
Translational Prospects and Challenges in Human Induced Pluripotent Stem Cell Research in Drug Discovery

Abstract
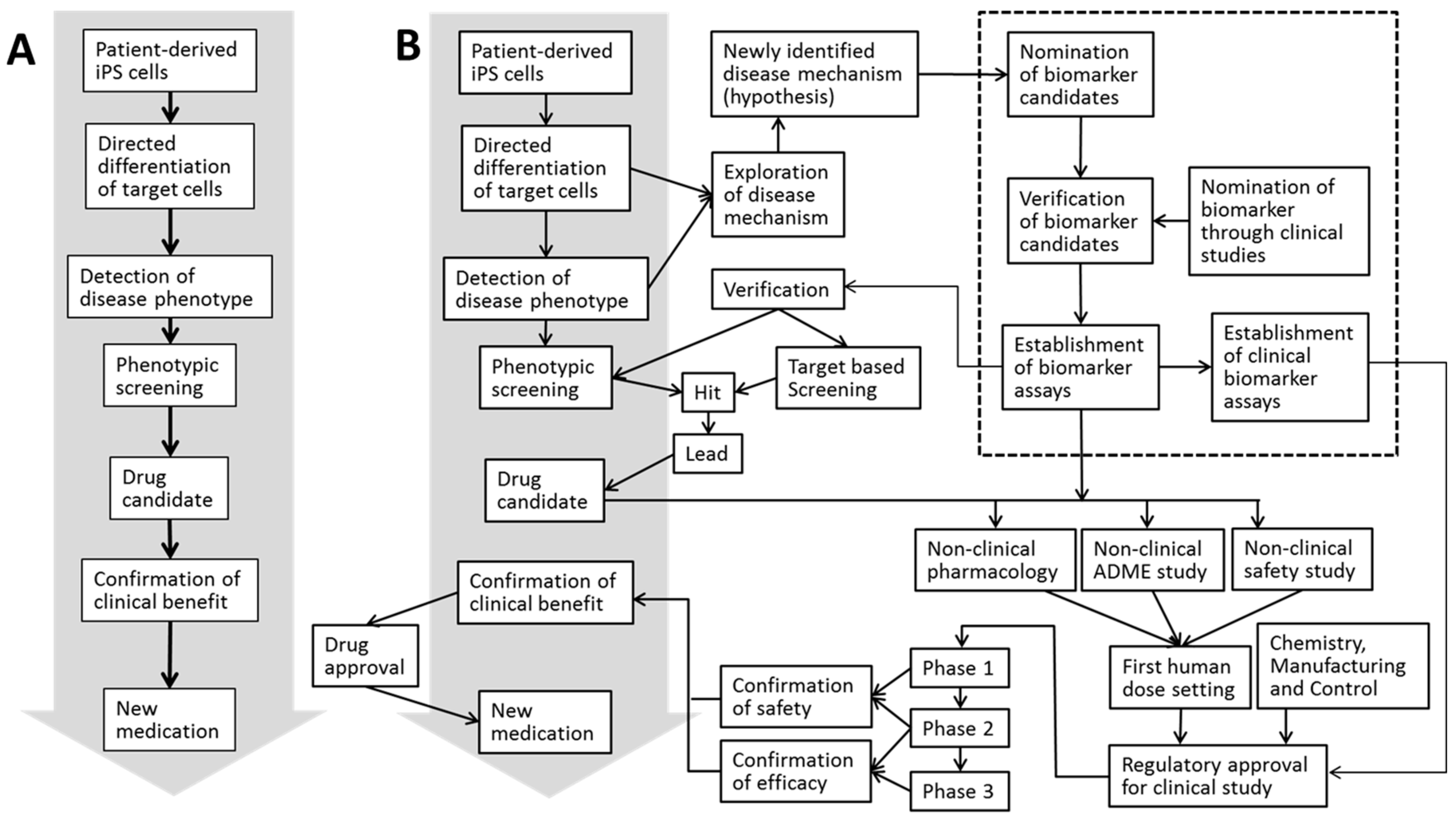
:1. Introduction
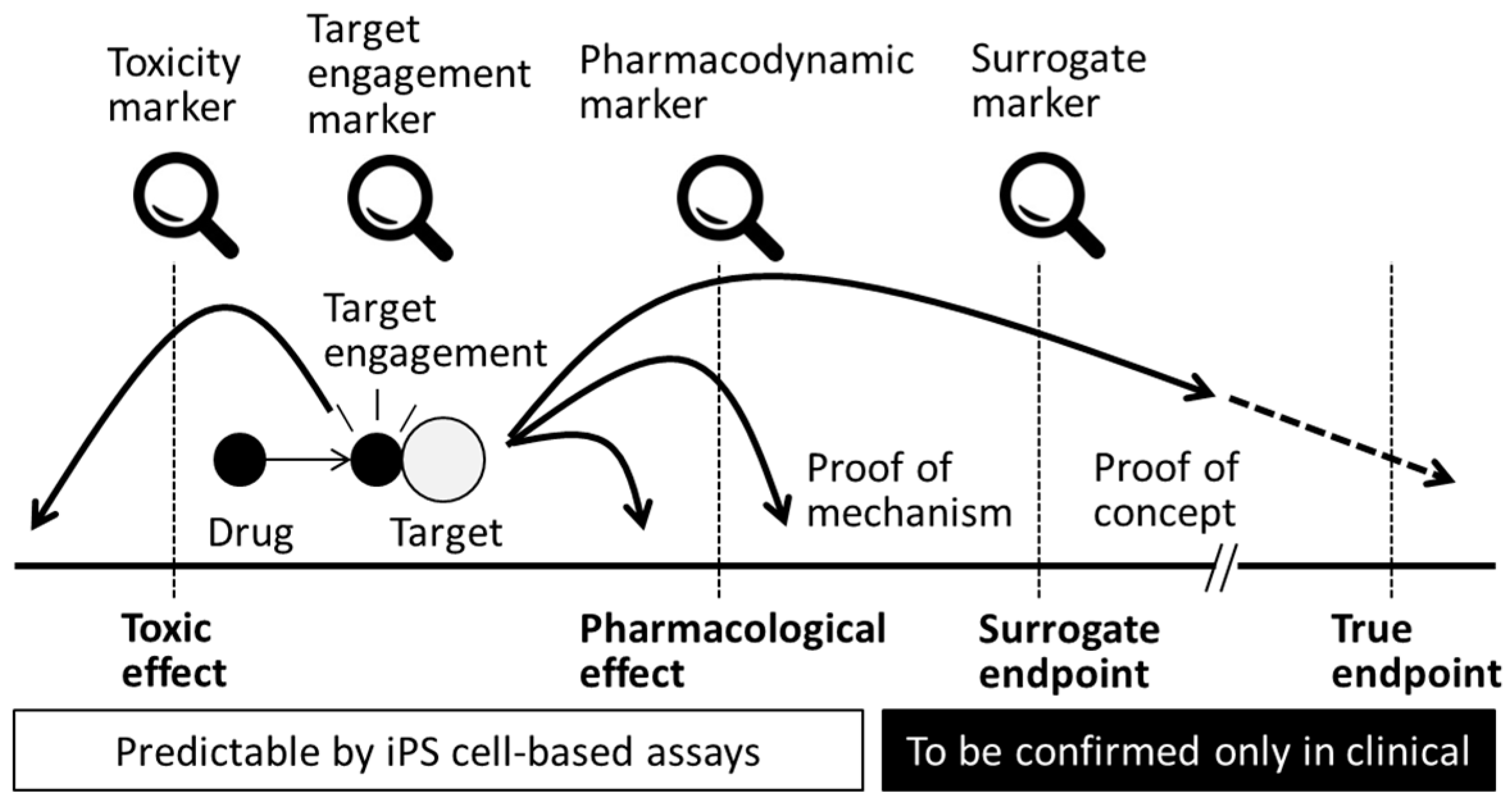
2. Biomarkers in Translational Research (TR)
3. Use of Induced Pluripotent Stem (iPS) Cells to Understand Diseases
3.1. Disease Mechanisms
3.2. Genetic Factors
3.3. Hypothesis Confirmation in Clinical Settings
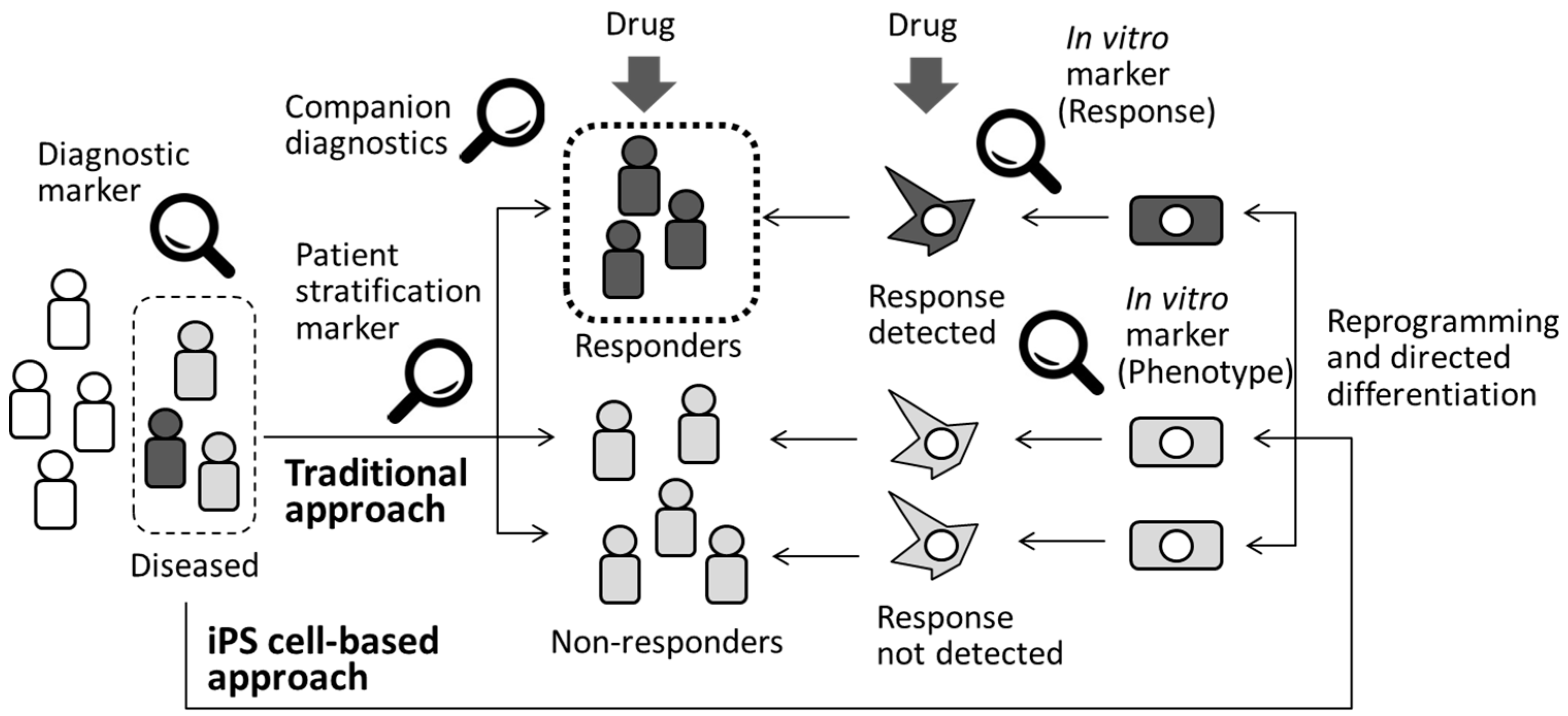
3.4. Patient Stratification and Precision Medicine
4. Pharmacological Studies Using iPS Cells
4.1. Validation of Cellular Models
4.2. Screening and Pharmacological Evaluation of Drug Candidates
4.3. Target Identification and Validation
4.4. Target Engagement
4.5. Pharmacokinetics and Pharmacodynamics
4.6. Clinical Endpoints
4.7. Integrative Approach
4.8. Benefits of iPS Cells in the Toxicological Assessment of Drugs Targeting Rare Diseases
5. Cytotoxicity
5.1. Hepatotoxicity
5.2. Cardiotoxicity
5.3. Nephrotoxicity
5.4. Neurotoxicity
6. Cell Therapy Using iPS Cells
7. Future Perspectives
8. Conclusions
Author Contributions
Conflicts of Interest
References
- Paul, G.; Li, J.Y.; Brundin, P. Stem cells: Hype or hope? Drug Discov. Today 2002, 7, 295–302. [Google Scholar] [CrossRef]
- Yu, J.; Thomson, J.A. Pluripotent stem cell lines. Genes Dev. 2008, 22, 1987–1997. [Google Scholar] [CrossRef] [PubMed]
- McNeish, J. Embryonic stem cells in drug discovery. Nat. Rev. Drug Discov. 2004, 3, 70–80. [Google Scholar] [CrossRef] [PubMed]
- De Wert, G.; Mummery, C. Human embryonic stem cells: Research, ethics and policy. Hum. Reprod. 2003, 18, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoi, T.; Yae, K.; Nakagawa, M.; Ichisaka, T.; Okita, K.; Takahashi, K.; Chiba, T.; Yamanaka, S. Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science 2008, 321, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Zhao, R.; West, J.A.; Yabuuchi, A.; Huo, H.; Ince, T.A.; Lerou, P.H.; Lensch, M.W.; Daley, G.Q. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 2008, 451, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Lowry, W.E.; Richter, L.; Yachechko, R.; Pyle, A.D.; Tchieu, J.; Sridharan, R.; Clark, A.T.; Plath, K. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc. Natl. Acad. Sci. USA 2008, 105, 2883–2888. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-specific induced pluripotent stem cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.S.; Malik, N. Assessing iPSC reprogramming methods for their suitability in translational medicine. J. Cell Biochem. 2012, 113, 3061–3068. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Otsu, M. Development of Sendai virus vectors and their potential applications in gene therapy and regenerative medicine. Curr. Gene. Ther. 2012, 12, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, B. Reprogramming barriers and enhancers: Strategies to enhance the efficiency and kinetics of induced pluripotency. Cell Regen. 2015, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Lund, R.J.; Narva, E.; Lahesmaa, R. Genetic and epigenetic stability of human pluripotent stem cells. Nat. Rev. Genet. 2012, 13, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Kyttälä, A.; Moraghebi, R.; Valensisi, C.; Kettunen, J.; Andrus, C.; Pasumarthy, K.K.; Nakanishi, M.; Nishimura, K.; Ohtaka, M.; Weltner, J.; et al. Genetic Variability Overrides the Impact of Parental Cell Type and Determines iPSC Differentiation Potential. Stem Cell Rep. 2016, 6, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Gage, F. The promise and the challenge of modelling human disease in a dish. EMBO Mol. Med. 2010, 2, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Hefti, F.F. Requirements for a lead compound to become a clinical candidate. BMC Neurosci. 2008, 9 (Suppl. S3), S7. [Google Scholar] [CrossRef] [PubMed]
- Kaitin, K.I. Deconstructing the drug development process: The new face of innovation. Clin. Pharmacol. Ther. 2010, 87, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Smietana, K.; Siatkowski, M.; Moller, M. Trends in clinical success rates. Nat. Rev. Drug. Discov. 2016, 15, 379–380. [Google Scholar] [CrossRef] [PubMed]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef] [PubMed]
- De Vos, J.; Bouckenheimer, J.; Sansac, C.; Lemaître, J.M.; Assou, S. Human induced pluripotent stem cells: A disruptive innovation. Curr. Res. Transl. Med. 2016, 64, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.; Bhatia, M. iPSC technology: Platform for drug discovery. Clin. Pharmacol. Ther. 2011, 89, 639–641. [Google Scholar] [CrossRef] [PubMed]
- Vitale, A.M.; Wolvetang, E.; Mackay-Sim, A. Induced pluripotent stem cells: A new technology to study human diseases. Int. J. Biochem. Cell Biol. 2011, 43, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Fishburn, C.S. Translational research: The changing landscape of drug discovery. Drug Discov. Today 2013, 18, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Mullane, K.; Winquist, R.J.; Williams, M. Translational paradigms in pharmacology and drug discovery. Biochem. Pharmacol. 2014, 87, 189–210. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; Rothstein, J.D.; Pouladi, M.A. Preclinical models: Needed in translation? A Pro/Con debate. Mov. Disord. 2014, 29, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Chen, G.; Hu, B. Induced pluripotency for translational research. Genom. Proteom. Bioinform. 2013, 11, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Modur, V.; Carayannopoulos, L.N.; Laterza, O.F. Biomarkers in pharmaceutical research. Clin. Chem. 2015, 61, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Strimbu, K.; Tavel, J.A. What are biomarkers? Curr. Opin. HIV AIDS 2010, 5, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Ward, T.H.; Greystoke, A.; Ranson, M.; Dive, C. Biomarker method validation in anticancer drug development. Br. J. Pharmacol. 2008, 153, 646–656. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, K.; He, J.; Taranova, O.; Liang, G.; D’Alessio, A.C.; Zhang, Y. Generation of insulin-secreting islet-like clusters from human skin fibroblasts. J. Biol. Chem. 2008, 283, 31601–31607. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Cai, J.; Liu, Y.; Zhao, D.; Yong, J.; Duo, S.; Song, X.; Guo, Y.; Zhao, Y.; Qin, H.; et al. Efficient generation of hepatocyte-like cells from human induced pluripotent stem cells. Cell Res. 2009, 19, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wilson, G.F.; Soerens, A.G.; Koonce, C.H.; Yu, J.; Palecek, S.P.; Thomson, J.A.; Kamp, T.J. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ. Res. 2009, 104, e30–e41. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.F.; Potter, W.Z.; Brasic, J.R. Proof of concept: Functional models for drug development in humans. In Neuropsychopharmacology: The Fifth Generation of Progress; Davis, K.L., Charney, D., Coyle, J.T., Nemeroff, C., Eds.; Lippincott, Williams & Wilkins: Philadelphia, PA, USA, 2002; pp. 457–473. [Google Scholar]
- Wehling, M. Assessing the translatability of drug projects: What needs to be scored to predict success? Nat. Rev. Drug Discov. 2009, 8, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Wendler, A.; Wehling, M. Translatability scoring in drug development: Eight case studies. J. Transl. Med. 2012, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Saha, K.; Jaenisch, R. Technical challenges in using human induced pluripotent stem cells to model disease. Cell Stem Cell 2009, 5, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Ebert, A.D.; Yu, J.; Rose, F.F., Jr.; Mattis, V.B.; Lorson, C.L.; Thomson, J.A.; Svendsen, C.N. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 2009, 457, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Papapetrou, E.P.; Kim, H.; Chambers, S.M.; Tomishima, M.J.; Fasano, C.A.; Ganat, Y.M.; Menon, J.; Shimizu, F.; Viale, A.; et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature 2009, 461, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Stacey, G.N.; Crook, J.M.; Hei, D.; Ludwig, T. Banking human induced pluripotent stem cells: Lessons learned from embryonic stem cells? Cell Stem Cell 2013, 13, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Gao, W.Q. Concise review: Patient-derived stem cell research for monogenic disorders. Stem Cells 2016, 34, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Jozefczuk, J.; Kashofer, K.; Ummanni, R.; Henjes, F.; Rehman, S.; Geenen, S.; Wruck, W.; Regenbrecht, C.; Daskalaki, A.; Wierling, C.; et al. A systems biology approach to deciphering the etiology of steatosis employing patient-derived dermal fibroblasts and iPS cells. Front. Physiol. 2012, 3, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetto, M.C.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Kim, J.; Zhou, L.; Wengert, E.; Zhang, L.; Wu, Z.; Carromeu, C.; Muotri, A.R.; Marchetto, M.C.; Gage, F.H.; et al. KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc. Natl. Acad. Sci. USA 2016, 113, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Takahashi, K.; Yamane, M.; Tomida, S.; Nakamura, S.; Oshima, K.; Niwa, A.; Nishikomori, R.; Kambe, N.; Hara, H.; et al. Induced pluripotent stem cells from CINCA syndrome patients as a model for dissecting somatic mosaicism and drug discovery. Blood 2012, 120, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Ho, R.; Sances, S.; Gowin, G.; Amoroso, M.W.; O’Rourke, J.G.; Sahabian, A.; Wichterle, H.; Baloh, R.H.; Sareen, D.; Svendsen, C.N. ALS disrupts spinal motor neuron maturation and aging pathways within gene co-expression networks. Nat. Neurosci. 2016, 19, 1256–1267. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Soldner, F.; Beard, C.; Gao, Q.; Mitalipova, M.; DeKelver, R.C.; Katibah, G.E.; Amora, R.; Boydston, E.A.; Zeitler, B.; et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 2009, 27, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Suzuki, K.; Qu, J.; Sancho-Martinez, I.; Yi, F.; Li, M.; Kumar, S.; Nivet, E.; Kim, J.; Soligalla, R.D.; et al. Targeted gene correction of laminopathy-associated LMNA mutations in patient-specific iPSCs. Cell Stem Cell 2011, 8, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Grobarczyk, B.; Franco, B.; Hanon, K.; Malgrange, B. Generation of isogenic human iPS cell line precisely corrected by genome editing using the CRISPR/Cas9 system. Stem Cell Rev. 2015, 11, 774–787. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Hrabovsky, A.; Pedrosa, E.; Wang, T.; Zheng, D.; Lachman, H.M. Allele-biased expression in differentiating human neurons: Implications for neuropsychiatric disorders. PLoS ONE 2012, 7, e44017. [Google Scholar] [CrossRef] [PubMed]
- Chae, J.I.; Kim, D.W.; Lee, N.; Jeon, Y.J.; Jeon, I.; Kwon, J.; Kim, J.; Soh, Y.; Lee, D.S.; Seo, K.S.; et al. Quantitative proteomic analysis of induced pluripotent stem cells derived from a human Huntington’s disease patient. Biochem. J. 2012, 446, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Harris, V.K.; Sadiq, S.A. Biomarkers of therapeutic response in multiple sclerosis: Current status. Mol. Diagn. Ther. 2014, 18, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Bala, S.; Petrasek, J.; Mundkur, S.; Catalano, D.; Levin, I.; Ward, J.; Alao, H.; Kodys, K.; Szabo, G. Circulating microRNAs in exosomes indicate hepatocyte injury and inflammation in alcoholic, drug-induced, and inflammatory liver diseases. Hepatology 2012, 56, 1946–1957. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.D.; Ganat, Y.M.; Kishinevsky, S.; Bowman, R.L.; Liu, B.; Tu, E.Y.; Mandal, P.K.; Vera, E.; Shim, J.W.; Kriks, S.; et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 2013, 13, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Carrigan, P.; Krahn, T. Impact of biomarkers on personalized medicine. Handb. Exp. Pharmacol. 2016, 232, 285–311. [Google Scholar] [PubMed]
- Limpitikul, W.B.; Dick, I.E.; Tester, D.; Boczek, N.J.; Limphong, P.; Yang, W.; Choi, M.H.; Babich, J.; DiSilvestre, D.; Kanter, R.J.; et al. A Precision Medicine Approach to the Rescue of Function on Malignant Calmodulinopathic Long QT Syndrome. Circ. Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Wang, Q.W.; Kim, Y.; Yu, D.X.; Pham, S.; Yang, B.; Zheng, Y.; Diffenderfer, K.E.; Zhang, J.; Soltani, S.; et al. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature 2015, 527, 95–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, Y.; Oda, H.; Ito, J.; Niwa, A.; Tanaka, T.; Hijikata, A.; Seki, R.; Nagahashi, A.; Osawa, M.; Asaka, I.; et al. Pluripotent cell-based phenotypic dissection identifies a high-frequency somatic NLRC4 mutation as a cause of autoinflammation. Arthritis Rheumatol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Nagata, N.; Kurokawa, H.; Yamanaka, S. iPS cells: A game changer for future medicine. EMBO J. 2014, 33, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Swinney, D.C.; Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011, 10, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.; Do-Ha, D.; Muñoz, S.S.; Ooi, L. Common pitfalls of stem cell differentiation: A guide to improving protocols for neurodegenerative disease models and research. Cell. Mol. Life Sci. 2016, 73, 3693–3709. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.T.; Corbineau, S.; Hannan, N.; Marciniak, S.J.; Miranda, E.; Alexander, G.; Huang-Doran, I.; Griffin, J.; Ahrlund-Richter, L.; Skepper, J.; et al. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J. Clin. Investig. 2010, 120, 3127–3136. [Google Scholar] [CrossRef] [PubMed]
- Janzen, W.P. Screening technologies for small molecule discovery: The state of the art. Chem. Biol. 2014, 21, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, I.; Gasparri, F. The development of high-content screening (HCS) technology and its importance to drug discovery. Expert Opin. Drug Discov. 2016, 11, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Xie, M.; Cao, N.; Ding, S. Patient-specific induced pluripotent stem cells for disease modeling and phenotypic drug discovery. J. Med. Chem. 2016, 59, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.Y.; Milton, M.N. The determination and interpretation of the therapeutic index in drug development. Nat. Rev. Drug Discov. 2012, 11, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, J.W. Target identification for biologically active small molecules using chemical biology approaches. Arch. Pharm. Res. 2016, 39, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Wagner, B.K.; Schreiber, S.L. The power of sophisticated phenotypic screening and modern mechanism-of-action methods. Cell Chem. Biol. 2016, 23, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Belda, I.; Penas, G.; Alonso, A.; Marquina, D.; Navascués, E.; Santos, A. Biotech patents and science policy: The Spanish experience. Nat. Biotechnol. 2014, 32, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, M.; Schuffenhauer, A.; Fruh, I.; Klein, J.; Thiemeyer, A.; Rigo, P.; Gomez-Mancilla, B.; Heidinger-Millot, V.; Bouwmeester, T.; Schopfer, U.; et al. High-Throughput Screening Using iPSC-Derived Neuronal Progenitors to Identify Compounds Counteracting Epigenetic Gene Silencing in Fragile X Syndrome. J. Biomol. Screen. 2015, 20, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, E.; Tresadern, G.; Bembenek, S.; Wroblowski, B.; Buyck, C.; Neefs, J.M.; Rassokhin, D.; Poncelet, A.; Hunt, J.; van Vlijmen, H. Extending kinome coverage by analysis of kinase inhibitor broad profiling data. Drug Discov. Today 2015, 20, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Durham, T.B.; Blanco, M.J. Target engagement in lead generation. Bioorg. Med. Chem. Lett. 2015, 25, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Morizane, A.; Doi, D.; Onoe, H.; Hayashi, T.; Kawasaki, T.; Saiki, H.; Miyamoto, S.; Takahashi, J. Survival of human induced pluripotent stem cell-derived midbrain dopaminergic neurons in the brain of a primate model of Parkinson’s disease. J. Parkinson’s Dis. 2011, 1, 395–412. [Google Scholar]
- Lippert, J.; Burghaus, R.; Kuepfer, L.; Ploeger, B.; Schaller, S.; Schmitt, W.; Willmann, S. Modeling and Simulation of In Vivo Drug Effects. Handb. Exp. Pharmacol. 2016, 232, 313–329. [Google Scholar] [PubMed]
- Iwao, T.; Toyota, M.; Miyagawa, Y.; Okita, H.; Kiyokawa, N.; Akutsu, H.; Umezawa, A.; Nagata, K.; Matsunaga, T. Differentiation of human induced pluripotent stem cells into functional enterocyte-like cells using a simple method. Drug Metab. Pharmacokinet. 2014, 29, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Minami, H.; Tashiro, K.; Okada, A.; Hirata, N.; Yamaguchi, T.; Takayama, K.; Mizuguchi, H.; Kawabata, K. Generation of Brain Microvascular Endothelial-Like Cells from Human Induced Pluripotent Stem Cells by Co-Culture with C6 Glioma Cells. PLoS ONE 2015, 10, e0128890. [Google Scholar] [CrossRef] [PubMed]
- Katt, M.E.; Xu, Z.S.; Gerecht, S.; Searson, P.C. Human Brain Microvascular Endothelial Cells Derived from the BC1 iPS Cell Line Exhibit a Blood-Brain Barrier Phenotype. PLoS ONE 2016, 11, e0152105. [Google Scholar] [CrossRef] [PubMed]
- Takasato, M.; Er, P.X.; Chiu, H.S.; Maier, B.; Baillie, G.J.; Ferguson, C.; Parton, R.G.; Wolvetang, E.J.; Roost, M.S.; Chuva de Sousa Lopes, S.M.; Little, M.H. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 2015, 526, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Otsu, M.; Matsuzaka, E.; Konishi, C.; Takagi, H.; Hanada, S.; Mochizuki, S.; Nakauchi, H.; Imai, K.; Tsuji, K.; et al. Screening of drugs to treat 8p11 myeloproliferative syndrome using patient-derived induced pluripotent stem cells with fusion gene CEP110-FGFR1. PLoS ONE 2015, 10, e0120841. [Google Scholar] [CrossRef] [PubMed]
- Aronson, J.K. Biomarkers and surrogate endpoints. Br. J. Clin. Pharmacol. 2005, 59, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Luni, C.; Serena, E.; Elvassore, N. Human-on-chip for therapy development and fundamental science. Curr. Opin. Biotechnol. 2014, 25, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Shupe, T.; Atala, A. Organoid-on-a-chip and body-on-a-chip systems for drug screening and disease modeling. Drug Discov. Today 2016, 21, 1399–1411. [Google Scholar] [CrossRef] [PubMed]
- Wendler, A.; Wehling, M. The translatability of animal models for clinical development: Biomarkers and disease models. Curr. Opin. Pharmacol. 2010, 10, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.M. Drug-induced hepatotoxicity. N. Engl. J. Med. 2003, 349, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Kaplowitz, N. Idiosyncratic drug hepatotoxicity. Nat. Rev. Drug Discov. 2005, 4, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Roth, R.A.; Ganey, P.E. Intrinsic versus idiosyncratic drug-induced hepatotoxicity—Two villains or one? J. Pharmacol. Exp. Ther. 2010, 332, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Gerets, H.H.; Tilmant, K.; Gerin, B.; Chanteux, H.; Depelchin, B.O.; Dhalluin, S.; Atienzar, F.A. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biol. Toxicol. 2012, 28, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Kanebratt, K.P.; Andersson, T.B. HepaRG cells as an in vitro model for evaluation of cytochrome P450 induction in humans. Drug Metab. Dispos. 2008, 36, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Kanebratt, K.P.; Andersson, T.B. Evaluation of HepaRG cells as an in vitro model for human drug metabolism studies. Drug Metab. Dispos. 2008, 36, 1444–1452. [Google Scholar] [CrossRef] [PubMed]
- Anson, B.D.; Kolaja, K.L.; Kamp, T.J. Opportunities for use of human iPS cells in predictive toxicology. Clin. Pharmacol. Ther. 2011, 89, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Choi, Y.J.; Kim, J.W.; Chun, H.S.; Im, I.; Yoon, S.; Han, Y.M.; Song, C.W.; Kim, H. Differences in the Epigenetic Regulation of Cytochrome P450 Genes between Human Embryonic Stem Cell-Derived Hepatocytes and Primary Hepatocytes. PLoS ONE 2015, 10, e0132992. [Google Scholar] [CrossRef] [PubMed]
- Tanimizu, N.; Nishikawa, M.; Saito, H.; Tsujimura, T.; Miyajima, A. Isolation of hepatoblasts based on the expression of Dlk/Pref-1. J. Cell Sci. 2003, 116, 1775–1786. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.D. Human Drug Metabolism: An Introduction, 2nd ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2010. [Google Scholar]
- Mann, D.A. Human induced pluripotent stem cell-derived hepatocytes for toxicology testing. Expert Opin. Drug. Metab. Toxicol. 2015, 11, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Einhorn, S.; Venkatarangan, L.; Miller, M.; Mann, D.A.; Watkins, P.B.; LeCluyse, E. Morphological and Functional Characterization and Assessment of iPSC-Derived Hepatocytes for In Vitro Toxicity Testing. Toxicol. Sci. 2015, 147, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Tasnim, F.; Toh, Y.C.; Qu, Y.; Li, H.; Phan, D.; Narmada, B.C.; Ananthanarayanan, A.; Mittal, N.; Meng, R.Q.; Yu, H. Functionally Enhanced Human Stem Cell Derived Hepatocytes in Galactosylated Cellulosic Sponges for Hepatotoxicity Testing. Mol. Pharm. 2016, 13, 1947–1957. [Google Scholar] [CrossRef] [PubMed]
- Ware, B.R.; Berger, D.R.; Khetani, S.R. Prediction of Drug-Induced Liver Injury in Micropatterned Co-cultures Containing iPSC-Derived Human Hepatocytes. Toxicol. Sci. 2015, 145, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Espejel, S.; Roll, G.R.; McLaughlin, K.J.; Lee, A.Y.; Zhang, J.Y.; Laird, D.J.; Okita, K.; Yamanaka, S.; Willenbring, H. Induced pluripotent stem cell-derived hepatocytes have the functional and proliferative capabilities needed for liver regeneration in mice. J. Clin. Investig. 2010, 120, 3120–3126. [Google Scholar] [CrossRef] [PubMed]
- Schomaker, S.; Warner, R.; Bock, J.; Johnson, K.; Potter, D.; van Winkle, J.; Aubrecht, J. Assessment of emerging biomarkers of liver injury in human subjects. Toxicol. Sci. 2013, 132, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Pirola, C.J.; Fernández Gianotti, T.; Castaño, G.O.; Mallardi, P.; San Martino, J.; Ledesma, M.M.G.L.; Flichman, D.; Mirshahi, F.; Sanyal, A.J.; Sookoian, S. Circulating microRNA signature in non-alcoholic fatty liver disease: From serum non-coding RNAs to liver histology and disease pathogenesis. Gut 2015, 64, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Enache, L.S.; Enache, E.L.; Ramière, C.; Diaz, O.; Bancu, L.; Sin, A.; André, P. Circulating RNA molecules as biomarkers in liver disease. Int. J. Mol. Sci. 2014, 15, 17644–17666. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Zhang, R.R.; Koike, H.; Kimura, M.; Yoshizawa, E.; Enomura, M.; Koike, N.; Sekine, K.; Taniguchi, H. Generation of a vascularized and functional human liver from an iPSC-derived organ bud transplant. Nat. Protoc. 2014, 9, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.R.; Takebe, T.; Miyazaki, L.; Takayama, M.; Koike, H.; Kimura, M.; Enomura, M.; Zheng, Y.W.; Sekine, K.; Taniguchi, H. Efficient hepatic differentiation of human induced pluripotent stem cells in a three-dimensional microscale culture. Methods Mol. Biol. 2014, 1210, 131–141. [Google Scholar] [PubMed]
- Ogawa, S.; Surapisitchat, J.; Virtanen, C.; Ogawa, M.; Niapour, M.; Sugamori, K.S.; Wang, S.; Tamblyn, L.; Guillemette, C.; Hoffmann, E.; et al. Three-dimensional culture and cAMP signaling promote the maturation of human pluripotent stem cell-derived hepatocytes. Development 2013, 140, 3285–3296. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Kawabata, K.; Nagamoto, Y.; Kishimoto, K.; Tashiro, K.; Sakurai, F.; Tachibana, M.; Kanda, K.; Hayakawa, T.; Furue, M.K.; et al. 3D spheroid culture of hESC/hiPSC-derived hepatocyte-like cells for drug toxicity testing. Biomaterials 2013, 34, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- McCarty, W.J.; Usta, O.B.; Yarmush, M.L. A Microfabricated Platform for Generating Physiologically-Relevant Hepatocyte Zonation. Sci. Rep. 2016, 6, 26868. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Vernetti, L.A.; Senutovitch, N.; Boltz, R.; DeBiasio, R.; Shun, T.Y.; Gough, A.; Taylor, D.L. A human liver microphysiology platform for investigating physiology, drug safety, and disease models. Exp. Biol. Med. 2016, 241, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Potta, S.P.; Šarić, T.; Heke, M.; Bahudhanapati, H.; Hescheler, J. Human Pluripotent Stem Cell Applications in Drug Discovery and Toxicology—An overview. In Pluripotent Stem Cell Biology—Advances in Mechanisms, Methods and Models; Atwood, C.S., Meethal, S.V., Eds.; InTech: Rijeka, Croatia, 2014. [Google Scholar]
- Cheng, H.; Kar, G.; Dicker, A.P.; Rodeck, U.; Koch, W.J.; Force, T. A novel preclinical strategy for identifying cardiotoxic kinase inhibitors and mechanisms of cardiotoxicity. Circ. Res. 2011, 109, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Force, T.; Kolaja, K.L. Cardiotoxicity of kinase inhibitors: The prediction and translation of preclinical models to clinical outcomes. Nat. Rev. Drug Discov. 2011, 10, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, Y.; Ohtsuki, A.; Oka, T.; Shimizu, T. Electrophysiological analysis of mammalian cells expressing hERG using automated 384-well-patch-clamp. BMC Pharmacol. Toxicol. 2015, 16, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhari, U.; Nemade, H.; Wagh, V.; Gaspar, J.A.; Ellis, J.K.; Srinivasan, S.P.; Spitkovski, D.; Nguemo, F.; Louisse, J.; Bremer, S.; et al. Identification of genomic biomarkers for anthracycline-induced cardiotoxicity in human iPSC-derived cardiomyocytes: An in vitro repeated exposure toxicity approach for safety assessment. Arch. Toxicol. 2016, 90, 2763–2777. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.; Lan, F.; Lee, A.S.; Gong, T.; Sanchez-Freire, V.; Wang, Y.; Diecke, S.; Sallam, K.; Knowles, J.W.; Wang, P.J.; et al. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation 2013, 127, 1677–1691. [Google Scholar] [CrossRef] [PubMed]
- Miki, K.; Endo, K.; Takahashi, S.; Funakoshi, S.; Takei, I.; Katayama, S.; Toyoda, T.; Kotaka, M.; Takaki, T.; Umeda, M.; et al. Efficient Detection and Purification of Cell Populations Using Synthetic MicroRNA Switches. Cell Stem Cell 2015, 16, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V.; Vaidya, V.S.; Schmouder, R.; Feig, P.; Dieterle, F. Next-generation biomarkers for detecting kidney toxicity. Nat. Biotechnol. 2010, 28, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Tiong, H.Y.; Huang, P.; Xiong, S.; Li, Y.; Vathsala, A.; Zink, D. Drug-induced nephrotoxicity: Clinical impact and preclinical in vitro models. Mol. Pharm. 2014, 11, 1933–1948. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, K.; Chuah, J.K.; Su, R.; Huang, P.; Eng, K.G.; Xiong, S.; Li, Y.; Chia, C.S.; Loo, L.H.; Zink, D. Prediction of drug-induced nephrotoxicity and injury mechanisms with human induced pluripotent stem cell-derived cells and machine learning methods. Sci. Rep. 2015, 5, 12337. [Google Scholar] [CrossRef] [PubMed]
- Van Meer, L.; Moerland, M.; Cohen, A.F.; Burggraaf, J. Urinary kidney biomarkers for early detection of nephrotoxicity in clinical drug development. Br. J. Clin. Pharmacol. 2014, 77, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.C.; Hewitt, P. Biomarkers for drug-induced renal damage and nephrotoxicity—An overview for applied toxicology. AAPS J. 2011, 13, 615–631. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.A. Self-organized Kidney Rudiments: Prospects for Better in vitro Nephrotoxicity Assays. Biomark. Insights 2015, 10 (Suppl. S1), 117–123. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.X.; Kaeslin, G.; Ranal, M.V.; Blaskovich, M.A.; Becker, B.; Butler, M.S.; Little, M.H.; Lash, L.H.; Cooper, M.A. Evaluation of biomarkers for in vitro prediction of drug-induced nephrotoxicity: Comparison of HK-2, immortalized human proximal tubule epithelial, and primary cultures of human proximal tubular cells. Pharmacol. Res. Perspect. 2015, 3, e00148. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.X.; Blaskovich, M.A.; Cooper, M.A. Cell- and biomarker-based assays for predicting nephrotoxicity. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1621–1635. [Google Scholar] [CrossRef] [PubMed]
- Human Renal Cells (Normal & Diseased, Lonza, Basel, Switzerland). Available online: http://www.lonza.com/products-services/bio-research/primary-cells/human-cells-and-media/renal-cells-and-media/human-renal-cells.aspx (accessed on 27 November 2016).
- Taguchi, A.; Kaku, Y.; Ohmori, T.; Sharmin, S.; Ogawa, M.; Sasaki, H.; Nishinakamura, R. Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell 2014, 14, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, A.; Nishinakamura, R. Nephron reconstitution from pluripotent stem cells. Kidney Int. 2015, 87, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Takasato, M.; Er, P.X.; Becroft, M.; Vanslambrouck, J.M.; Stanley, E.G.; Elefanty, A.G.; Little, M.H. Directing human embryonic stem cell differentiation towards a renal lineage generates a self-organizing kidney. Nat. Cell Biol. 2014, 16, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Morizane, R.; Lam, A.Q.; Freedman, B.S.; Kishi, S.; Valerius, M.T.; Bonventre, J.V. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat. Biotechnol. 2015, 33, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Nieskens, T.T.; Wilmer, M.J. Kidney-on-a-chip technology for renal proximal tubule tissue reconstruction. Eur. J. Pharmacol. 2016, 790, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Wilmer, M.J.; Ng, C.P.; Lanz, H.L.; Vulto, P.; Suter-Dick, L.; Masereeuw, R. Kidney-on-a-Chip Technology for Drug-Induced Nephrotoxicity Screening. Trends Biotechnol. 2016, 34, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Tukker, A.M.; de Groot, M.W.; Wijnolts, F.M.; Kasteel, E.E.; Hondebrink, L.; Westerink, R.H. Is the time right for in vitro neurotoxicity testing using human iPSC-derived neurons? ALTEX 2016, 33, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Bal-Price, A.K.; Hogberg, H.T.; Buzanska, L.; Lenas, P.; van Vliet, E.; Hartung, T. In vitro developmental neurotoxicity (DNT) testing: Relevant models and endpoints. Neurotoxicology 2010, 31, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Bal-Price, A.K.; Hogberg, H.T.; Buzanska, L.; Coecke, S. Relevance of in vitro neurotoxicity testing for regulatory requirements: Challenges to be considered. Neurotoxicol. Teratol. 2010, 32, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Aday, S.; Cecchelli, R.; Hallier-Vanuxeem, D.; Dehouck, M.P.; Ferreira, L. Stem Cell-Based Human Blood-Brain Barrier Models for Drug Discovery and Delivery. Trends Biotechnol. 2016, 34, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Zehendner, C.M.; White, R.; Hedrich, J.; Luhmann, H.J. A neurovascular blood-brain barrier in vitro model. Methods Mol. Biol. 2014, 1135, 403–413. [Google Scholar] [PubMed]
- Wheeler, H.E.; Wing, C.; Delaney, S.M.; Komatsu, M.; Dolan, M.E. Modeling chemotherapeutic neurotoxicity with human induced pluripotent stem cell-derived neuronal cells. PLoS ONE 2015, 10, e0118020. [Google Scholar] [CrossRef] [PubMed]
- Kamao, H.; Mandai, M.; Okamoto, S.; Sakai, N.; Suga, A.; Sugita, S.; Kiryu, J.; Takahashi, M. Characterization of human induced pluripotent stem cell-derived retinal pigment epithelium cell sheets aiming for clinical application. Stem Cell Rep. 2014, 2, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Sayed, N.; Liu, C.; Wu, J.C. Translation of Human-Induced Pluripotent Stem Cells: From Clinical Trial in a Dish to Precision Medicine. J. Am. Coll. Cardiol. 2016, 67, 2161–2176. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Chaudhury, S. Statistics without tears: Populations and samples. Ind. Psychiatry J. 2010, 19, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Kunisada, Y.; Tsubooka-Yamazoe, N.; Shoji, M.; Hosoya, M. Small molecules induce efficient differentiation into insulin-producing cells from human induced pluripotent stem cells. Stem Cell Res. 2012, 8, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Baden, M.Y.; Fukui, K.; Hosokawa, Y.; Iwahashi, H.; Imagawa, A.; Shimomura, I. Examination of a Viral Infection Mimetic Model in Human iPS Cell-Derived Insulin-Producing Cells and the Anti-Apoptotic Effect of GLP-1 Analogue. PLoS ONE 2015, 10, e0144606. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Author/Year/Title | Expression of Stage-Specific Markers in Human iPS Cells | Toxic Compounds Tested | Biomarkers Tested | Comments | |
|---|---|---|---|---|---|
| Hepatotoxicity | Ware et al. 2015 [98]; Prediction of Drug-Induced Liver Injury in Micropatterned Co-cultures Containing iPSC-Derived Human Hepatocytes | CYP3A4 activity corresponding to ~90% of primary hepatocytes cultured for 24 h in vitro Albumin to alpha-fetoprotein ratio 12.2 at day 21 of culture | 47 compounds were segregated into three groups based on previous study performed on primary hepatocytes (hepatotoxic, non-hepatotoxic, and compounds previously incorrectly classified as non-toxic). | Albumin secretion (ELISA); Urea production | Micropatterned co-culture system of iPSC-hepatocytes with fibroblasts prolonged liver hepatic functions up to 4 weeks when compared with single culture condition. Co-cultured micropatterned hepatic cells showed predictive DILI capabilities of 65-70% and 100% for sensitivity and specificity, respectively. Changes in urea production was the most sensitive assay endpoint |
| Tasnim et al. 2016 [97]; Functionally Enhanced Human Stem Cell Derived Hepatocytes in Galactosylated Cellulosic Sponges for Hepatotoxicity Testing | AFP, ALB, AAT, HNF4a, CYP3A4, CYP3A7, CYP1A1, CYP1A2, ASGPR, MRP2 (qRT-PCR) Urea and Albumin production CYP induction (LC-MS) | APAP, Troglitazone, Methotrexate (24 h exposure) | Cell viability | Cellulosic scaffolds used during final stage of maturation enhanced hPSC-hepatocyte functions, including CYP activity and sensitivity to methotrexate Expression of alpha-fetoprotein was higher than albumin at Day 20 and 32 in both 2D and 3D cultures, however the albumin to alpha-fetoprotein ratio was the highest at days 32 in the 3D cultured | |
| Cardiotoxicity | Liang et al. 2013 [115]; Drug Screening Using a Library of Human Induced Pluripotent Stem Cell–Derived Cardiomyocytes Reveals Disease-Specific Patterns of Cardiotoxicity | Troponin T (FACS) Expression of cardiac ion channel: SCN5A, KCND3, CACNA1C, KCNH2, KCNQ1, KCNA5, HCN2, HCN4, KCNJ2, KCNJ3, KCNJ5, KCNJ11, KCNE1, KChIP2 (qRT-PCR) | Verpamil, Alfuzosin, Cisapride Nicorandil | CM/AP assay (compound muscle action potential) | hiPSC-derived cardiomyocytes were shown to model cardiotoxicity more accurately than commercially available hERG cell lines |
| Nephrotoxicity | Morizane et al. 2015 [129]; Nephron organoids derived from human pluripotent stem cells model kidney development and injury | NPCs: 90% of NPC were positive for SIX2, SALL1, WT1, and PAX2; NPC-derived renal vesicles : 76% were positive for PAX8 and LHX1 segmental markers in nephron—like continuous structures: (A) glomerular podocytes: NPHS1 and PODXL; (B) proximal tubules: LTL and CDH2, (C) loops of Henle/distal tubules: E-Cad/CDH1, UMOD and BRN1 | Nephrotoxicants tested on hESCs-derived 3D kidney organoids: Gentamycin (48 h, at 5 mg/mL) Cisplatin (2,6,24 or 48 h; at 5 μM, 50 μM) | KIM-1, LTL, E-Cad (CDH1) (ICC, qRT-PCR) | In gentamicin-treated organoids KIM-1 was expressed at the luminal surface of LTL-positive tubules but was not detected in E-Cad/CDH1-positive cells. qRT-PCR analysis showed gentamycin-caused dose-dependent upregulation of this marker. Cisplatin upregulated KIM-1 expression in LTL-positive cells but also suppressed E-Cad/CDH1 expression, indicating both proximal and distal tubular toxicity |
| Kandasamy et al. 2015 [119]; Prediction of drug-induced nephrotoxicity and injury mechanisms with human induced pluripotent stem cell-derived cells and machine learning methods | Proximal tubular-like cells expressed e.g., SIX2, WT1, GDNF, HOXD11, KSP-CAD, AQP1, OAT3, GGT, and other markers expressed along proximal tubular cell development; however some of the main stemness markers were highly also expressed | Nephrotoxicants tested: Aristolochic acid, Arsenic (III) oxide, Bismuth (III) oxide, Cadmium chloride, Cephalosporin C, Cisplatin, Citrinin, Copper (II) chloride, 5-Fluorouracil, Gentamicin, Gold (I) chloride, Lead acetate, Paraquat, Potassium dichromate, Puromycin, Rifampicin, Tacrolimus, Tobramycin; (16 h exposure, at 1, 10, 1000 μg/mL) | IL-6, IL-8 (qRT-PCR, normalized to GAPDH and PPIA) | Nephrotoxicity response in iPSC-derived HPTC-like cells were compared to the corresponding dataset from previous study on cultured human primary HPTC cells Comparative automated unbiased data analysis showed 99.8% and 87.0% training balanced accuracy and test balanced accuracy, respectively | |
| Neurotoxicity | Wheeler et al. 2015 [137]; Modeling Chemotherapeutic Neurotoxicity with Human Induced Pluripotent Stem Cell-Derived Neuronal Cells | Cortical neurons were defined as Tuj1-positive and Nestin-negative (ICC) | 4 chemotherapeutics: Cisplatin, Paclitaxel, Vincristine, Hydroxyurea; (72 h exposure, at 0.001–100 μM) | Neurite outgrowth response upon chemotherapeutic treatment | The differences between selected paclitaxel-resistant and paclitaxel-sensitive LCL-derived neurons were significant but only partially correlated with the patient’s initial sensitivity to this chemotherapeutic Reduced TUBB2A sensitized iPSC-derived neurons to paclitaxel |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hosoya, M.; Czysz, K. Translational Prospects and Challenges in Human Induced Pluripotent Stem Cell Research in Drug Discovery. Cells 2016, 5, 46. https://doi.org/10.3390/cells5040046
Hosoya M, Czysz K. Translational Prospects and Challenges in Human Induced Pluripotent Stem Cell Research in Drug Discovery. Cells. 2016; 5(4):46. https://doi.org/10.3390/cells5040046
Chicago/Turabian StyleHosoya, Masaki, and Katherine Czysz. 2016. "Translational Prospects and Challenges in Human Induced Pluripotent Stem Cell Research in Drug Discovery" Cells 5, no. 4: 46. https://doi.org/10.3390/cells5040046
APA StyleHosoya, M., & Czysz, K. (2016). Translational Prospects and Challenges in Human Induced Pluripotent Stem Cell Research in Drug Discovery. Cells, 5(4), 46. https://doi.org/10.3390/cells5040046