia |
|
| HPLC-MS/MS | [89] |
| |||||
| Human | Adult patients with ASD (90 ASD patients and 104 controls) |
|
| LC-MS/MS | [90] |
| |||||
| Sprague-Dawley adult, male rats | Controls |
| - | HPLC-MS/MS | [74] |
| After LPS challenge |
| - | |||
| C57BL/6 adult, male mice | Experimentally evoked autoimmuneencephalomyelitis |
| - | HPLC-MS/MS | [74] |
| C57BL/6 adult, male mice | Controls |
|
| UPLC-MS/MS | [88] |
| CA (0.25mg/kg, ip) acutely injected |
| ||||
| Female mice (Stc2+/+, C57BL/6 background) | Controls |
|
| HPLC-MS/MS | [94] |
4. In Vitro Studies
5. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Richard, D.M.; Dawes, M.A.; Mathias, C.W.; Acheson, A.; Hill-Kapturczak, N.; Dougherty, D.M. L-Tryptophan: Basic Metabolic Functions, Behavioral Research and Therapeutic Indications. Int. J. Tryptophan. Res. 2009, 2, 45–60. [Google Scholar] [CrossRef]
- Zhao, D.; Yu, Y.; Shen, Y.; Liu, Q.; Zhao, Z.; Sharma, R.; Reiter, R.J. Melatonin Synthesis and Function: Evolutionary History in Animals and Plants. Front. Endocrinol. 2019, 10, 249. [Google Scholar] [CrossRef]
- Correia, A.S.; Vale, N. Tryptophan Metabolism in Depression: A Narrative Review with a Focus on Serotonin and Kynurenine Pathways. Int. J. Mol. Sci. 2022, 23, 8493. [Google Scholar] [CrossRef] [PubMed]
- Marszalek-Grabska, M.; Walczak, K.; Gawel, K.; Wicha-Komsta, K.; Wnorowska, S.; Wnorowski, A.; Turski, W.A. Kynurenine emerges from the shadows Current knowledge on its fate and function. Pharmacol. Ther. 2021, 225, 107845. [Google Scholar] [CrossRef] [PubMed]
- Turska, M.; Paluszkiewicz, P.; Turski, W.A.; Parada-Turska, J. A Review of the Health Benefits of Food Enriched with Kynurenic Acid. Nutrients 2022, 14, 4182. [Google Scholar] [CrossRef] [PubMed]
- Walczak, K.; Wnorowski, A.; Turski, W.A.; Plech, T. Kynurenic acid and cancer: Facts and controversies. Cell. Mol. Life. Sci. 2020, 77, 1531–1550. [Google Scholar] [CrossRef] [PubMed]
- Hestad, K.; Alexander, J.; Rootwelt, H.; Aaseth, J.O. The Role of Tryptophan Dysmetabolism and Quinolinic Acid in Depressive and Neurodegenerative Diseases. Biomolecules 2022, 12, 998. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Fazio, F.; Lionetto, L.; Curto, M.; Iacovelli, L.; Copeland, C.S.; Neale, S.A.; Bruno, V.; Battaglia, G.; Salt, T.E.; Nicoletti, F. Cinnabarinic acid and xanthurenic acid: Two kynurenine metabolites that interact with metabotropic glutamate receptors. Neuropharmacology 2017, 112 Pt B, 365–372. [Google Scholar] [CrossRef]
- Mehler, A.H.; Knox, W.E. The conversion of tryptophan to kynurenine in liver. II. The enzymatic hydrolysis of formylkynurenine. J. Biol. Chem. 1950, 187, 431–438. [Google Scholar] [CrossRef]
- Knox, W.E.; Mehler, A.H. The conversion of tryptophan to kynurenine in liver. I. The coupled tryptophan peroxidase-oxidase system forming formylkynurenine. J. Biol. Chem. 1950, 187, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Mándi, Y.; Vécsei, L. The kynurenine system and immunoregulation. J. Neural. Transm. 2012, 119, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Takikawa, O. Biochemical and medical aspects of the indoleamine 2,3-dioxygenase-initiated L-tryptophan metabolism. Biochem. Biophys. Res. Commun. 2005, 338, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Dykens, J.A.; Sullivan, S.G.; Stern, A. Glucose metabolism and hemoglobin reactivity in human red blood cells exposed to the tryptophan metabolites 3-hydroxyanthranilate, quinolinate and picolinate. Biochem. Pharmacol. 1989, 38, 1555–1562. [Google Scholar] [CrossRef]
- Tanaka, M.; Vécsei, L. Monitoring the kynurenine system: Concentrations, ratios or what else? Adv. Clin. Exp. Med. 2021, 30, 775–778. [Google Scholar] [CrossRef]
- Perkins, M.N.; Stone, T.W. An iontophoretic investigation of the actions of convulsant kynurenines and their interaction with the endogenous excitant quinolinic acid. Brain. Res. 1982, 247, 184–187. [Google Scholar] [CrossRef]
- Perkins, M.N.; Stone, T.W. Actions of kynurenic acid and quinolinic acid in the rat hippocampus in vivo. Exp. Neurol. 1985, 88, 570–579. [Google Scholar] [CrossRef]
- Stone, T.W. Neuropharmacology of quinolinic and kynurenic acids. Pharmacol. Rev. 1993, 45, 309–379. [Google Scholar]
- Hilmas, C.; Pereira, E.F.; Alkondon, M.; Rassoulpour, A.; Schwarcz, R.; Albuquerque, E.X. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: Physiopathological implications. J. Neurosci. 2001, 21, 7463–7473. [Google Scholar] [CrossRef]
- Stone, T.W. Does kynurenic acid act on nicotinic receptors? An assessment of the evidence. J. Neurochem. 2020, 152, 627–649. [Google Scholar] [CrossRef]
- Wang, J.; Simonavicius, N.; Wu, X.; Swaminath, G.; Reagan, J.; Tian, H.; Ling, L. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J. Biol. Chem. 2006, 281, 22021–22028. [Google Scholar] [CrossRef]
- Kapolka, N.J.; Isom, D.G. HCAR3: An underexplored metabolite sensor. Nat. Rev. Drug. Discov. 2020, 199, 745. [Google Scholar] [CrossRef]
- Bartlett, R.D.; Esslinger, C.S.; Thompson, C.M.; Bridges, R.J. Substituted quinolines as inhibitors of L-glutamate transport into synaptic vesicles. Neuropharmacology 1998, 37, 839–846. [Google Scholar] [CrossRef]
- Haruki, H.; Hovius, R.; Pedersen, M.G.; Johnsson, K. Tetrahydrobiopterin Biosynthesis as a Potential Target of the Kynurenine Pathway Metabolite Xanthurenic Acid. J. Biol. Chem. 2016, 291, 652–657. [Google Scholar] [CrossRef]
- Copeland, C.S.; Neale, S.A.; Salt, T.E. Actions of Xanthurenic acid, a putative endogenous Group II metabotropic glutamate receptor agonist, on sensory transmission in the thalamus. Neuropharmacology 2013, 66, 133–142. [Google Scholar] [CrossRef]
- Stone, T.W.; Perkins, M.N. Quinolinic acid: A potent endogenous excitant at amino acid receptors in CNS. Eur. J. Pharmacol. 1981, 72, 411–412. [Google Scholar] [CrossRef]
- Guillemin, G.J. Quinolinic acid, the inescapable neurotoxin. FEBS J. 2012, 279, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Ocampo, J.; Ramírez-Ortega, D.; Cervantes, G.I.; Pineda, B.; Balderas, P.M.; González-Esquivel, D.; Sánchez-Chapul, L.; Lugo-Huitrón, R.; Silva-Adaya, D.; Ríos, C.; et al. Mitochondrial dysfunction related to cell damage induced by 3-hydroxykynurenine and 3-hydroxyanthranilic acid: Non-dependent-effect of early reactive oxygen species production. Neurotoxicology 2015, 50, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Walczak, K.; Dąbrowski, W.; Langner, E.; Zgrajka, W.; Piłat, J.; Kocki, T.; Rzeski, W.; Turski, W.A. Kynurenic acid synthesis and kynurenine aminotransferases expression in colon derived normal and cancer cells. Scand. J. Gastroenterol. 2011, 46, 903–912. [Google Scholar] [CrossRef]
- Walczak, K.; Zurawska, M.; Kiś, J.; Starownik, R.; Zgrajka, W.; Bar, K.; Turski, W.A.; Rzeski, W. Kynurenic acid in human renal cell carcinoma: Its antiproliferative and antimigrative action on Caki-2 cells. Amino Acids 2012, 43, 1663–1670. [Google Scholar] [CrossRef]
- Wang, S.; van Schooten, F.J.; Jin, H.; Jonkers, D.; Godschalk, R. The Involvement of Intestinal Tryptophan Metabolism in Inflammatory Bowel Disease Identified by a Meta-Analysis of the Transcriptome and a Systematic Review of the Metabolome. Nutrients 2023, 15, 2886. [Google Scholar] [CrossRef] [PubMed]
- Dudzińska, E.; Szymona, K.; Kloc, R.; Gil-Kulik, P.; Kocki, T.; Świstowska, M.; Bogucki, J.; Kocki, J.; Urbanska, E.M. Increased expression of kynurenine aminotransferases mRNA in lymphocytes of patients with inflammatory bowel disease. Therap. Adv. Gastroenterol. 2019, 12, 1756284819881304. [Google Scholar] [CrossRef] [PubMed]
- Fiedorowicz, M.; Choragiewicz, T.; Thaler, S.; Schuettauf, F.; Nowakowska, D.; Wojtunik, K.; Reibaldi, M.; Avitabile, T.; Kocki, T.; Turski, W.A.; et al. Tryptophan and Kynurenine Pathway Metabolites in Animal Models of Retinal and Optic Nerve Damage: Different Dynamics of Changes. Front. Physiol. 2019, 10, 1254. [Google Scholar] [CrossRef] [PubMed]
- Fiedorowicz, M.; Choragiewicz, T.; Turski, W.A.; Kocki, T.; Nowakowska, D.; Wertejuk, K.; Kamińska, A.; Avitabile, T.; Wełniak-Kaminska, M.; Grieb, P.; et al. Tryptophan Pathway Abnormalities in a Murine Model of Hereditary Glaucoma. Int. J. Mol. Sci. 2021, 22, 1039. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.S.; Owe-Larsson, B.; Asp, L.; Kocki, T.; Adler, M.; Hetta, J.; Gardner, R.; Lundkvist, G.B.; Urbanska, E.M.; Karlsson, H. Activation of kynurenine pathway in ex vivo fibroblasts from patients with bipolar disorder or schizophrenia: Cytokine challenge increases production of 3-hydroxykynurenine. Psychiatr. Res. 2013, 47, 1815–1823. [Google Scholar] [CrossRef]
- Szymona, K.; Zdzisińska, B.; Karakuła-Juchnowicz, H.; Kocki, T.; Kandefer-Szerszeń, M.; Flis, M.; Rosa, W.; Urbańska, E.M. Correlations of Kynurenic Acid, 3-Hydroxykynurenine, sIL-2R, IFN-α, and IL-4 with Clinical Symptoms During Acute Relapse of Schizophrenia. Neurotox. Res. 2017, 32, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Myint, A.M.; Kim, Y.K.; Verkerk, R.; Scharpé, S.; Steinbusch, H.; Leonard, B. Kynurenine pathway in major depression: Evidence of impaired neuroprotection. J. Affect. Disord. 2007, 98, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Oxenkrug, G.; Turski, W.; Zgrajka, W.; Weinstock, J.; Ruthazer, R.; Summergrad, P. Disturbances of Tryptophan Metabolism and Risk of Depression in HCV Patients Treated with IFN-Alpha. J. Infect. Dis. Ther. 2014, 2, 131. [Google Scholar] [CrossRef]
- Bakker, L.; Köhler, S.; Eussen, S.J.P.M.; Choe, K.; van den Hove, D.L.A.; Kenis, G.; Rutten, B.P.F.; Ulvik, A.; Ueland, P.M.; Verhey, F.R.J.; et al. Correlations between kynurenines in plasma and CSF, and their relation to markers of Alzheimer’s disease pathology. Brain. Behav. Immun. 2023, 111, 312–319. [Google Scholar] [CrossRef]
- Zakrocka, I.; Targowska-Duda, K.M.; Wnorowski, A.; Kocki, T.; Jóźwiak, K.; Turski, W.A. Angiotensin II Type 1 Receptor Blockers Inhibit KAT II Activity in the Brain-Its Possible Clinical Applications. Neurotox. Res. 2017, 32, 639–648. [Google Scholar] [CrossRef]
- Breda, C.; Sathyasaikumar, K.V.; Sograte Idrissi, S.; Notarangelo, F.M.; Estranero, J.G.; Moore, G.G.; Green, E.W.; Kyriacou, C.P.; Schwarcz, R.; Giorgini, F. Tryptophan-2,3-dioxygenase (TDO) inhibition ameliorates neurodegeneration by modulation of kynurenine pathway metabolites. Proc. Natl. Acad. Sci. USA 2016, 113, 5435–5440. [Google Scholar] [CrossRef] [PubMed]
- Campesan, S.; Green, E.W.; Breda, C.; Sathyasaikumar, K.V.; Muchowski, P.J.; Schwarcz, R.; Kyriacou, C.P.; Giorgini, F. The kynurenine pathway modulates neurodegeneration in a Drosophila model of Huntington’s disease. Curr. Biol. 2011, 21, 961–966. [Google Scholar] [CrossRef]
- Samadi, P.; Grégoire, L.; Rassoulpour, A.; Guidetti, P.; Izzo, E.; Schwarcz, R.; Bédard, P.J. Effect of kynurenine 3-hydroxylase inhibition on the dyskinetic and antiparkinsonian responses to levodopa in Parkinsonian monkeys. Mov. Disord. 2005, 20, 792–802. [Google Scholar] [CrossRef]
- Rejdak, K.; Bartosik-Psujek, H.; Dobosz, B.; Kocki, T.; Grieb, P.; Giovannoni, G.; Turski, W.A.; Stelmasiak, Z. Decreased level of kynurenic acid in cerebrospinal fluid of relapsing-onset multiple sclerosis patients. Neurosci. Lett. 2002, 331, 63–65. [Google Scholar] [CrossRef]
- Rejdak, K.; Petzold, A.; Kocki, T.; Kurzepa, J.; Grieb, P.; Turski, W.A.; Stelmasiak, Z. Astrocytic activation in relation to inflammatory markers during clinical exacerbation of relapsing-remitting multiple sclerosis. J. Neural. Transm. 2007, 114, 1011–1015. [Google Scholar] [CrossRef] [PubMed]
- Kamiński, R.M.; Zielińska, E.; Dekundy, A.; van Luijtelaar, G.; Turski, W. Deficit of endogenous kynurenic acid in the frontal cortex of rats with a genetic form of absence epilepsy. Pol. J. Pharmacol. 2003, 55, 741–746. [Google Scholar] [PubMed]
- Żarnowska, I.; Wróbel-Dudzińska, D.; Tulidowicz-Bielak, M.; Kocki, T.; Mitosek-Szewczyk, K.; Gasior, M.; Turski, W.A. Changes in tryptophan and kynurenine pathway metabolites in the blood of children treated with ketogenic diet for refractory epilepsy. Seizure 2019, 69, 265–272. [Google Scholar] [CrossRef]
- Butenandt, A.; Biekert, E.; Baumann, U. Ommochromes. XI. Model experiments on the constitution of ommochromes; oxidative degradation of 3-amino-4, 5-diacetylphenoxazone-2]. Arch. Biochem. Biophys. 1957, 69, 100–105. [Google Scholar] [CrossRef]
- Fazio, F.; Zappulla, C.; Notartomaso, S.; Busceti, C.; Bessede, A.; Scarselli, P.; Vacca, C.; Gargaro, M.; Volpi, C.; Allegrucci, M.; et al. Cinnabarinic acid, an endogenous agonist of type-4 metabotropic glutamate receptor, suppresses experimental autoimmune encephalomyelitis in mice. Neuropharmacology 2014, 81, 237–243. [Google Scholar] [CrossRef]
- PubChem National Library of Medicine. 2023. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Cinnabarinic-acid (accessed on 23 November 2023).
- Ishiguro, I.; Nagamura, Y.; Hara, A. Studies on the formation of phenoxazine-pigment from o-aminophenol derivatives by hemoglobin. I. Conversion of 3-OH-anthranilic acid into cinnabarinic acid in the presence of Mn]. Yakugaku Zasshi 1971, 91, 760–765. [Google Scholar] [CrossRef]
- Meng, D.; Shao, X.; Luo, S.P.; Tian, Q.P.; Liao, X.R. Pigment production by a newly isolated strain Pycnoporus sanguineus SYBC-L7 in solid-state fermentation. Front. Microbiol. 2022, 13, 1015913. [Google Scholar] [CrossRef] [PubMed]
- Yue, S.J.; Song, C.; Li, S.; Huang, P.; Guo, S.Q.; Hu, H.B.; Wang, W.; Zhang, X.H. Synthesis of cinnabarinic acid by metabolically engineered Pseudomonas chlororaphis GP72. Biotechnol. Bioeng. 2019, 116, 3072–3083. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Piñeiro, R.J.; Dali, M.; Mansuy, D.; Boucher, J.L. Unstability of cinnabarinic acid, an endogenous metabolite of tryptophan, under situations mimicking physiological conditions. Biochimie 2022, 199, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Li, M.J.; Jiang, G.F.; Wang, W. Metabolite Changes in Orange Dead Leaf Butterfly Kallima inachus during Ontogeny and Diapause. Metabolites 2022, 12, 804. [Google Scholar] [CrossRef] [PubMed]
- Dias, D.A.; Urban, S. HPLC and NMR studies of phenoxazone alkaloids from Pycnoporus cinnabarinus. Nat. Prod. Commun. 2009, 4, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Eggert, C. Laccase-catalyzed formation of cinnabarinic acid is responsible for antibacterial activity of Pycnoporus cinnabarinus. Microbiol. Res. 1997, 152, 315–318. [Google Scholar] [CrossRef]
- Lin, W.; Jia, G.; Sun, H.; Sun, T.; Hou, D. Genome sequence of the fungus Pycnoporus sanguineus, which produces cinnabarinic acid and pH- and thermo- stable laccases. Gene 2020, 742, 144586. [Google Scholar] [CrossRef]
- Temp, U.; Zierold, U.; Eggert, C. Cloning and characterization of a second laccase gene from the lignin-degrading basidiomycete Pycnoporus cinnabarinus. Gene 1999, 236, 169–177. [Google Scholar] [CrossRef]
- Göçenoğlu Sarıkaya, A.; Osman, B.; Kara, A.; Pazarlioglu, N.; Beşirli, N. Adsorption of cinnabarinic acid from culture fluid with magnetic microbeads. Biomed. Chromatogr. 2016, 30, 88–96. [Google Scholar] [CrossRef]
- Temp, U.; Eggert, C. Novel interaction between laccase and cellobiose dehydrogenase during pigment synthesis in the white rot fungus Pycnoporus cinnabarinus. Appl. Environ. Microbiol. 1999, 65, 389–395. [Google Scholar] [CrossRef]
- Christen, S.; Southwell-Keely, P.T.; Stocker, R. Oxidation of 3-hydroxyanthranilic acid to the phenoxazinone cinnabarinic acid by peroxyl radicals and by compound I of peroxidases or catalase. Biochemistry 1992, 31, 8090–8097. [Google Scholar] [CrossRef]
- Manthey, M.K.; Pyne, S.G.; Truscott, R.J. Mechanism of reaction of 3-hydroxyanthranilic acid with molecular oxygen. Biochim. Biophys. Acta 1990, 1034, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, O.; Lerch, K. Catalytic oxidation of 2-aminophenols and ortho hydroxylation of aromatic amines by tyrosinase. Biochemistry 1987, 26, 8567–8571. [Google Scholar] [CrossRef]
- Iwahashi, H.; Ishii, T.; Sugata, R.; Kido, R. Superoxide dismutase enhances the formation of hydroxyl radicals in the reaction of 3-hydroxyanthranilic acid with molecular oxygen. Biochem. J. 1988, 251, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Iwahashi, H.J. 3-Hydroxyanthranilic acid-derived compounds formed through electrochemical oxidation. Chromatogr. B Biomed. Sci. Appl. 1999, 736, 237–245. [Google Scholar] [CrossRef]
- Ogawa, H.; Nagamura, Y.; Ishiguro, I. Cinnabarinic acid formation in Malpighian tubules of the silkworm, Bombyx mori. Participation of catalase in cinnabarinic acid formation in the presence of manganese ion. Hoppe. Seylers. Z. Physiol. Chem. 1983, 364, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.V.; Vaidyanathan, C.S. Enzymic conversion of 3-hydroxyanthranilic acid to cinnabarinic acid by the leaves of Tecoma stans. Arch. Biochem. Biophys. 1966, 115, 27–34. [Google Scholar] [CrossRef]
- Turska, M.; Pelak, J.; Turski, M.P.; Kocki, T.; Dukowski, P.; Plech, T.; Turski, W. Fate and distribution of kynurenic acid administered as beverage. Pharmacol. Rep. 2018, 70, 1089–1096. [Google Scholar] [CrossRef]
- Turska, M.; Rutyna, R.; Paluszkiewicz, M.; Terlecka, P.; Dobrowolski, A.; Pelak, J.; Turski, M.P.; Muszyńska, B.; Dabrowski, W.; Kocki, T.; et al. Presence of kynurenic acid in alcoholic beverages—Is this good news, or bad news? Med. Hypotheses 2019, 122, 200–205. [Google Scholar] [CrossRef]
- Turski, M.P.; Turska, M.; Zgrajka, W.; Bartnik, M.; Kocki, T.; Turski, W.A. Distribution, synthesis, and absorption of kynurenic acid in plants. Planta Med. 2011, 77, 858–864. [Google Scholar] [CrossRef]
- Turski, M.P.; Turska, M.; Zgrajka, W.; Kuc, D.; Turski, W.A. Presence of kynurenic acid in food and honeybee products. Amino Acids 2009, 36, 75–80. [Google Scholar] [CrossRef]
- Turski, M.P.; Kamiński, P.; Zgrajka, W.; Turska, M.; Turski, W.A. Potato- an important source of nutritional kynurenic acid. Plant Foods Hum. Nutr. 2012, 67, 17–23. [Google Scholar] [CrossRef]
- Fazio, F.; Lionetto, L.; Molinaro, G.; Bertrand, H.O.; Acher, F.; Ngomba, R.T.; Notartomaso, S.; Curini, M.; Rosati, O.; Scarselli, P.; et al. Cinnabarinic acid, an endogenous metabolite of the kynurenine pathway, activates type 4 metabotropic glutamate receptors. Mol. Pharmacol. 2012, 81, 643–656. [Google Scholar] [CrossRef]
- Nicoletti, F.; Bockaert, J.; Collingridge, G.L.; Conn, P.J.; Ferraguti, F.; Schoepp, D.D.; Wroblewski, J.T.; Pin, J.P. Metabotropic glutamate receptors: From the workbench to the bedside. Neuropharmacology 2011, 60, 1017–1041. [Google Scholar] [CrossRef]
- Pasceri, R.; Siegel, D.; Ross, D.; Moody, C.J. Aminophenoxazinones as inhibitors of indoleamine 2,3-dioxygenase (IDO). Synthesis of exfoliazone and chandrananimycin A. J. Med. Chem. 2013, 56, 3310–3317. [Google Scholar] [CrossRef]
- Carr, G.; Tay, W.; Bottriell, H.; Andersen, S.K.; Mauk, A.G.; Andersen, R.J. Plectosphaeroic acids A, B, and C, indoleamine 2,3-dioxygenase inhibitors produced in culture by a marine isolate of the fungus Plectosphaerella cucumerina. Org. Lett. 2009, 11, 2996–2999. [Google Scholar] [CrossRef]
- Lowe, M.M.; Mold, J.E.; Kanwar, B.; Huang, Y.; Louie, A.; Pollastri, M.P.; Wang, C.; Patel, G.; Franks, D.G.; Schlezinger, J.; et al. Identification of cinnabarinic acid as a novel endogenous aryl hydrocarbon receptor ligand that drives IL-22 production. PLoS ONE 2014, 9, e87877. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, J.; Ihara, K.; Nakayama, H.; Hikino, S.; Satoh, K.; Kubo, N.; Iida, T.; Fujii, Y.; Hara, T. Characteristic expression of aryl hydrocarbon receptor repressor gene in human tissues: Organ-specific distribution and variable induction patterns in mononuclear cells. Life Sci. 2004, 74, 1039–1049. [Google Scholar] [CrossRef]
- Wang, G.Z.; Zhang, L.; Zhao, X.C.; Gao, S.H.; Qu, L.W.; Yu, H.; Fang, W.F.; Zhou, Y.C.; Liang, F.; Zhang, C.; et al. The Aryl hydrocarbon receptor mediates tobacco-induced PD-L1 expression and is associated with response to immunotherapy. Nat. Commun. 2019, 10, 1125. [Google Scholar] [CrossRef]
- Stevens, E.A.; Mezrich, J.D.; Bradfield, C.A. The aryl hydrocarbon receptor: A perspective on potential roles in the immune system. Immunology 2009, 127, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Zollner, H. Effects of cinnabarinic acid on mitochondrial respiration. Biochem. Pharmacol. 1976, 25, 643–648. [Google Scholar] [CrossRef]
- Boyland, E. The biochemistry of cancer of the bladder. Br. Med. Bull. 1958, 14, 153–158. [Google Scholar] [CrossRef]
- Nagamura, Y.; Uesugi, K.; Naito, J.; Ishiguro, I. Cinnabarinic acid was formed in damaged mitochondria and its effect on mitochondrial respiration. Adv. Exp. Med. Biol. 1999, 467, 419–423. [Google Scholar] [CrossRef]
- Hiramatsu, R.; Hara, T.; Akimoto, H.; Takikawa, O.; Kawabe, T.; Isobe, K.; Nagase, F. Cinnabarinic acid generated from 3-hydroxyanthranilic acid strongly induces apoptosis in thymocytes through the generation of reactive oxygen species and the induction of caspase. J. Cell Biochem. 2008, 103, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.D.; Hossain, E.; Elferink, C.J. Epigenetic Regulation by Agonist-Specific Aryl Hydrocarbon Receptor Recruitment of Metastasis-Associated Protein 2 Selectively Induces Stanniocalcin 2 Expression. Mol. Pharmacol. 2017, 92, 366–374. [Google Scholar] [CrossRef]
- Patil, N.Y.; Rus, I.; Joshi, A.D. Role of ERK1/2 Signaling in Cinnabarinic Acid-Driven Stanniocalcin 2-Mediated Protection against Alcohol-Induced Apoptosis. J. Pharmacol. Exp. Ther. 2023, 387, 111–120. [Google Scholar] [CrossRef]
- Ulivieri, M.; Wierońska, J.M.; Lionetto, L.; Martinello, K.; Cieslik, P.; Chocyk, A.; Curto, M.; Di Menna, L.; Iacovelli, L.; Traficante, A.; et al. The Trace Kynurenine, Cinnabarinic Acid, Displays Potent Antipsychotic-Like Activity in Mice and Its Levels Are Reduced in the Prefrontal Cortex of Individuals Affected by Schizophrenia. Schizophr. Bull. 2020, 46, 1471–1481. [Google Scholar] [CrossRef]
- Shilov, Y.E.; Baymeeva, N.V.; Brusov, O.S.; Oleichik, I.V.; Sizov, S.V.; Tyurin, I.A. Cinnabarinic acid as a potential prognostic marker of schizophrenia. ZhNevrol. Psikhiatr. Im. SS Korsakova 2022, 122, 138–142. [Google Scholar] [CrossRef]
- Launay, J.M.; Delorme, R.; Pagan, C.; Callebert, J.; Leboyer, M.; Vodovar, N. Impact of IDO activation and alterations in the kynurenine pathway on hyperserotonemia, NAD+ production, and AhR activation in autism spectrum disorder. Transl. Psychiatry 2023, 13, 380. [Google Scholar] [CrossRef] [PubMed]
- Notartomaso, S.; Boccella, S.; Antenucci, N.; Ricciardi, F.; Fazio, F.; Liberatore, F.; Scarselli, P.; Scioli, M.; Mascio, G.; Bruno, V.; et al. Analgesic Activity of Cinnabarinic Acid in Models of Inflammatory and Neuropathic Pain. Front. Mol. Neurosci. 2022, 15, 892870. [Google Scholar] [CrossRef] [PubMed]
- Vilar, B.; Busserolles, J.; Ling, B.; Laffray, S.; Ulmann, L.; Malhaire, F.; Chapuy, E.; Aissouni, Y.; Etienne, M.; Bourinet, E.; et al. Alleviating pain hypersensitivity through activation of type 4 metabotropic glutamate receptor. J. Neurosci. 2013, 33, 18951–18965. [Google Scholar] [CrossRef]
- Zussy, C.; Gómez-Santacana, X.; Rovira, X.; De Bundel, D.; Ferrazzo, S.; Bosch, D.; Asede, D.; Malhaire, F.; Acher, F.; Giraldo, J.; et al. Dynamic modulation of inflammatory pain-related affective and sensory symptoms by optical control of amygdala metabotropic glutamate receptor 4. Mol. Psychiatry 2018, 23, 509–520. [Google Scholar] [CrossRef]
- Hu, S.; Hu, C.; Luo, L.; Zhang, H.; Zhao, S.; Liu, Z.; Zeng, L. Pu-erh tea increases the metabolite Cinnabarinic acid to improve circadian rhythm disorder-induced obesity. Food Chem. 2022, 394, 133500. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.D.; Thinakaran, G.; Elferink, C.J. Cinnabarinic Acid-Induced Stanniocalcin 2 Confers Cytoprotection against Alcohol-Induced Liver Injury. Pharmacol. Exp. Ther. 2022, 381, 1–11. [Google Scholar] [CrossRef]
- Milart, P.; Paluszkiewicz, P.; Dobrowolski, P.; Tomaszewska, E.; Smolinska, K.; Debinska, I.; Gawel, K.; Walczak, K.; Bednarski, J.; Turska, M.; et al. Kynurenic acid as the neglected ingredient of commercial baby formulas. Sci. Rep. 2019, 9, 6108. [Google Scholar] [CrossRef] [PubMed]
- Patil, N.Y.; Rus, I.; Downing, E.; Mandala, A.; Friedman, J.E.; Joshi, A.D. Cinnabarinic Acid Provides Hepatoprotection Against Nonalcoholic Fatty Liver Disease. J. Pharmacol. Exp. Ther. 2022, 383, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Majewski, M.; Kasica, N.; Jakimiuk, A.; Podlasz, P. Toxicity and cardiac effects of acute exposure to tryptophan metabolites on the kynurenine pathway in early developing zebrafish (Danio rerio) embryos. Toxicol. Appl. Pharmacol. 2018, 341, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Marszalek-Grabska, M.; Gawel, K.; Kosheva, N.; Kocki, T.; Turski, W.A. Developmental Exposure to Kynurenine Affects Zebrafish and Rat Behavior. Cells 2023, 12, 2224. [Google Scholar] [CrossRef]
- Kocki, T.; Kocki, J.; Wielosz, M.; Turski, W.A.; Urbanska, E.M. Carbamazepine enhances brain production of kynurenic acid in vitro. Eur. J. Pharmacol. 2004, 498, 325–326. [Google Scholar] [CrossRef]
- Kocki, T.; Wielosz, M.; Turski, W.A.; Urbanska, E.M. Enhancement of brain kynurenic acid production by anticonvulsants--novel mechanism of antiepileptic activity? Eur. J. Pharmacol. 2006, 541, 147–151. [Google Scholar] [CrossRef]
- Maciejak, P.; Szyndler, J.; Turzyńska, D.; Sobolewska, A.; Płaźnik, A. Kynurenic acid: A new effector of valproate action? Pharmacol. Rep. 2011, 63, 1569–1573. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Okada, M. Effects of levetiracetam on astroglial release of kynurenine-pathway metabolites. Br. J. Pharmacol. 2018, 175, 4253–4265. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Tanahashi, S.; Hoshikawa, M.; Shinagawa, R.; Okada, M. Zonisamide regulates basal ganglia transmission via astroglial kynurenine pathway. Neuropharmacology 2014, 76 Pt A, 137–145. [Google Scholar] [CrossRef]
- Kakoti, M.; Dullah, S.; Hazarika, D.J.; Barooah, M.; Boro, R.C. Cinnabarinic acid from Trametes coccinea fruiting bodies exhibits antibacterial activity through inhibiting the biofilm formation. Arch. Microbiol. 2022, 204, 173. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gawel, K. A Review on the Role and Function of Cinnabarinic Acid, a “Forgotten” Metabolite of the Kynurenine Pathway. Cells 2024, 13, 453. https://doi.org/10.3390/cells13050453
Gawel K. A Review on the Role and Function of Cinnabarinic Acid, a “Forgotten” Metabolite of the Kynurenine Pathway. Cells. 2024; 13(5):453. https://doi.org/10.3390/cells13050453
Chicago/Turabian StyleGawel, Kinga. 2024. "A Review on the Role and Function of Cinnabarinic Acid, a “Forgotten” Metabolite of the Kynurenine Pathway" Cells 13, no. 5: 453. https://doi.org/10.3390/cells13050453
APA StyleGawel, K. (2024). A Review on the Role and Function of Cinnabarinic Acid, a “Forgotten” Metabolite of the Kynurenine Pathway. Cells, 13(5), 453. https://doi.org/10.3390/cells13050453