Dupuytren’s and Ledderhose Diseases in a Family with LMNA-Related Cardiomyopathy and a Novel Variant in the ASTE1 Gene

Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Family
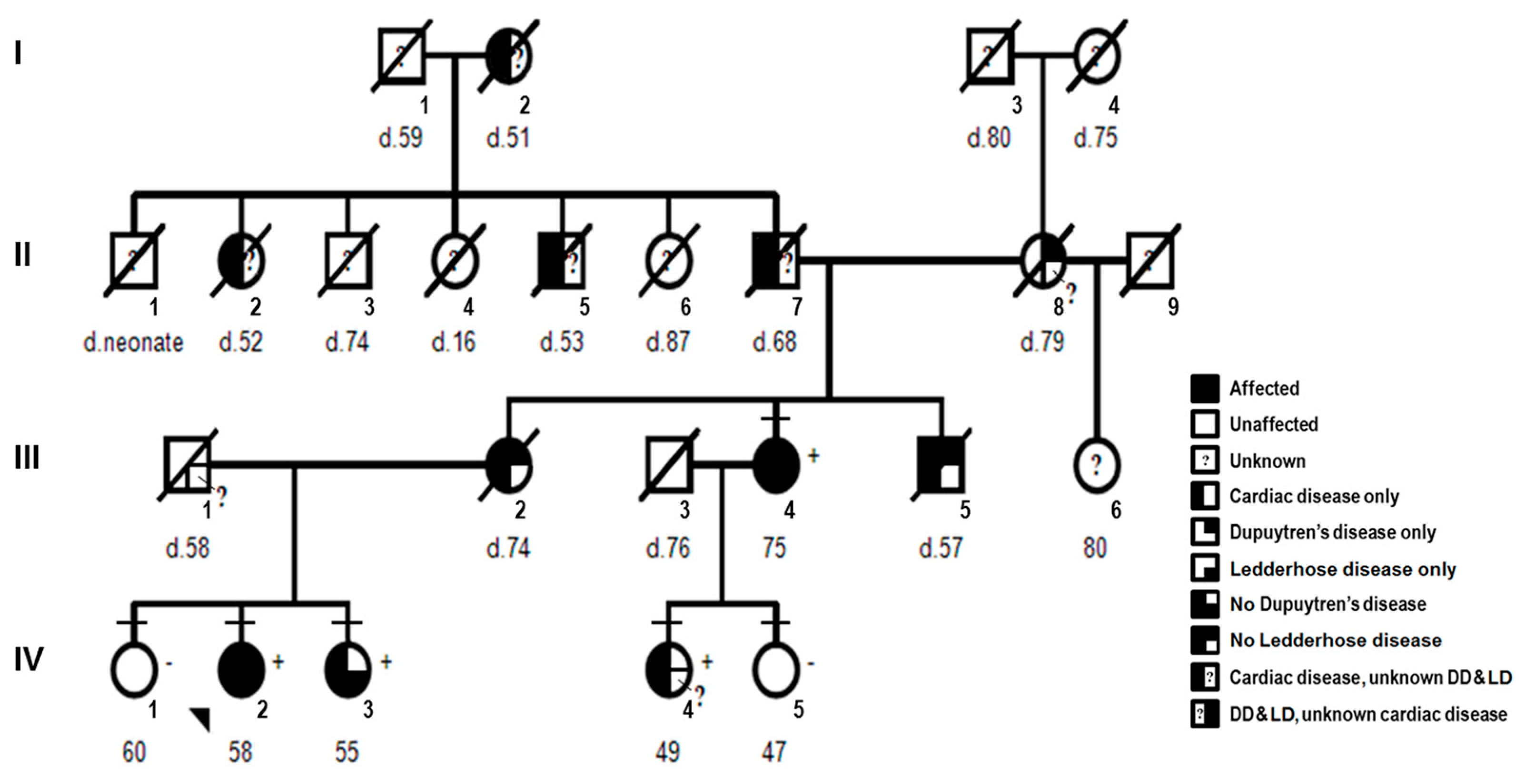
2.1.1. Pedigree and Consent
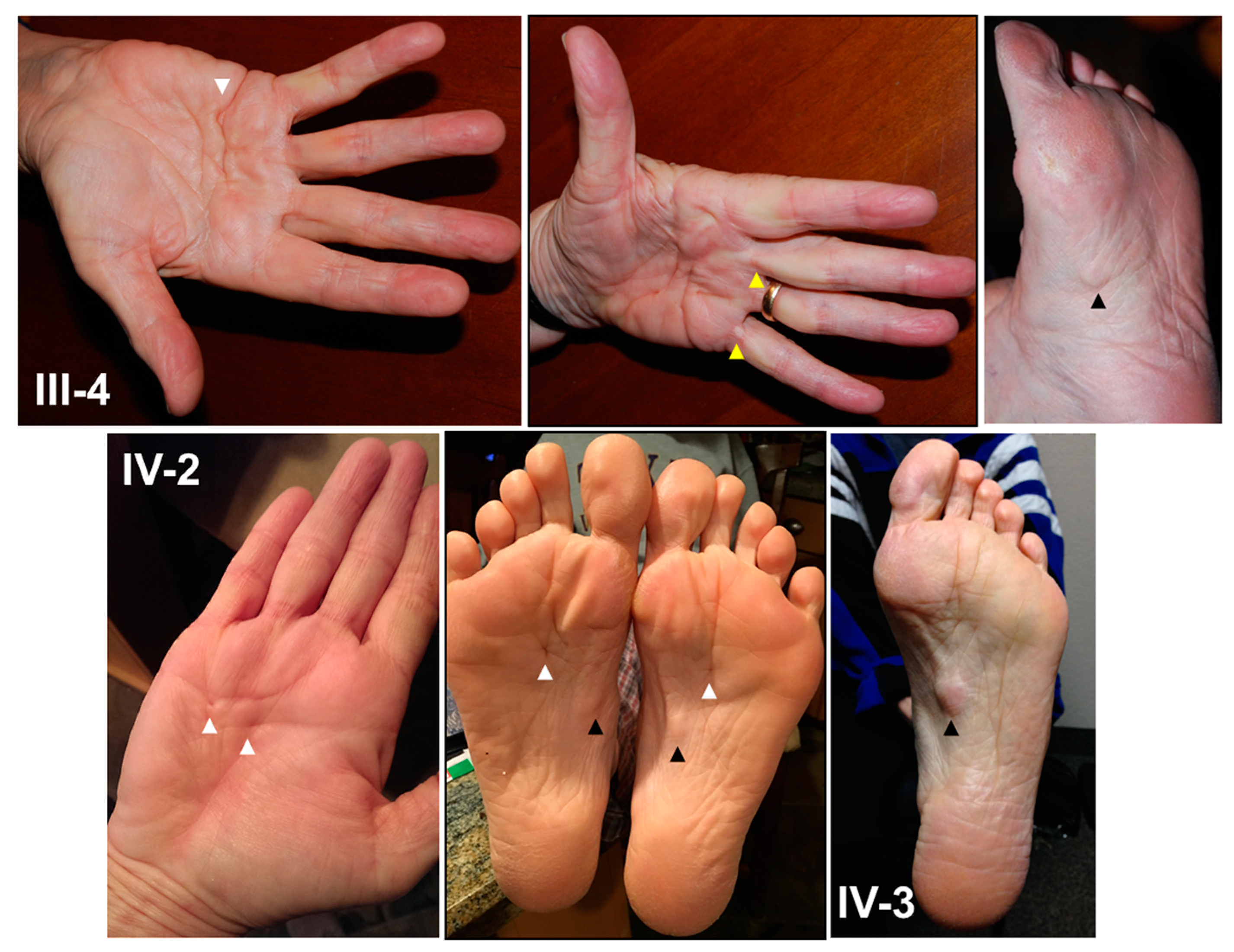
2.1.2. Clinical Features
2.1.3. Proband
2.1.4. Affected Family Members
2.2. Molecular Studies
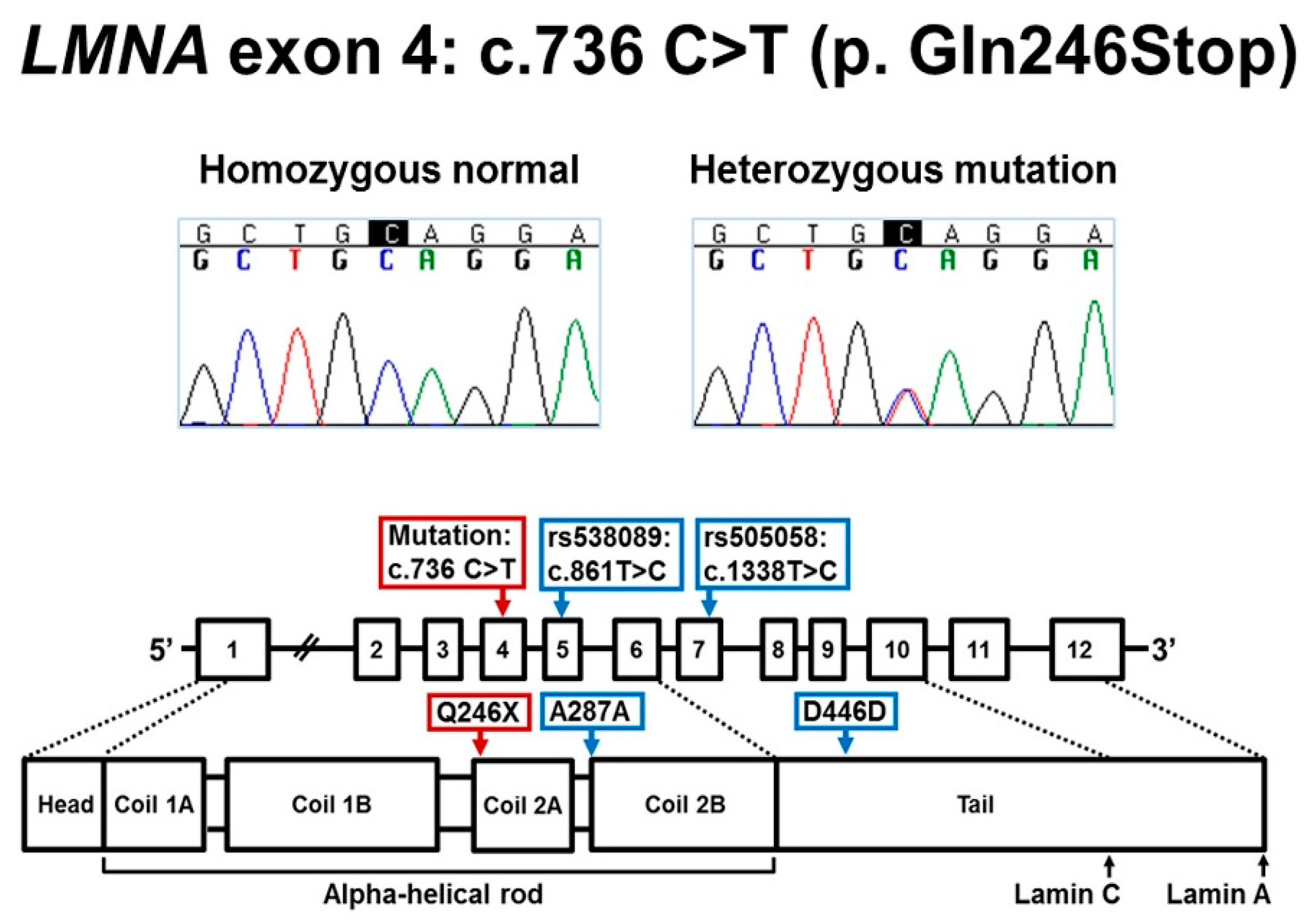
2.2.1. Sanger Sequencing for LMNA Exon 4
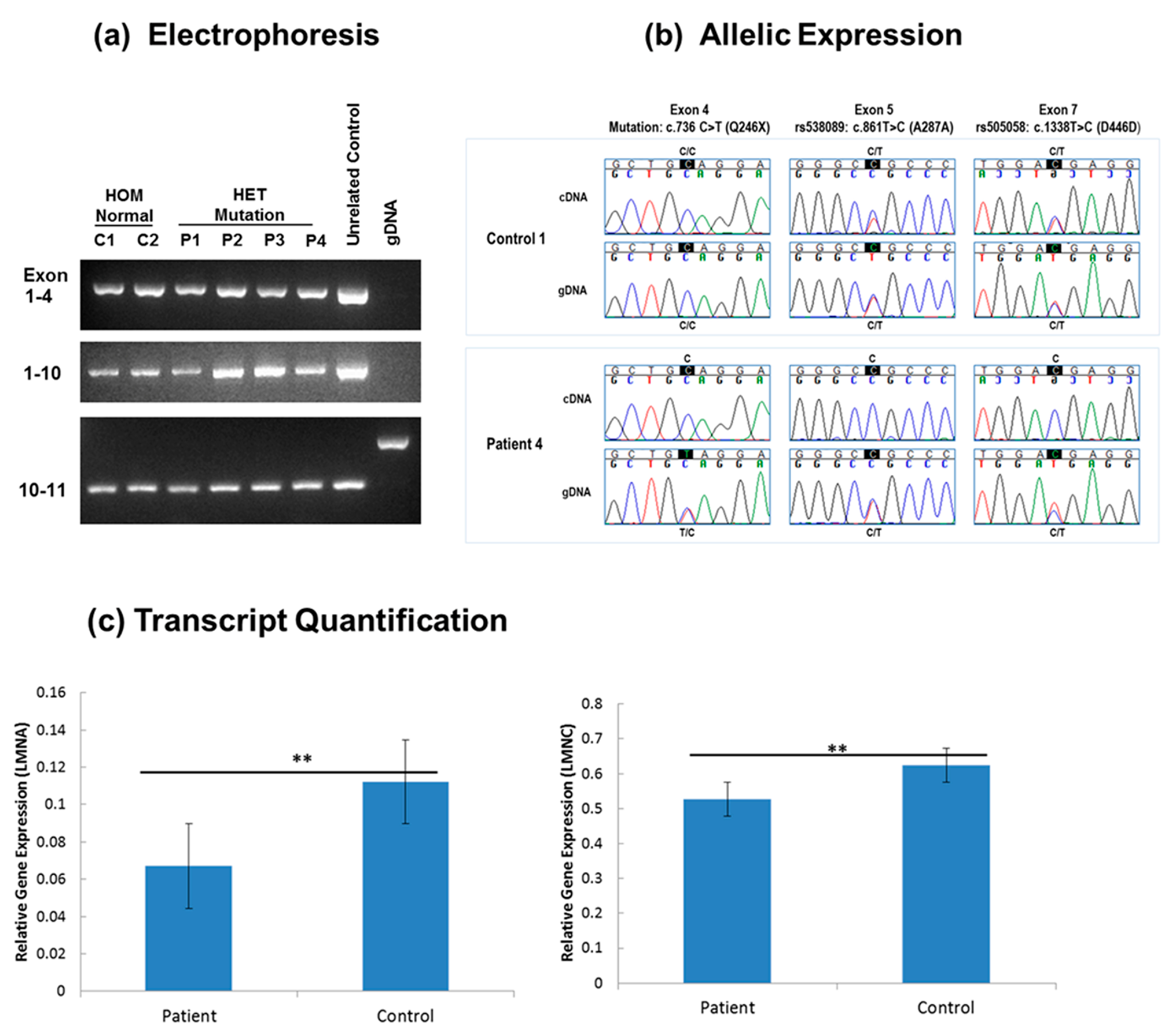
2.2.2. LMNA RNA and DNA Studies in Fibroblast
2.2.3. Exome Sequencing, Bioinformatics Analysis, and Variant Filtering
2.2.4. Family Studies
3. Results
3.1. LMNA DNA and RNA Studies
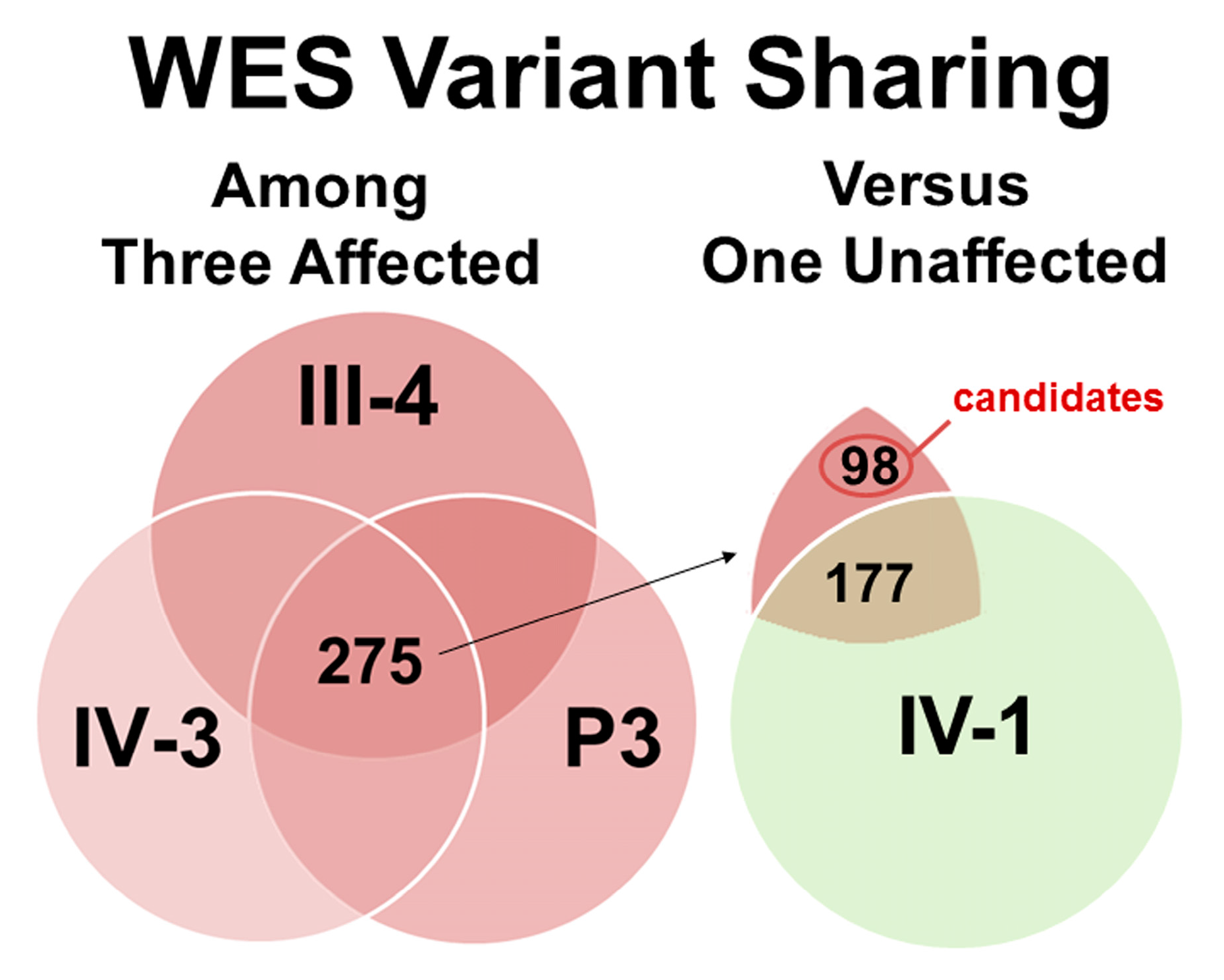
3.2. Exome Sequencing and Co-Segregation for DD/LD-Associated Variants
4. Discussion
4.1. Molecular Mechanism of LMNA c.736C>T (p.Gln246Stop)
4.2. Dupuytren’s and Ledderhose Diseases as Primary Features of Laminopathies?
4.3. Identification of a Candidate Susceptibility Variant for DD and LD in the ASTE1 Gene
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dupuytren, G. Clinical Lectures on Surgery delivered by Baron Dupuytren, during the Session of 1833. Lancet 1834, 2, 222–225. [Google Scholar]
- Tripoli, M.; Cordova, A.; Moschella, F. Update on the role of molecular factors and fibroblasts in the pathogenesis of Dupuytren’s disease. J. Cell Commun. Signal. 2016, 10, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Akhavani, M.A.; McMurtrie, A.; Webb, M.; Muir, L. A review of the classification of Dupuytren’s disease. J. Hand Surg. 2015, 40, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Ledderhose, G. Über Zerreisungen der Plantarfascie. Arch. Klin. Chir. 1894, 48, 853–856. [Google Scholar]
- Pickren, J.W.; Smith, A.G.; Stevenson, T.W., Jr.; Stout, A.P. Fibromatosis of the plantar fascia. Cancer 1951, 4, 846–856. [Google Scholar] [CrossRef]
- Veith, N.T.; Tschernig, T.; Histing, T.; Madry, H. Plantar fibromatosis—Topical review. Foot Ankle Int. 2013, 34, 1742–1746. [Google Scholar] [CrossRef] [PubMed]
- Michou, L.; Lermusiaux, J.L.; Teyssedou, J.P.; Bardin, T.; Beaudreuil, J.; Petit-Teixeira, E. Genetics of Dupuytren’s disease. Jt. Bone Spine 2012, 79, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.T.; Van Dyne, T.A. Familial plantar fibromatosis. J. Surg. Oncol. 1985, 29, 240–241. [Google Scholar] [CrossRef] [PubMed]
- De Palma, L.; Santucci, A.; Gigante, A.; Di Giulio, A.; Carloni, S. Plantar fibromatosis: An immunohistochemical and ultrastructural study. Foot Ankle Int. 1999, 20, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.Z.; Nystrom, A.; Ahmed, A.; Palmquist, M.; Dopico, R.; Mossberg, I.; Gladitz, J.; Rayner, M.; Post, J.C.; Ehrlich, G.D.; et al. Mapping of an autosomal dominant gene for Dupuytren’s contracture to chromosome 16q in a Swedish family. Clin. Genet. 2005, 68, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Worman, H.J. Nuclear lamins and laminopathies. J. Pathol. 2012, 226, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Dechat, T.; Adam, S.A.; Taimen, P.; Shimi, T.; Goldman, R.D. Nuclear lamins. Cold Spring Harb. Perspect. Biol. 2010, 2, a000547. [Google Scholar] [CrossRef] [PubMed]
- Perovanovic, J.; Dell’Orso, S.; Gnochi, V.F.; Jaiswal, J.K.; Sartorelli, V.; Vigouroux, C.; Mamchaoui, K.; Mouly, V.; Bonne, G.; Hoffman, E.P. Laminopathies disrupt epigenomic developmental programs and cell fate. Sci. Transl. Med. 2016, 8, 335ra358. [Google Scholar] [CrossRef] [PubMed]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008, 453, 948–951. [Google Scholar] [CrossRef] [PubMed]
- Muchir, A.; Pavlidis, P.; Decostre, V.; Herron, A.J.; Arimura, T.; Bonne, G.; Worman, H.J. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J. Clin. Investig. 2007, 117, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Van Berlo, J.H.; Voncken, J.W.; Kubben, N.; Broers, J.L.; Duisters, R.; van Leeuwen, R.E.; Crijns, H.J.; Ramaekers, F.C.; Hutchison, C.J.; Pinto, Y.M. A-type lamins are essential for TGF-beta1 induced PP2A to dephosphorylate transcription factors. Hum. Mol. Genet. 2005, 14, 2839–2849. [Google Scholar] [CrossRef] [PubMed]
- Le Dour, C.; Macquart, C.; Sera, F.; Homma, S.; Bonne, G.; Morrow, J.P.; Worman, H.J.; Muchir, A. Decreased WNT/beta-catenin signalling contributes to the pathogenesis of dilated cardiomyopathy caused by mutations in the lamin a/C gene. Hum. Mol. Genet. 2017, 26, 333–343. [Google Scholar] [PubMed]
- Luo, Y.B.; Mastaglia, F.L. Wilton SD Normal and aberrant splicing of LMNA. J. Med. Genet. 2014, 51, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Renard, D.; Fourcade, G.; Milhaud, D.; Bessis, D.; Esteves-Vieira, V.; Boyer, A.; Roll, P.; Bourgeois, P.; Levy, N.; De Sandre-Giovannoli, A. Novel LMNA mutation in atypical Werner syndrome presenting with ischemic disease. Stroke 2009, 40, e11–e14. [Google Scholar] [CrossRef] [PubMed]
- Lace, B.; Inashkina, I.; Micule, I.; Vasiljeva, I.; Naudina, M.S.; Strautmanis, J.; Stavusis, J.; Jankevics, E. Dupuytren’s Contracture Cosegregation with Limb-Girdle Muscle Dystrophy. Case Rep. Neurol. Med. 2013, 2013, 254950. [Google Scholar] [CrossRef] [PubMed]
- Zaragoza, M.V.; Fung, L.; Jensen, E.; Oh, F.; Cung, K.; McCarthy, L.A.; Tran, C.K.; Hoang, V.; Hakim, S.A.; Grosberg, A. Exome Sequencing Identifies a Novel LMNA Splice-Site Mutation and Multigenic Heterozygosity of Potential Modifiers in a Family with Sick Sinus Syndrome, Dilated Cardiomyopathy, and Sudden Cardiac Death. PLoS ONE 2016, 11, e0155421. [Google Scholar] [CrossRef] [PubMed]
- Zaragoza, M.V.; Hakim, S.A.; Hoang, V. Elliott AM Heart-hand syndrome IV: A second family with LMNA-related cardiomyopathy and brachydactyly. Clin. Genet. 2017, 91, 499–500. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC browser: Displaying reference data information from over 60,000 exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef] [PubMed]
- Pasotti, M.; Klersy, C.; Pilotto, A.; Marziliano, N.; Rapezzi, C.; Serio, A.; Mannarino, S.; Gambarin, F.; Favalli, V.; Grasso, M.; et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J. Am. Coll. Cardiol. 2008, 52, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Saj, M.; Bilinska, Z.T.; Tarnowska, A.; Sioma, A.; Bolongo, P.; Sobieszczanska-Malek, M.; Michalak, E.; Golen, D.; Mazurkiewicz, L.; Malek, L.; et al. LMNA mutations in Polish patients with dilated cardiomyopathy: Prevalence, clinical characteristics, and in vitro studies. BMC Med. Genet. 2013, 14, 55. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Morales, A. Dilated Cardiomyopathy Overview. In GeneReviews(R); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mefford, H.C., Stephens, K., Amemiya, A., Ledbetter, N., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Narula, N.; Favalli, V.; Tarantino, P.; Grasso, M.; Pilotto, A.; Bellazzi, R.; Serio, A.; Gambarin, F.I.; Charron, P.; Meder, B.; et al. Quantitative expression of the mutated lamin A/C gene in patients with cardiolaminopathy. J. Am. Coll. Cardiol. 2012, 60, 1916–1920. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.C.; Muchir, A.; Wu, W.; Iwata, S.; Homma, S.; Morrow, J.P.; Worman, H.J. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci. Transl. Med. 2012, 4, 144ra102. [Google Scholar] [CrossRef] [PubMed]
- Chatzifrangkeskou, M.; Le Dour, C.; Wu, W.; Morrow, J.P.; Joseph, L.C.; Beuvin, M.; Sera, F.; Homma, S.; Vignier, N.; Mougenot, N.; et al. ERK1/2 directly acts on CTGF/CCN2 expression to mediate myocardial fibrosis in cardiomyopathy caused by mutations in the lamin A/C gene. Hum. Mol. Genet. 2016, 25, 2220–2233. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, P.; Chaturvedi, P.; Kumaran, R.I.; Kumar, S.; Parnaik, V.K. Lamin A/C haploinsufficiency modulates the differentiation potential of mouse embryonic stem cells. PLoS ONE 2013, 8, e57891. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1–30. [Google Scholar]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Apweiler, R.; Alpi, E.; Antunes, R.; Arganiska, J.; Bely, B.; Bingley, M.; et al. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar]
- Kotarski, M.A.; Leonard, D.A.; Bennett, S.A.; Bishop, C.P.; Wahn, S.D.; Sedore, S.A.; Shrader, M. The Drosophila gene asteroid encodes a novel protein and displays dosage-sensitive interactions with Star and Egfr. Genome 1998, 41, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.Y.; Dosztányi, Z.; El-Gebali, S.; Fraser, M.; et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef] [PubMed]
- OMIM Online Mendelian Inheritance in Man. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University: Baltimore, MD, USA. Available online: https://omim.org/ (accessed on 6 October 2017).
- Tougeron, D.; Fauquembergue, E.; Rouquette, A.; Le Pessot, F.; Sesboue, R.; Laurent, M.; Berthet, P.; Mauillon, J.; Di Fiore, F.; Sabourin, J.C.; et al. Tumor-infiltrating lymphocytes in colorectal cancers with microsatellite instability are correlated with the number and spectrum of frameshift mutations. Mod. Pathol. 2009, 22, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- Malnic, B.; Godfrey, P.A.; Buck, L.B. The human olfactory receptor gene family. Proc. Natl. Acad. Sci. USA 2004, 101, 2584–2589. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Shilo, B.Z. SnapShot: EGFR signaling pathway. Cell 2007, 131, 1018. [Google Scholar] [CrossRef] [PubMed]
- Magro, G.; Lanzafame, S.; Micali, G. Co-ordinate expression of alpha 5 beta 1 integrin and fibronectin in Dupuytren’s disease. Acta Histochem. 1995, 97, 229–233. [Google Scholar] [CrossRef]
- Kloen, P.; Jennings, C.L.; Gebhardt, M.C.; Springfield, D.S.; Mankin, H.J. Transforming growth factor-beta: Possible roles in Dupuytren’s contracture. J. Hand Surg. Am. 1995, 20, 101–108. [Google Scholar] [CrossRef]
- Augoff, K.; Tabola, R.; Kula, J.; Gosk, J.; Rutowski, R. Epidermal growth factor receptor (EGF-R) in Dupuytren’s disease. J. Hand Surg. Br. 2005, 30, 570–573. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene b | Position (hg38) | DNA Variant | Protein Change | dbSNP c | ExAC Frequency d | Family Studies: Genotype e | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| III-4 | IV-2 | IV-3 | P3 | IV-1 | ||||||
| ASTE1 | Chr3:131025077 | c.230T>C | p.Val77Ala | novel | No data | −/+ | −/+ | −/+ | −/+ | −/− |
| FZD2 | Chr17:44558223 | c.535A>C | p.Thr179Pro | novel | No data | −/− | −/− | −/− | −/− | −/− |
| FSD1 | Chr19:4311959 | c.608G>A | p.Arg203Gln | novel | No data | −/+ | −/− | −/+ | −/+ | −/− |
| LMNA | Chr1:156134901 | c.736C>T | p.Gln246Ter | rs267607587 | No data | −/+ | −/+ | −/+ | −/+ | −/− |
| OR51A7 | Chr11:4908132 | c.763A>G | p.Ile255Val | rs144609747 | 0.0040 | −/+ | −/+ | −/+ | −/+ | −/− |
| SYPL2 | Chr1:109476793 | c.272A>T | p.Tyr91Phe | rs79613472 | 0.0086 | −/+ | −/+ | −/+ | −/+ | −/+ |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaragoza, M.V.; Nguyen, C.H.H.; Widyastuti, H.P.; McCarthy, L.A.; Grosberg, A. Dupuytren’s and Ledderhose Diseases in a Family with LMNA-Related Cardiomyopathy and a Novel Variant in the ASTE1 Gene. Cells 2017, 6, 40. https://doi.org/10.3390/cells6040040
Zaragoza MV, Nguyen CHH, Widyastuti HP, McCarthy LA, Grosberg A. Dupuytren’s and Ledderhose Diseases in a Family with LMNA-Related Cardiomyopathy and a Novel Variant in the ASTE1 Gene. Cells. 2017; 6(4):40. https://doi.org/10.3390/cells6040040
Chicago/Turabian StyleZaragoza, Michael V., Cecilia H. H. Nguyen, Halida P. Widyastuti, Linda A. McCarthy, and Anna Grosberg. 2017. "Dupuytren’s and Ledderhose Diseases in a Family with LMNA-Related Cardiomyopathy and a Novel Variant in the ASTE1 Gene" Cells 6, no. 4: 40. https://doi.org/10.3390/cells6040040
APA StyleZaragoza, M. V., Nguyen, C. H. H., Widyastuti, H. P., McCarthy, L. A., & Grosberg, A. (2017). Dupuytren’s and Ledderhose Diseases in a Family with LMNA-Related Cardiomyopathy and a Novel Variant in the ASTE1 Gene. Cells, 6(4), 40. https://doi.org/10.3390/cells6040040