
Phosphodiesterases 4B and 4D Differentially Regulate cAMP Signaling in Calcium Handling Microdomains of Mouse Hearts

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cardiomyocyte Isolation
2.3. Live Cell Imaging
2.4. Single Cell Contractility Measurements
2.5. Immunofluorescence Staining and STED Microscopy
2.6. Langendorff-Perfused Whole Heart Stimulation
2.7. Immunoblot Analysis and Co-Immunoprecipication
2.8. Statistics
3. Results
3.1. Impact of PDE4B and PDE4D Ablation on PDE Expression
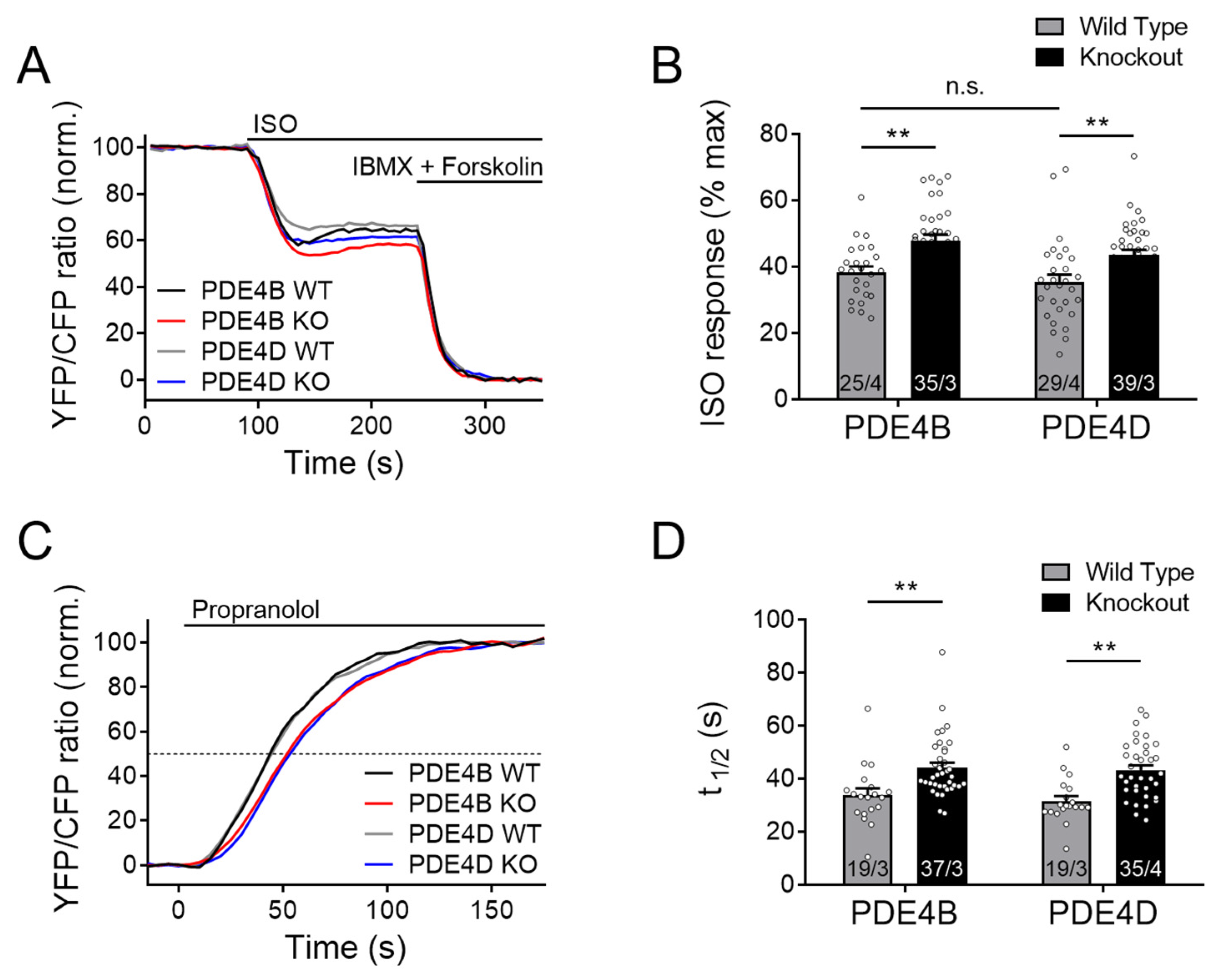
3.2. PDE4B and PDE4D Are Both Involved in the Regulation of cAMP Dynamics in the Caveolin-Rich Plasma Membrane
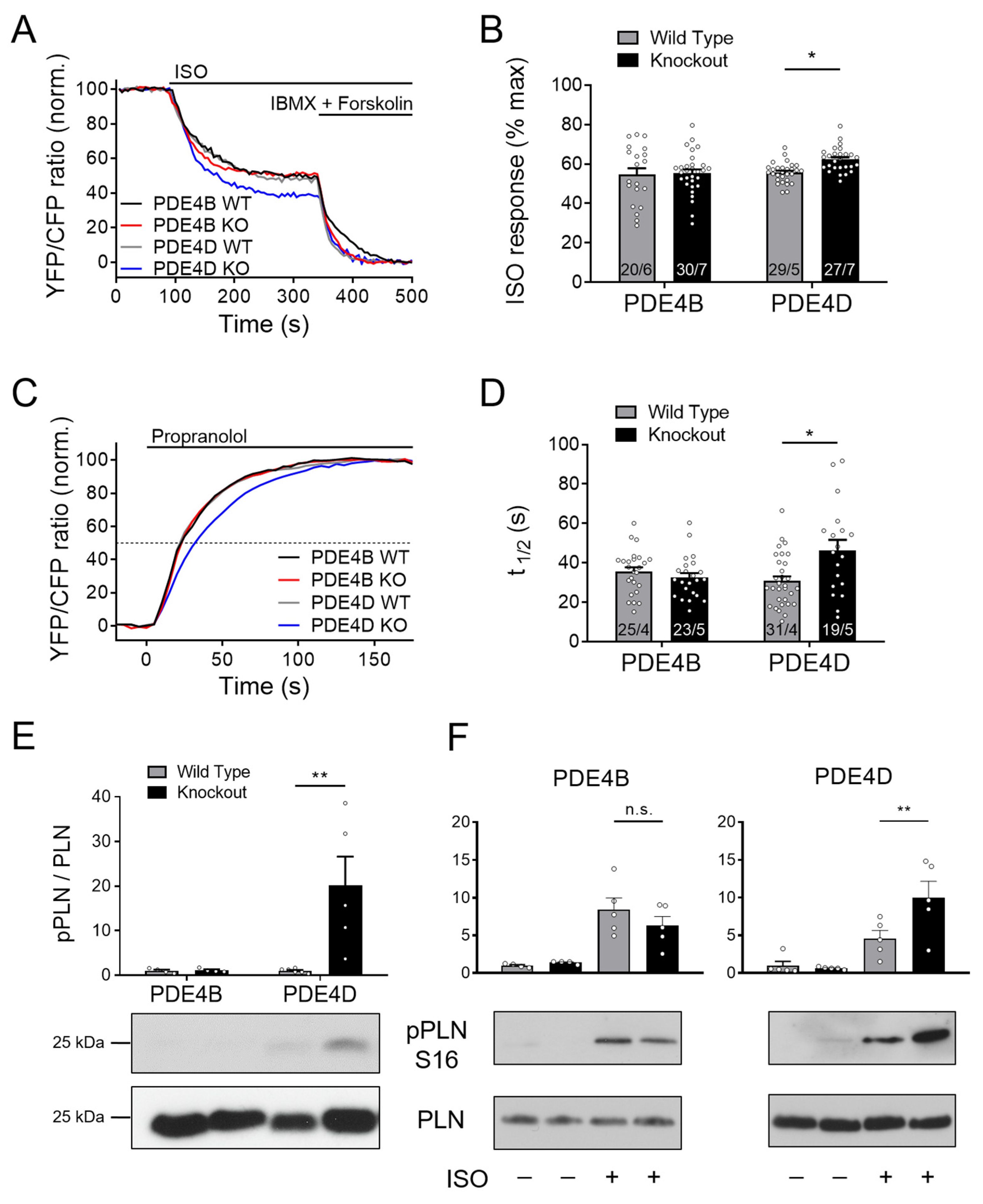
3.3. SERCA2a Microdomain Is Predomimantly Regulated by PDE4D
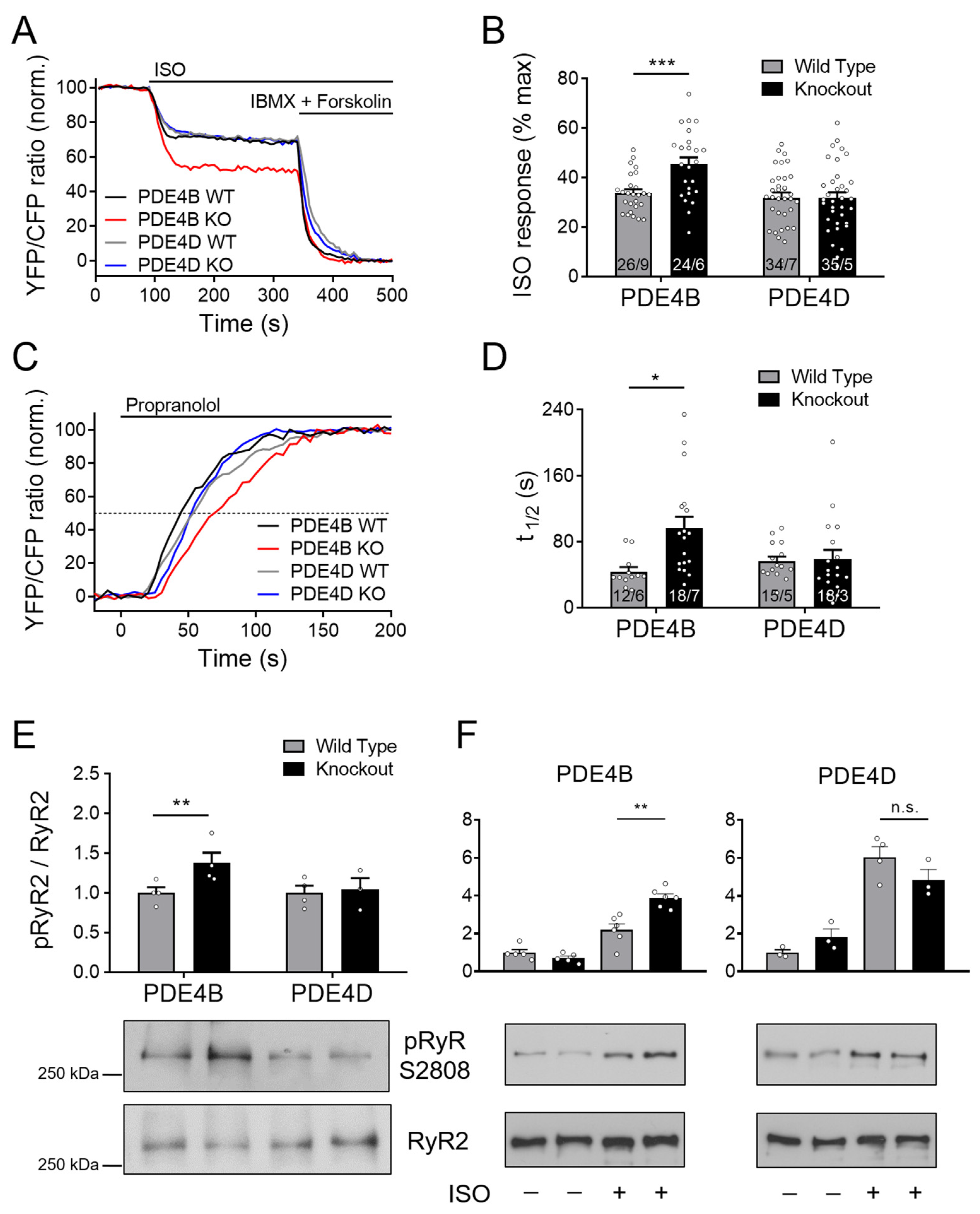
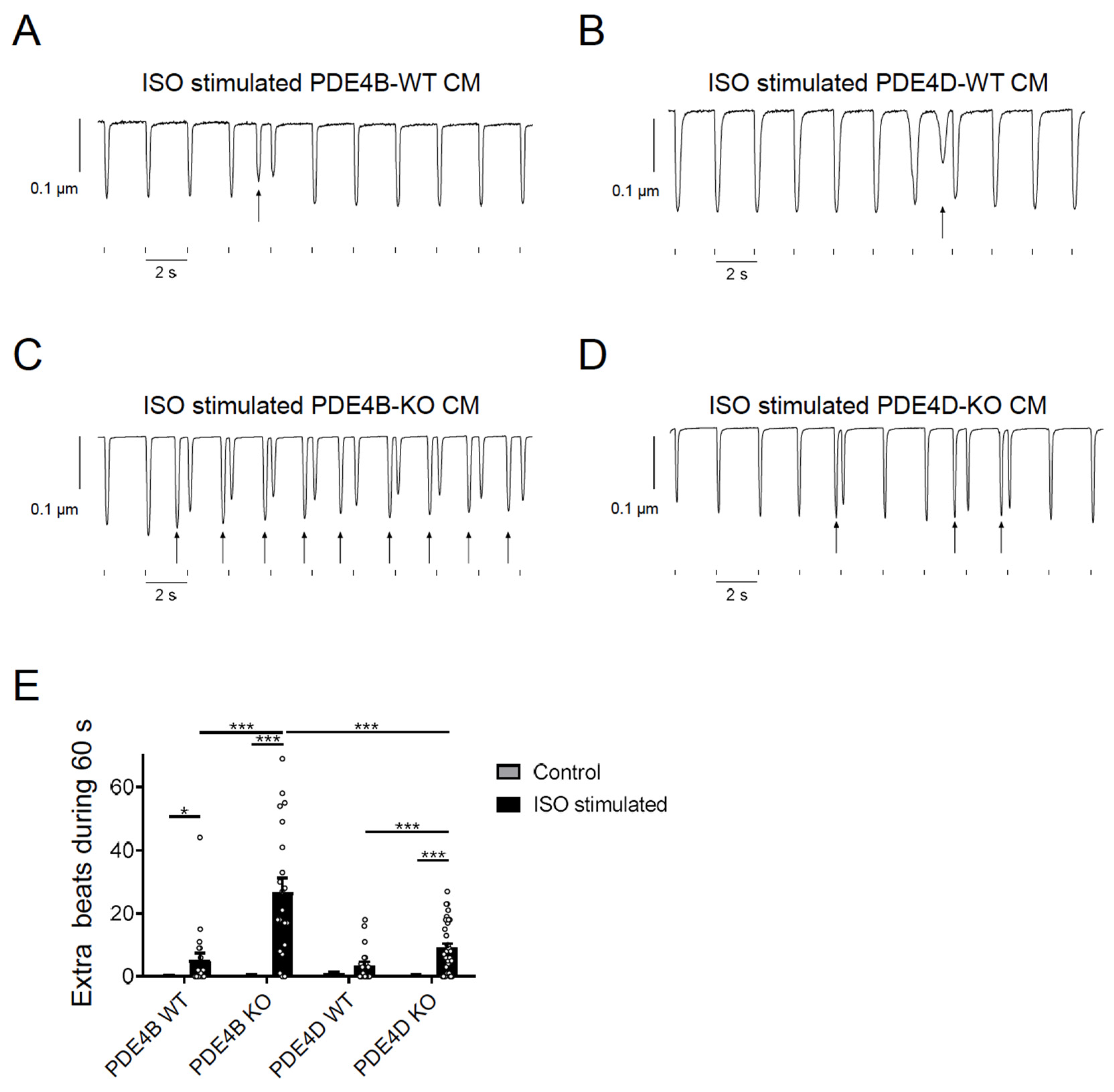
3.4. cAMP Dynamics at the RyR2 Complex Are Predominantly Regulated by PDE4B
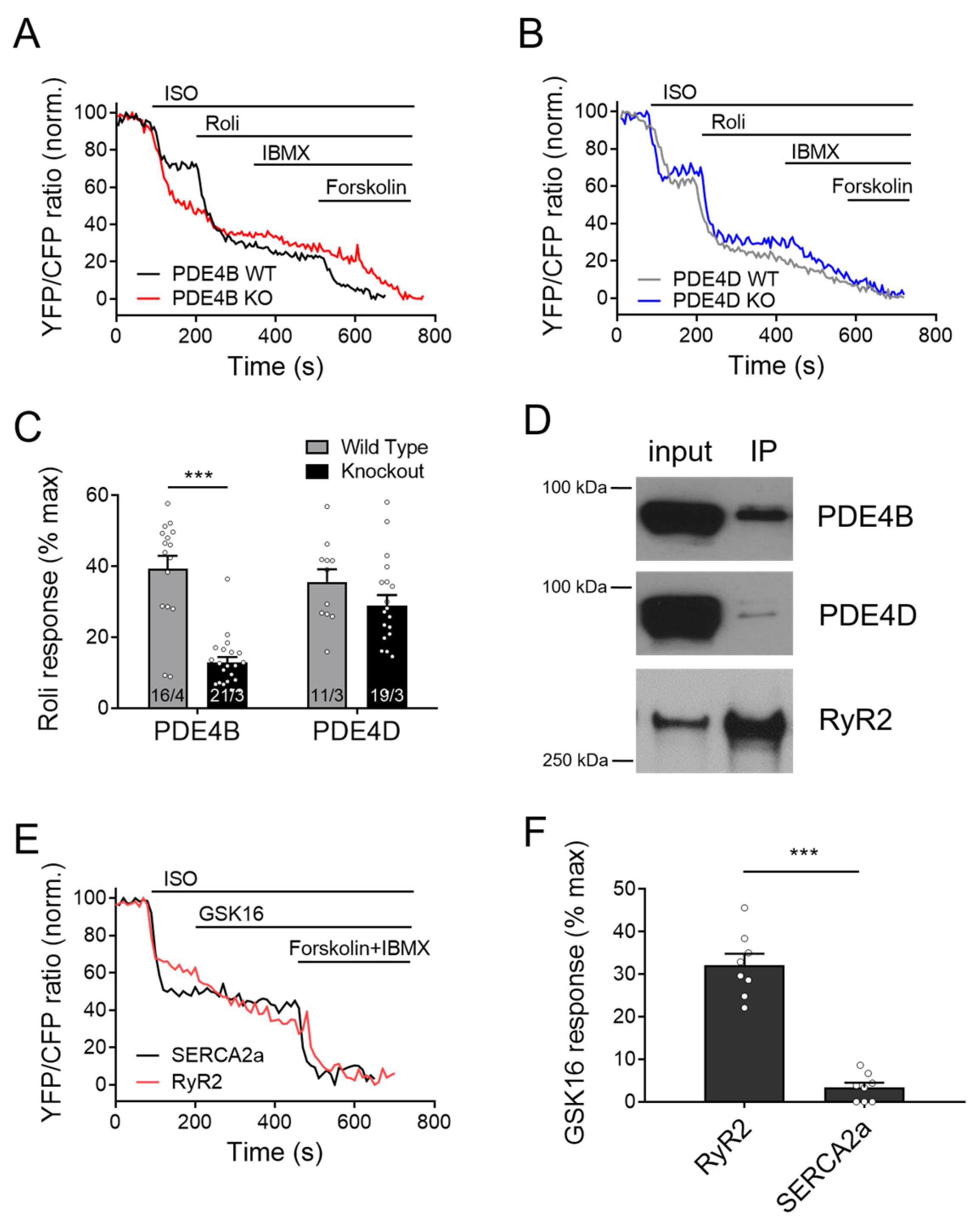
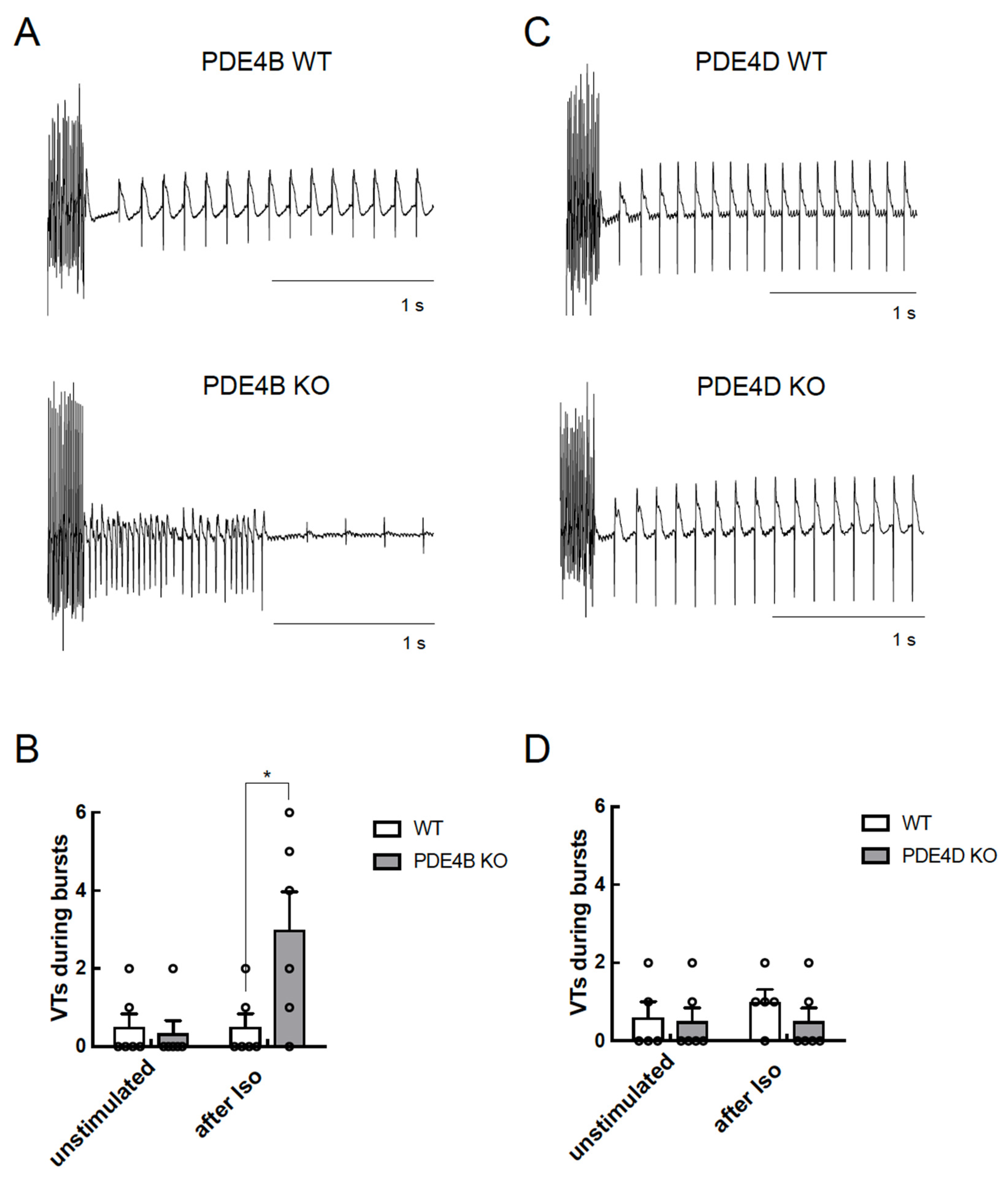
3.5. PDE4B Physically and Functionally Interacts with RyR2
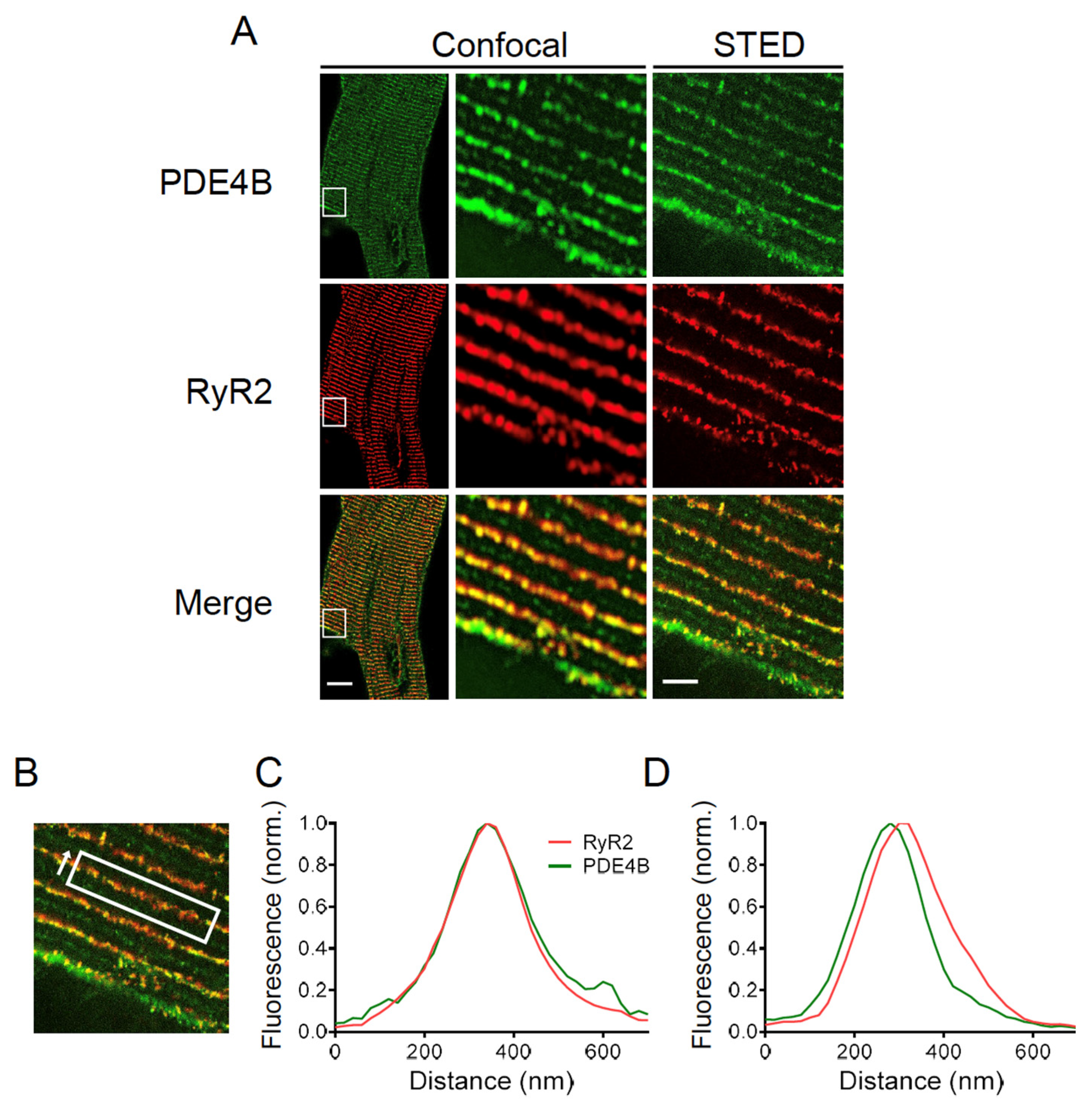
3.6. PDE4B Co-Localizes with RyR2
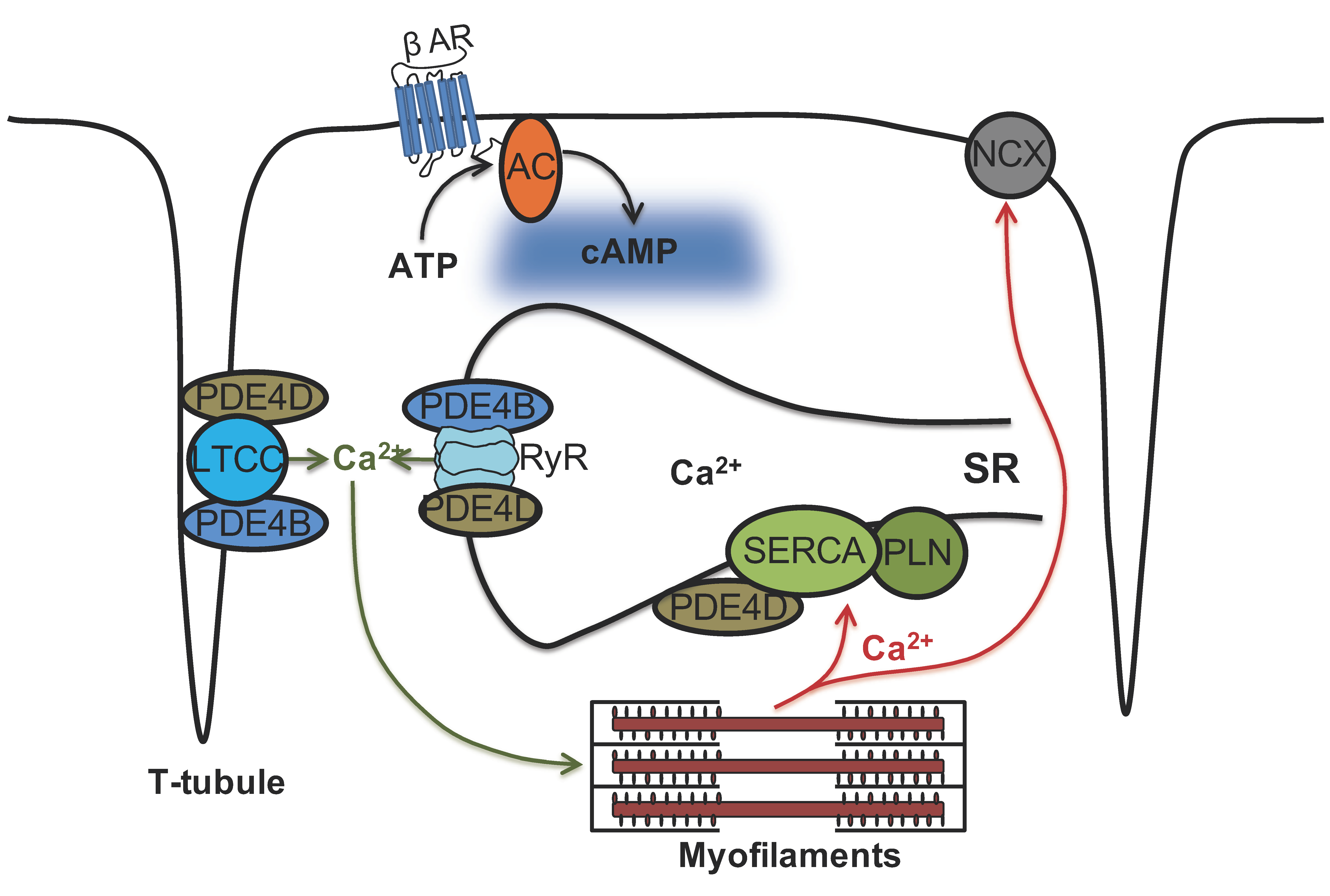
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zuo, H.; Cattani-Cavalieri, I.; Musheshe, N.; Nikolaev, V.O.; Schmidt, M. Phosphodiesterases as therapeutic targets for respiratory diseases. Pharmacol. Ther. 2019, 197, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Bodor, J.; Bopp, T.; Vaeth, M.; Klein, M.; Serfling, E.; Hünig, T.; Becker, C.; Schild, H.; Schmitt, E. Cyclic AMP underpins suppression by regulatory T cells. Eur. J. Immunol. 2012, 42, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Torres-Quesada, O.; Mayrhofer, J.E.; Stefan, E. The many faces of compartmentalized PKA signalosomes. Cell. Signal. 2017, 37, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Boularan, C.; Gales, C. Cardiac cAMP: Production, hydrolysis, modulation and detection. Front. Pharmacol. 2015, 6, 203. [Google Scholar] [CrossRef] [PubMed]
- Dodge-Kafka, K.L.; Langeberg, L.; Scott, J.D. Compartmentation of cyclic nucleotide signaling in the heart. Circ. Res. 2006, 98, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Ercu, M.; Klussmann, E. Roles of A-Kinase Anchoring Proteins and Phosphodiesterases in the Cardiovascular System. J. Cardiovasc. Dev. Dis. 2018, 5, 14. [Google Scholar] [CrossRef]
- Bers, D.M.; Xiang, Y.K.; Zaccolo, M. Whole-cell cAMP and PKA activity are epiphenomena, nanodomain signaling matters. Physiology 2019, 34, 240–249. [Google Scholar] [CrossRef]
- Schleicher, K.; Zaccolo, M. Using cAMP Sensors to Study Cardiac Nanodomains. J. Cardiovasc. Dev. Dis. 2018, 5, 17. [Google Scholar] [CrossRef]
- Zaccolo, M.; Zerio, A.; Lobo, M.J. Subcellular organization of the cAMP signaling pathway. Pharmacol. Rev. 2021, 73, 278–309. [Google Scholar] [CrossRef]
- Mika, D.; Leroy, J.; Vandecasteele, G.; Fischmeister, R. PDEs create local domains of cAMP signaling. J. Mol. Cell. Cardiol. Gen Physiol. 2012, 52, 323–329. [Google Scholar] [CrossRef]
- Preedy, M.E.J. Cardiac Cyclic Nucleotide Phosphodiesterases: Roles and Therapeutic Potential in Heart Failure. Cardiovasc. Drugs. Ther. 2020, 34, 401–417. [Google Scholar] [CrossRef]
- Musheshe, N.; Schmidt, M.; Zaccolo, M. cAMP: From Long-Range Second Messenger to Nanodomain Signalling. Trends Pharmacol. Sci. 2018, 39, 209–222. [Google Scholar] [CrossRef]
- Kamel, R.; Leroy, J.; Vandecasteele, G.; Fischmeister, R. Cyclic nucleotide phosphodiesterases as therapeutic targets in cardiac hypertrophy and heart failure. Nat. Rev. Cardiol. 2023, 20, 90–108. [Google Scholar] [CrossRef]
- Miller, C.L.; Yan, C. Targeting cyclic nucleotide phosphodiesterase in the heart: Therapeutic implications. J. Cardiovasc. Transl. Res. 2010, 3, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Fertig, B.A.; Baillie, G.S. PDE4-Mediated cAMP signalling. J. Cardiovasc. Dev. Dis. 2018, 5, 8. [Google Scholar] [CrossRef]
- Liu, G.; Papa, A.; Katchman, A.N.; Zakharov, S.I.; Roybal, D.; Hennessey, J.A.; Kushner, J.; Yang, L.; Chen, B.X.; Kushnir, A.; et al. Mechanism of adrenergic CaV1.2 stimulation revealed by proximity proteomics. Nature 2020, 577, 695–700. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Oz, S.; Benmocha, A.; Dascal, N. Regulation of Cardiac L-Type Ca2+ Channel CaV1.2 Via the β-Adrenergic-cAMP-Protein Kinase A Pathway. Circ. Res. 2013, 113, 617–631. [Google Scholar] [CrossRef]
- Leroy, J.; Richter, W.; Mika, D.; Castro, L.R.V.; Abi-Gerges, A.; Xie, M.; Scheitrum, C.; Lefebvre, F.; Schittl, J.; Mateo, P.; et al. Phosphodiesterase 4B in the cardiac L-type Ca2+ channel complex regulates Ca2+ current and protects against ventricular arrhythmias in mice. J. Clin. Investig. 2011, 121, 2651–2661. [Google Scholar] [CrossRef] [PubMed]
- Lehnart, S.E.; Wehrens, X.H.T.; Reiken, S.; Warrier, S.; Belevych, A.E.; Harvey, R.D.; Richter, W.; Jin, S.L.C.; Conti, M.; Marks, A.R. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 2005, 123, 25–35. [Google Scholar] [CrossRef]
- Lompré, A.M.; Hajjar, R.J.; Harding, S.E.; Kranias, E.G.; Lohse, M.J.; Marks, A.R. Ca2+ cycling and new therapeutic approaches for heart failure. Circulation 2010, 121, 822–830. [Google Scholar] [CrossRef]
- Chao, Y.C.; Surdo, N.C.; Pantano, S.; Zaccolo, M. Imaging cAMP nanodomains in the heart. Biochem. Soc. Trans. 2019, 47, 1383–1392. [Google Scholar] [CrossRef]
- Gorelik, J.; Yang, L.Q.; Zhang, Y.; Lab, M.; Korchev, Y.; Harding, S.E. A novel Z-groove index characterizing myocardial surface structure. Cardiovasc. Res. 2006, 72, 422–429. [Google Scholar] [CrossRef]
- West, T.M.; Wang, Q.; Deng, B.; Zhang, Y.; Barbagallo, F.; Reddy, G.R.; Chen, D.; Phan, K.S.; Xu, B.; Isidori, A.; et al. Phosphodiesterase 5 associates with β2 adrenergic receptor to modulate cardiac function in type 2 diabetic hearts. J. Am. Heart. Assoc. 2019, 8, e012273. [Google Scholar] [CrossRef]
- Perera, R.K.; Sprenger, J.; Steinbrecher, J.H.; Hübscher, D.; Lehnart, S.E.; Abesser, M.; Schuh, K.; El-Armouche, A.; Nikolaev, V.O. Microdomain switch of cGMP-regulated phosphodiesterases leads to ANP-induced augmentation of beta-adrenoceptor-stimulated contractility in early cardiac hypertrophy. Circ. Res. 2015, 116, 1304–1311. [Google Scholar] [CrossRef]
- Sprenger, J.U.; Perera, R.K.; Steinbrecher, J.H.; Lehnart, S.E.; Maier, L.S.; Hasenfuss, G.; Nikolaev, V.O. In vivo model with targeted cAMP biosensor reveals changes in receptor microdomain communication in cardiac disease. Nat. Commun. 2015, 6, 6965. [Google Scholar] [CrossRef]
- Berisha, F.; Götz, K.R.; Wegener, J.W.; Brandenburg, S.; Subramanian, H.; Molina, C.E.; Rüffer, A.; Petersen, J.; Bernhardt, A.; Girdauskas, E.; et al. cAMP imaging at ryanodine receptors reveals β2-adrenoceptor driven arrhythmias. Circ. Res. 2021, 129, 81–94. [Google Scholar] [CrossRef]
- Jin, S.-L.C.; Conti, M. Induction of the cyclic nucleotide phosphodiesterase PDE4B is essential for LPS-activated TNF-α responses. Proc. Natl. Acad. Sci. USA 2002, 99, 7628–7633. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.-L.C.; Richard, F.J.; Kuo, W.-P.; Ercole, A.J.; Conti, M. Impaired growth and fertility of cAMP-specific phosphodiesterase PDE4D-deficient mice. Proc. Natl. Acad. Sci. USA 1999, 96, 11998–12003. [Google Scholar] [CrossRef] [PubMed]
- Calebiro, D.; Nikolaev, V.O.; Gagliani, M.C.; de Filippis, T.; Dees, C.; Tacchetti, C.; Persani, L.; Lohse, M.J. Persistent cAMP-Signals Triggered by Internalized G-Protein–Coupled Receptors. PLoS Biol. 2009, 7, e1000172. [Google Scholar] [CrossRef] [PubMed]
- Kraft, A.E.; Nikolaev, V.O. FRET Microscopy for Real-Time Visualization of Second Messengers in Living Cell. Methods Mol. Biol. 2017, 1563, 85–90. [Google Scholar]
- Brette, F.; Komukai, K.; Orchard, C.H. Validation of formamide as a detubulation agent in isolated rat cardiac cells. Am. J. Physiol. Circ. Physiol. 2002, 283, H1720–H1728. [Google Scholar] [CrossRef]
- Wright, P.T.; Gorelik, J.; Harding, S.E. Electrophysiological Remodeling: Cardiac T-Tubules and ß-Adrenoceptors. Cells 2021, 10, 2456. [Google Scholar] [CrossRef] [PubMed]
- Mika, D.; Richter, W.; Westenbroek, R.E.; Catterall, W.A.; Conti, M. PDE4B mediates local feedback regulation of β₁-adrenergic cAMP signaling in a sarcolemmal compartment of cardiac myocytes. J. Cell. Sci. 2014, 127, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Wang, Y.; Bahriz, S.M.F.M.; Zhao, M.; Zhu, C.; Xiang, Y.K. Probing spatiotemporal PKA activity at the ryanodine receptor and SERCA2a nanodomains in cardomyocytes. Cell. Commun. Signal. 2022, 20, 143. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.; Gorelik, J.; Yacoub, M.H.; Terracciano, C.M. The structure and function of cardiac t-tubules in health and disease. Proc. R. Soc. B Biol. Sci. 2011, 278, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Beca, S.; Helli, P.B.; Simpson, J.A.; Zhao, D.; Farman, G.P.; Jones, P.P.; Tian, X.; Wilson, L.S.; Ahmad, F.; Chen, S.R.W.; et al. Phosphodiesterase 4D regulates baseline sarcoplasmic reticulum Ca2+ release and cardiac contractility.; independently of L-type Ca2+ current. Circ. Res. 2011, 109, 1024–1030. [Google Scholar] [CrossRef]
- Beca, S.; Ahmad, F.; Shen, W.; Liu, J.; Makary, S.; Polidovitch, N.; Sun, J.; Hockman, S.; Chung, Y.W.; Movsesian, M.; et al. Phosphodiesterase type 3A regulates basal myocardial contractility through interacting with sarcoplasmic reticulum calcium ATPase type 2a Signaling complexes in mouse heart. Circ. Res. 2013, 112, 289–297. [Google Scholar] [CrossRef]
- Skogestad, J.; Albert, I.; Hougen, K.; Lothe, G.B.; Lunde, M.; Sovik Eken, O.; Veras, I.; Thi-Huynh, N.T.; Borstad, M.; Marshall, S.; et al. Disruption of phosphodiesterase 3A binding to SERCA2 increases SERCA2 activity and reduces mortality in mice with chronic heart failure. Circulation 2023, 147, 1221–1236. [Google Scholar] [CrossRef]
- Beca, S.; Aschars-Sobbi, R.; Panama, B.K.; Backx, P.H. Regulation of murine cardiac function by phosphodiesterases type 3 and 4. Curr. Opin. Pharmacol. 2011, 11, 714–719. [Google Scholar] [CrossRef]
- Karam, S.; Margaria, J.P.; Bourcier, A.; Mika, D.; Varin, A.; Bedioune, I.; Lindner, M.; Bouadjel, K.; Dessillons, M.; Gaudin, F.; et al. Cardiac Overexpression of PDE4B Blunts β-Adrenergic Response and Maladaptive Remodeling in Heart Failure. Circulation 2020, 142, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Patrucco, E.; Albergine, M.S.; Santana, L.F.; Beavo, J.A. Phosphodiesterase 8A (PDE8A) regulates excitation contraction coupling in ventricular myocytes. J. Mol. Cell. Cardiol. 2010, 49, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, B.; Yan, W.; Yang, Z.; Peng, X.; Lin, D.; Weng, X.; Ye, T.; Qu, J. Resolution improvement in STED super-resolution microscopy at low power using a phasor plot approach. Nanoscale 2018, 10, 16252–16260. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kraft, A.E.; Bork, N.I.; Subramanian, H.; Pavlaki, N.; Failla, A.V.; Zobiak, B.; Conti, M.; Nikolaev, V.O. Phosphodiesterases 4B and 4D Differentially Regulate cAMP Signaling in Calcium Handling Microdomains of Mouse Hearts. Cells 2024, 13, 476. https://doi.org/10.3390/cells13060476
Kraft AE, Bork NI, Subramanian H, Pavlaki N, Failla AV, Zobiak B, Conti M, Nikolaev VO. Phosphodiesterases 4B and 4D Differentially Regulate cAMP Signaling in Calcium Handling Microdomains of Mouse Hearts. Cells. 2024; 13(6):476. https://doi.org/10.3390/cells13060476
Chicago/Turabian StyleKraft, Axel E., Nadja I. Bork, Hariharan Subramanian, Nikoleta Pavlaki, Antonio V. Failla, Bernd Zobiak, Marco Conti, and Viacheslav O. Nikolaev. 2024. "Phosphodiesterases 4B and 4D Differentially Regulate cAMP Signaling in Calcium Handling Microdomains of Mouse Hearts" Cells 13, no. 6: 476. https://doi.org/10.3390/cells13060476
APA StyleKraft, A. E., Bork, N. I., Subramanian, H., Pavlaki, N., Failla, A. V., Zobiak, B., Conti, M., & Nikolaev, V. O. (2024). Phosphodiesterases 4B and 4D Differentially Regulate cAMP Signaling in Calcium Handling Microdomains of Mouse Hearts. Cells, 13(6), 476. https://doi.org/10.3390/cells13060476