Mitochondrial Quality Control Mechanisms and the PHB (Prohibitin) Complex


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mitochondrial Stress Responses
2.1. Mitochondrial Dynamics
2.2. Mitophagy
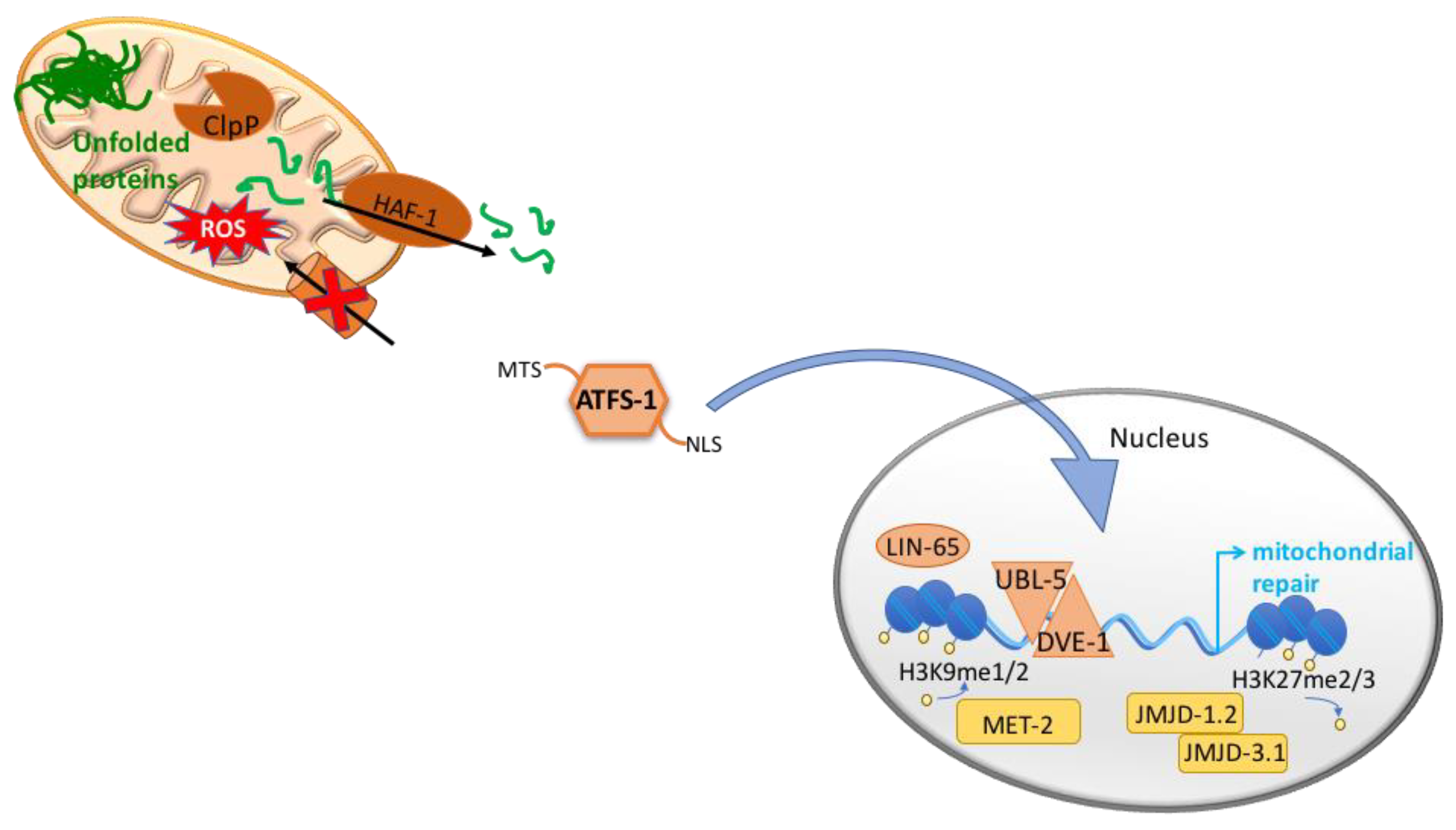
2.3. The Mitochondrial Unfolded Protein Response, UPRmt
2.4. Cytosolic Responses Reacting to Mitochondrial Proteotoxic Stress
3. Mitochondrial Unfolded Protein Response and Its Impact on Aging
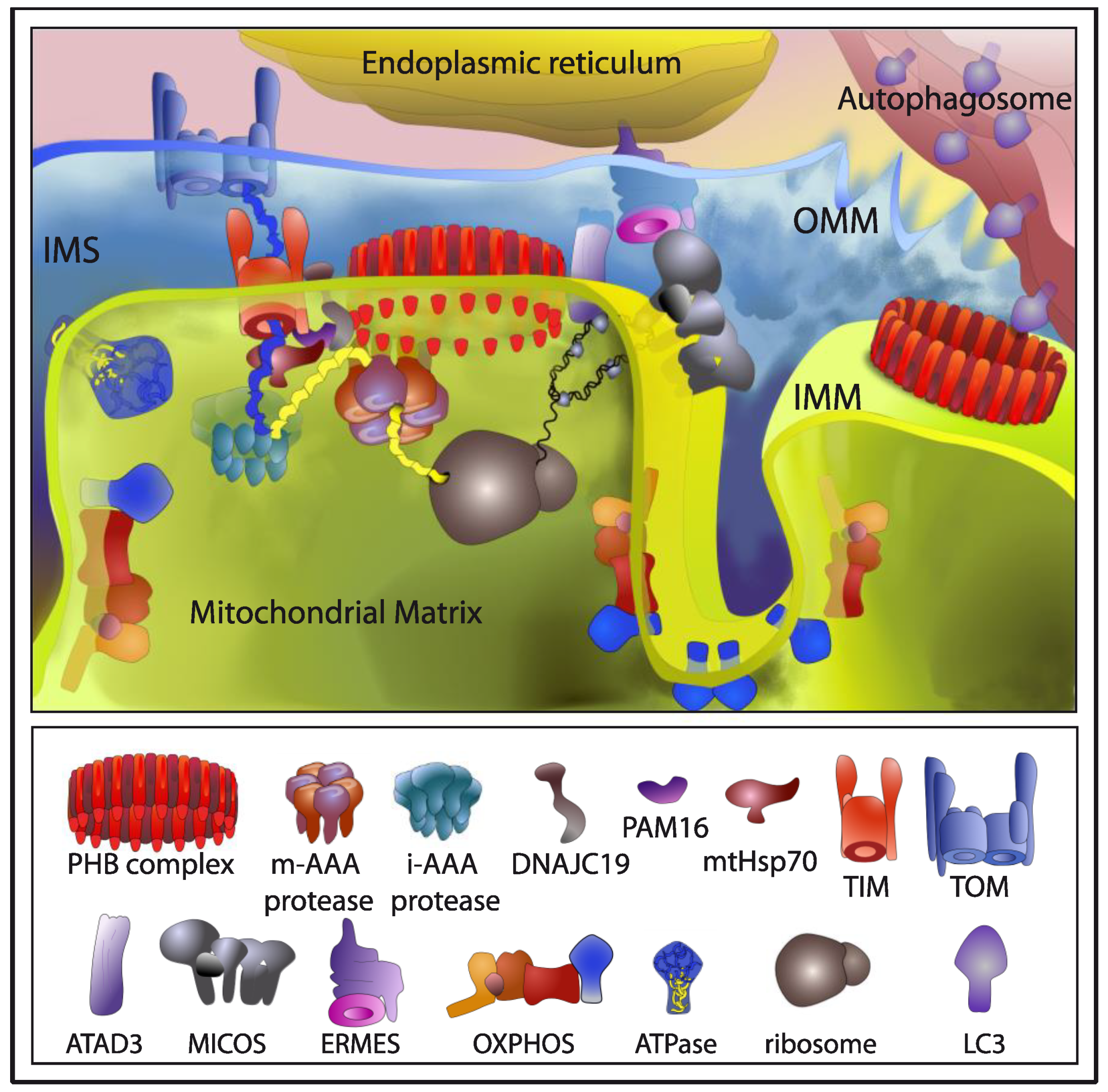
4. Mitochondrial Prohibitins, Key Players in Mitochondrial Quality Control Mechanisms
4.1. The PHB Complex in Mitochondrial Turnover
4.2. The PHB Complex Responds to Mitochondrial Stress
4.3. The PHB Complex and Lifespan
5. Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Eisenberg-Bord, M.; Shai, N.; Schuldiner, M.; Bohnert, M. A tether is a tether is a tether: Tethering at membrane contact sites. Dev. Cell 2016, 39, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.C.; Uchiyama, L.F.; Nunnari, J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 2016, 353, aaf5549. [Google Scholar] [CrossRef] [PubMed]
- Diogo, C.V.; Yambire, K.F.; Mosquera, L.F.; Branco, F.T.; Raimundo, N. Mitochondrial adventures at the organelle society. Biochem. Biophys. Res. Commun. 2018, 500, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.B.; Louie, S.M.; Daniele, J.R.; Tran, Q.; Dillin, A.; Zoncu, R.; Nomura, D.K.; Olzmann, J.A. DGAT1-Dependent Lipid Droplet Biogenesis Protects Mitochondrial Function during Starvation-Induced Autophagy. Dev. Cell 2017, 42, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, A.; Mattie, S.; Prudent, J.; McBride, H.M. Newly born peroxisomes are a hybrid of mitochondrial and ER-derived pre-peroxisomes. Nature 2017, 542, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A. Biogenesis of iron-sulfur clusters in mammalian cells: new insights and relevance to human disease. Dis. Model Mech. 2012, 5, 155–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca(2+) homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C. Mitochondrial metabolites: Undercover signalling molecules. Interface Focus 2017, 7, 20160100. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Quiros, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Harbauer, A.B.; Zahedi, R.P.; Sickmann, A.; Pfanner, N.; Meisinger, C. The protein import machinery of mitochondria-a regulatory hub in metabolism, stress, and disease. Cell Metab. 2014, 19, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Tatsuta, T.; Scharwey, M.; Langer, T. Mitochondrial lipid trafficking. Trends Cell Biol. 2014, 24, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Tavernarakis, N.; Driscoll, M.; Kyrpides, N.C. The SPFH domain: Implicated in regulating targeted protein turnover in stomatins and other membrane-associated proteins. Trends Biochem. Sci. 1999, 24, 425–427. [Google Scholar] [CrossRef]
- Browman, D.T.; Hoegg, M.B.; Robbins, S.M. The SPFH domain-containing proteins: More than lipid raft markers. Trends Cell Biol. 2007, 17, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Artal-Sanz, M.; Tavernarakis, N. Prohibitin and mitochondrial biology. Trends Endocrinol. Metab. 2009, 20, 394–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artal-Sanz, M.; Tavernarakis, N. Prohibitin couples diapause signalling to mitochondrial metabolism during ageing in C. elegans. Nature 2009, 461, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, A.B.; Munoz-Jimenez, C.; Venegas-Caleron, M.; Artal-Sanz, M. Analysis of the effect of the mitochondrial prohibitin complex, a context-dependent modulator of longevity, on the C. elegans metabolome. Biochim. Biophys. Acta 2015, 1847, 1457–1468. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Benedetti, C.; Urano, F.; Clark, S.G.; Harding, H.P.; Ron, D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J. Cell Sci. 2004, 117, 4055–4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, C.F.; Vander Wende, H.; Simko, M.; Klum, S.; Barfield, S.; Choi, H.; Pineda, V.V.; Kaeberlein, M. Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nat. Commun. 2014, 5, 3483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatsi, R.; Schulze, B.; Rodriguez-Palero, M.J.; Hernando-Rodriguez, B.; Baumeister, R.; Artal-Sanz, M. Prohibitin-mediated lifespan and mitochondrial stress implicate SGK-1, insulin/IGF and mTORC2 in C. elegans. PLoS ONE 2014, 9, e107671. [Google Scholar] [CrossRef] [PubMed]
- Hernando-Rodriguez, B.; Erinjeri, A.P.; Rodriguez-Palero, M.J.; Millar, V.; Gonzalez-Hernandez, S.; Olmedo, M.; Schulze, B.; Baumeister, R.; Munoz, M.J.; Askjaer, P.; et al. Combined flow cytometry and high-throughput image analysis for the study of essential genes in Caenorhabditis elegans. BMC Biol. 2018, 16, 36. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Labrousse, A.M.; Zappaterra, M.D.; Rube, D.A.; van der Bliek, A.M. C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol. Cell 1999, 4, 815–826. [Google Scholar] [CrossRef]
- Rolland, S.G.; Lu, Y.; David, C.N.; Conradt, B. The BCL-2-like protein CED-9 of C. elegans promotes FZO-1/Mfn1,2- and EAT-3/Opa1-dependent mitochondrial fusion. J. Cell Biol. 2009, 186, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, T.; Zappaterra, M.D.; Hasegawa, A.; Wright, A.P.; Newman-Smith, E.D.; Buttle, K.F.; McDonald, K.; Mannella, C.A.; van der Bliek, A.M. The C. elegans Opa1 homologue EAT-3 is essential for resistance to free radicals. PLoS Genet. 2008, 4, e1000022. [Google Scholar] [CrossRef] [PubMed]
- Luz, A.L.; Rooney, J.P.; Kubik, L.L.; Gonzalez, C.P.; Song, D.H.; Meyer, J.N. Mitochondrial Morphology and Fundamental Parameters of the Mitochondrial Respiratory Chain Are Altered in Caenorhabditis elegans Strains Deficient in Mitochondrial Dynamics and Homeostasis Processes. PLoS ONE 2015, 10, e0130940. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, S.N.; Kipreos, E.T. Increased mitochondrial fusion allows the survival of older animals in diverse C. elegans longevity pathways. Nat. Commun. 2017, 8, 182. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Chen, D.; Lee, S.S.; Walter, L. The dynamin-related protein DRP-1 and the insulin signaling pathway cooperate to modulate Caenorhabditis elegans longevity. Aging Cell 2011, 10, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.M.; Nargund, A.M.; Sun, T.; Haynes, C.M. Protective coupling of mitochondrial function and protein synthesis via the eIF2alpha kinase GCN-2. PLoS Genet. 2012, 8, e1002760. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.M.; Williams, J.A.; Ding, W.X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell Biol. 2016, 26, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 2015, 521, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Youle, R.J. PINK1 import regulation: A fine system to convey mitochondrial stress to the cytosol. BMC Biol. 2018, 16, 2. [Google Scholar] [CrossRef] [PubMed]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, M.; Sato, K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science 2011, 334, 1141–1144. [Google Scholar] [CrossRef] [PubMed]
- Al Rawi, S.; Louvet-Vallee, S.; Djeddi, A.; Sachse, M.; Culetto, E.; Hajjar, C.; Boyd, L.; Legouis, R.; Galy, V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 2011, 334, 1144–1147. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chiang, W.C.; Sumpter, R., Jr.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238. [Google Scholar] [CrossRef] [PubMed]
- Sharpley, M.S.; Marciniak, C.; Eckel-Mahan, K.; McManus, M.; Crimi, M.; Waymire, K.; Lin, C.S.; Masubuchi, S.; Friend, N.; Koike, M.; et al. Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell 2012, 151, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Politi, Y.; Gal, L.; Kalifa, Y.; Ravid, L.; Elazar, Z.; Arama, E. Paternal mitochondrial destruction after fertilization is mediated by a common endocytic and autophagic pathway in Drosophila. Dev. Cell 2014, 29, 305–320. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Li, H.; Li, H.; Nakagawa, A.; Lin, J.L.; Lee, E.S.; Harry, B.L.; Skeen-Gaar, R.R.; Suehiro, Y.; William, D.; et al. Mitochondrial endonuclease G mediates breakdown of paternal mitochondria upon fertilization. Science 2016, 353, 394–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLuca, S.Z.; O’Farrell, P.H. Barriers to male transmission of mitochondrial DNA in sperm development. Dev. Cell 2012, 22, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Kirienko, N.V.; Ausubel, F.M.; Ruvkun, G. Mitophagy confers resistance to siderophore-mediated killing by Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2015, 112, 1821–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chikka, M.R.; Anbalagan, C.; Dvorak, K.; Dombeck, K.; Prahlad, V. The Mitochondria-Regulated Immune Pathway Activated in the C. elegans Intestine Is Neuroprotective. Cell Rep. 2016, 16, 2399–2414. [Google Scholar] [CrossRef] [PubMed]
- Schiavi, A.; Maglioni, S.; Palikaras, K.; Shaik, A.; Strappazzon, F.; Brinkmann, V.; Torgovnick, A.; Castelein, N.; De Henau, S.; Braeckman, B.P.; et al. Iron-starvation-induced mitophagy mediates lifespan extension upon mitochondrial stress in C. elegans. Curr. Biol. 2015, 25, 1810–1822. [Google Scholar] [CrossRef] [PubMed]
- Munch, C. The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol. 2018, 16, 81. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.F.; Schulz, A.M.; Pellegrino, M.W.; Lu, Y.; Shaham, S.; Haynes, C.M. Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 2016, 533, 416–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houtkooper, R.H.; Mouchiroud, L.; Ryu, D.; Moullan, N.; Katsyuba, E.; Knott, G.; Williams, R.W.; Auwerx, J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013, 497, 451–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynes, C.M.; Petrova, K.; Benedetti, C.; Yang, Y.; Ron, D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 2007, 13, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Yang, Y.; Blais, S.P.; Neubert, T.A.; Ron, D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol. Cell 2010, 37, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, C.; Haynes, C.M.; Yang, Y.; Harding, H.P.; Ron, D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 2006, 174, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Nargund, A.M.; Fiorese, C.J.; Pellegrino, M.W.; Deng, P.; Haynes, C.M. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol. Cell 2015, 58, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Melo, J.A.; Ruvkun, G. Inactivation of conserved C. elegans genes engages pathogen- and xenobiotic-associated defenses. Cell 2012, 149, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Samuel, B.S.; Breen, P.C.; Ruvkun, G. Caenorhabditis elegans pathways that surveil and defend mitochondria. Nature 2014, 508, 406–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrino, M.W.; Nargund, A.M.; Kirienko, N.V.; Gillis, R.; Fiorese, C.J.; Haynes, C.M. Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature 2014, 516, 414–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunbar, T.L.; Yan, Z.; Balla, K.M.; Smelkinson, M.G.; Troemel, E.R. C. elegans detects pathogen-induced translational inhibition to activate immune signaling. Cell Host Microbe 2012, 11, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Merkwirth, C.; Jovaisaite, V.; Durieux, J.; Matilainen, O.; Jordan, S.D.; Quiros, P.M.; Steffen, K.K.; Williams, E.G.; Mouchiroud, L.; Tronnes, S.U.; et al. Two Conserved Histone Demethylases Regulate Mitochondrial Stress-Induced Longevity. Cell 2016, 165, 1209–1223. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Garcia, G.; Bian, Q.; Steffen, K.K.; Joe, L.; Wolff, S.; Meyer, B.J.; Dillin, A. Mitochondrial Stress Induces Chromatin Reorganization to Promote Longevity and UPR(mt). Cell 2016, 165, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Teperino, R.; Schoonjans, K.; Auwerx, J. Histone methyl transferases and demethylases; can they link metabolism and transcription? Cell Metab. 2010, 12, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Durieux, J.; Wolff, S.; Dillin, A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 2011, 144, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.W.; Niu, R.; Liu, Y. Neuropeptide signals cell non-autonomous mitochondrial unfolded protein response. Cell Res. 2016, 26, 1182–1196. [Google Scholar] [CrossRef] [PubMed]
- Berendzen, K.M.; Durieux, J.; Shao, L.W.; Tian, Y.; Kim, H.E.; Wolff, S.; Liu, Y.; Dillin, A. Neuroendocrine Coordination of Mitochondrial Stress Signaling and Proteostasis. Cell 2016, 166, 1553–1563. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wu, X.; Chen, P.; Liu, L.; Xin, N.; Tian, Y.; Dillin, A. The mitochondrial unfolded protein response is mediated cell-non-autonomously by retromer-dependent wnt signaling. Cell 2018, 174, 870–883. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Sieburth, D. Sphingosine Kinase Activates the Mitochondrial Unfolded Protein Response and Is Targeted to Mitochondria by Stress. Cell Rep. 2018, 24, 2932–2945. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, X.J. A cytosolic network suppressing mitochondria-mediated proteostatic stress and cell death. Nature 2015, 524, 481–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrobel, L.; Topf, U.; Bragoszewski, P.; Wiese, S.; Sztolsztener, M.E.; Oeljeklaus, S.; Varabyova, A.; Lirski, M.; Chroscicki, P.; Mroczek, S.; et al. Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature 2015, 524, 485–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safra, M.; Ben-Hamo, S.; Kenyon, C.; Henis-Korenblit, S. The ire-1 ER stress-response pathway is required for normal secretory-protein metabolism in C. elegans. J. Cell Sci. 2013, 126, 4136–4146. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.E.; Grant, A.R.; Simic, M.S.; Kohnz, R.A.; Nomura, D.K.; Durieux, J.; Riera, C.E.; Sanchez, M.; Kapernick, E.; Wolff, S.; et al. Lipid Biosynthesis Coordinates a Mitochondrial-to-Cytosolic Stress Response. Cell 2016, 166, 1539–1552. [Google Scholar] [CrossRef] [PubMed]
- Suhm, T.; Kaimal, J.M.; Dawitz, H.; Peselj, C.; Masser, A.E.; Hanzen, S.; Ambrozic, M.; Smialowska, A.; Bjorck, M.L.; Brzezinski, P.; et al. Mitochondrial Translation Efficiency Controls Cytoplasmic Protein Homeostasis. Cell Metab. 2018, 27, 1309–1322. [Google Scholar] [CrossRef] [PubMed]
- Jovaisaite, V.; Auwerx, J. The mitochondrial unfolded protein response-synchronizing genomes. Curr. Opin. Cell Biol. 2015, 33, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Lee, R.Y.; Fraser, A.G.; Kamath, R.S.; Ahringer, J.; Ruvkun, G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat. Genet. 2003, 33, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jiang, N.; Hughes, B.; Bigras, E.; Shoubridge, E.; Hekimi, S. Evolutionary conservation of the clk-1-dependent mechanism of longevity: Loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 2005, 19, 2424–2434. [Google Scholar] [CrossRef] [PubMed]
- Owusu-Ansah, E.; Song, W.; Perrimon, N. Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell 2013, 155, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Dillin, A.; Hsu, A.L.; Arantes-Oliveira, N.; Lehrer-Graiwer, J.; Hsin, H.; Fraser, A.G.; Kamath, R.S.; Ahringer, J.; Kenyon, C. Rates of behavior and aging specified by mitochondrial function during development. Science 2002, 298, 2398–2401. [Google Scholar] [CrossRef] [PubMed]
- Rea, S.L.; Ventura, N.; Johnson, T.E. Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol. 2007, 5, e259. [Google Scholar] [CrossRef] [PubMed]
- Schulz, A.M.; Haynes, C.M. UPR(mt)-mediated cytoprotection and organismal aging. Biochim. Biophys. Acta 2015, 1847, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Hekimi, S.; Lapointe, J.; Wen, Y. Taking a "good" look at free radicals in the aging process. Trends Cell Biol. 2011, 21, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, J.M.; Hekimi, S. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 2009, 5, e1000361. [Google Scholar] [CrossRef] [PubMed]
- Merkwirth, C.; Langer, T. Prohibitin function within mitochondria: Essential roles for cell proliferation and cristae morphogenesis. Biochim. Biophys. Acta 2009, 1793, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osman, C.; Haag, M.; Potting, C.; Rodenfels, J.; Dip, P.V.; Wieland, F.T.; Brugger, B.; Westermann, B.; Langer, T. The genetic interactome of prohibitins: Coordinated control of cardiolipin and phosphatidylethanolamine by conserved regulators in mitochondria. J. Cell Biol. 2009, 184, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Artal-Sanz, M.; Tsang, W.Y.; Willems, E.M.; Grivell, L.A.; Lemire, B.D.; van der Spek, H.; Nijtmans, L.G. The mitochondrial prohibitin complex is essential for embryonic viability and germline function in Caenorhabditis elegans. J. Biol. Chem. 2003, 278, 32091–32099. [Google Scholar] [CrossRef] [PubMed]
- Merkwirth, C.; Dargazanli, S.; Tatsuta, T.; Geimer, S.; Lower, B.; Wunderlich, F.T.; von Kleist-Retzow, J.C.; Waisman, A.; Westermann, B.; Langer, T. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 2008, 22, 476–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Back, J.W.; Sanz, M.A.; De Jong, L.; De Koning, L.J.; Nijtmans, L.G.; De Koster, C.G.; Grivell, L.A.; Van Der Spek, H.; Muijsers, A.O. A structure for the yeast prohibitin complex: Structure prediction and evidence from chemical crosslinking and mass spectrometry. Protein Sci. 2002, 11, 2471–2478. [Google Scholar] [CrossRef] [PubMed]
- Winter, A.; Kamarainen, O.; Hofmann, A. Molecular modeling of prohibitin domains. Proteins 2007, 68, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Berger, K.H.; Yaffe, M.P. Prohibitin family members interact genetically with mitochondrial inheritance components in Saccharomyces cerevisiae. Mol. Cell Biol. 1998, 18, 4043–4052. [Google Scholar] [CrossRef] [PubMed]
- Bogenhagen, D.F.; Wang, Y.; Shen, E.L.; Kobayashi, R. Protein components of mitochondrial DNA nucleoids in higher eukaryotes. Mol. Cell Proteom. 2003, 2, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Kasashima, K.; Sumitani, M.; Satoh, M.; Endo, H. Human prohibitin 1 maintains the organization and stability of the mitochondrial nucleoids. Exp. Cell Res. 2008, 314, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bogenhagen, D.F. Human mitochondrial DNA nucleoids are linked to protein folding machinery and metabolic enzymes at the mitochondrial inner membrane. J. Biol. Chem. 2006, 281, 25791–25802. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Cooper, H.M.; Reyes, A.; Di Re, M.; Sembongi, H.; Litwin, T.R.; Gao, J.; Neuman, K.C.; Fearnley, I.M.; Spinazzola, A.; et al. Mitochondrial nucleoid interacting proteins support mitochondrial protein synthesis. Nucleic Acids Res. 2012, 40, 6109–6121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerhold, J.M.; Cansiz-Arda, S.; Lohmus, M.; Engberg, O.; Reyes, A.; van Rennes, H.; Sanz, A.; Holt, I.J.; Cooper, H.M.; Spelbrink, J.N. Human Mitochondrial DNA-Protein Complexes Attach to a Cholesterol-Rich Membrane Structure. Sci. Rep. 2015, 5, 15292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peralta, S.; Goffart, S.; Williams, S.L.; Diaz, F.; Garcia, S.; Nissanka, N.; Area-Gomez, E.; Pohjoismaki, J.; Moraes, C.T. ATAD3 controls mitochondrial cristae structure in mouse muscle, influencing mtDNA replication and cholesterol levels. J. Cell Sci. 2018, 131, jcs.217075. [Google Scholar] [CrossRef] [PubMed]
- Baudier, J. ATAD3 proteins: Brokers of a mitochondria-endoplasmic reticulum connection in mammalian cells. Biol. Rev. Camb. Philos. Soc. 2018, 93, 827–844. [Google Scholar] [CrossRef] [PubMed]
- Nijtmans, L.G.; de Jong, L.; Sanz, M.A.; Coates, P.J.; Berden, J.A.; Back, J.W.; Muijsers, A.O.; van der Spek, H.; Grivell, L.A. Prohibitins act as a membrane-bound chaperone for the stabilization of mitochondrial proteins. EMBO J. 2000, 19, 2444–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strub, G.M.; Paillard, M.; Liang, J.; Gomez, L.; Allegood, J.C.; Hait, N.C.; Maceyka, M.; Price, M.M.; Chen, Q.; Simpson, D.C.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 2011, 25, 600–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miwa, S.; Jow, H.; Baty, K.; Johnson, A.; Czapiewski, R.; Saretzki, G.; Treumann, A.; von Zglinicki, T. Low abundance of the matrix arm of complex I in mitochondria predicts longevity in mice. Nat. Commun. 2014, 5, 3837. [Google Scholar] [CrossRef] [PubMed]
- Steglich, G.; Neupert, W.; Langer, T. Prohibitins regulate membrane protein degradation by the m-AAA protease in mitochondria. Mol. Cell Biol. 1999, 19, 3435–3442. [Google Scholar] [CrossRef] [PubMed]
- Osman, C.; Wilmes, C.; Tatsuta, T.; Langer, T. Prohibitins interact genetically with Atp23, a novel processing peptidase and chaperone for the F1Fo-ATP synthase. Mol. Biol. Cell 2007, 18, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Mitsopoulos, P.; Chang, Y.H.; Wai, T.; Konig, T.; Dunn, S.D.; Langer, T.; Madrenas, J. Stomatin-like protein 2 is required for in vivo mitochondrial respiratory chain supercomplex formation and optimal cell function. Mol. Cell Biol. 2015, 35, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Jian, C.; Xu, F.; Hou, T.; Sun, T.; Li, J.; Cheng, H.; Wang, X. Deficiency of PHB complex impairs respiratory supercomplex formation and activates mitochondrial flashes. J. Cell Sci. 2017, 130, 2620–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter-Dennerlein, R.; Korwitz, A.; Haag, M.; Tatsuta, T.; Dargazanli, S.; Baker, M.; Decker, T.; Lamkemeyer, T.; Rugarli, E.I.; Langer, T. DNAJC19, a mitochondrial cochaperone associated with cardiomyopathy, forms a complex with prohibitins to regulate cardiolipin remodeling. Cell Metab. 2014, 20, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Birner, R.; Nebauer, R.; Schneiter, R.; Daum, G. Synthetic lethal interaction of the mitochondrial phosphatidylethanolamine biosynthetic machinery with the prohibitin complex of Saccharomyces cerevisiae. Mol. Biol. Cell 2003, 14, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Klecker, T.; Wemmer, M.; Haag, M.; Weig, A.; Bockler, S.; Langer, T.; Nunnari, J.; Westermann, B. Interaction of MDM33 with mitochondrial inner membrane homeostasis pathways in yeast. Sci. Rep. 2015, 5, 18344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornmann, B.; Currie, E.; Collins, S.R.; Schuldiner, M.; Nunnari, J.; Weissman, J.S.; Walter, P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 2009, 325, 477–481. [Google Scholar] [CrossRef] [PubMed]
- van der Laan, M.; Bohnert, M.; Wiedemann, N.; Pfanner, N. Role of MINOS in mitochondrial membrane architecture and biogenesis. Trends Cell Biol. 2012, 22, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Oeljeklaus, S.; Reinartz, B.S.; Wolf, J.; Wiese, S.; Tonillo, J.; Podwojski, K.; Kuhlmann, K.; Stephan, C.; Meyer, H.E.; Schliebs, W.; et al. Identification of core components and transient interactors of the peroxisomal importomer by dual-track stable isotope labeling with amino acids in cell culture analysis. J. Proteome Res. 2012, 11, 2567–2580. [Google Scholar] [CrossRef] [PubMed]
- Brasaemle, D.L.; Dolios, G.; Shapiro, L.; Wang, R. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. J. Biol. Chem. 2004, 279, 46835–46842. [Google Scholar] [CrossRef] [PubMed]
- Garin, J.; Diez, R.; Kieffer, S.; Dermine, J.F.; Duclos, S.; Gagnon, E.; Sadoul, R.; Rondeau, C.; Desjardins, M. The phagosome proteome: Insight into phagosome functions. J. Cell Biol. 2001, 152, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.R.; Fan, Y.S.; Yang, W.X. Mitochondrial prohibitin and its ubiquitination during spermatogenesis of the swimming crab Charybdis japonica. Gene 2017, 627, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.C.; Wei, C.G.; Lu, C.P.; Gao, X.M.; Yang, W.X.; Zhu, J.Q. Prohibitin-mediated mitochondrial ubiquitination during spermiogenesis in Chinese mitten crab Eriocheir sinensis. Oncotarget 2017, 8, 98782–98797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, C.C.; Gao, X.M.; Ni, J.; Mu, D.L.; Yang, H.Y.; Liu, C.; Zhu, J.Q. The expression pattern and potential functions of PHB in the spermiogenesis of Phascolosoma esculenta. Gene 2018, 652, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, J.; von Stockum, S.; Marchesan, E.; Caicci, F.; Ferrari, V.; Rakovic, A.; Klein, C.; Antonini, A.; Bubacco, L.; Ziviani, E. USP14 inhibition corrects an in vivo model of impaired mitophagy. EMBO Mol. Med. 2018, 10, e9014. [Google Scholar] [CrossRef] [PubMed]
- Coates, P.J.; Nenutil, R.; McGregor, A.; Picksley, S.M.; Crouch, D.H.; Hall, P.A.; Wright, E.G. Mammalian prohibitin proteins respond to mitochondrial stress and decrease during cellular senescence. Exp. Cell Res. 2001, 265, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Perez-Martinez, X.; Butler, C.A.; Shingu-Vazquez, M.; Fox, T.D. Dual functions of Mss51 couple synthesis of Cox1 to assembly of cytochrome c oxidase in Saccharomyces cerevisiae mitochondria. Mol. Biol. Cell 2009, 20, 4371–4380. [Google Scholar] [CrossRef] [PubMed]
- Nijtmans, L.G.; Artal Sanz, M.; Bucko, M.; Farhoud, M.H.; Feenstra, M.; Hakkaart, G.A.; Zeviani, M.; Grivell, L.A. Shy1p occurs in a high molecular weight complex and is required for efficient assembly of cytochrome c oxidase in yeast. FEBS Lett. 2001, 498, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Tortelli, T.C.J.; de Godoy, L.M.F.; de Souza, G.A.; Bonatto, D.; Otake, A.H.; de Freitas Saito, R.; Rosa, J.C.; Greene, L.J.; Chammas, R. Accumulation of prohibitin is a common cellular response to different stressing stimuli and protects melanoma cells from ER stress and chemotherapy-induced cell death. Oncotarget 2017, 8, 43114–43129. [Google Scholar] [CrossRef]
- Thuaud, F.; Ribeiro, N.; Nebigil, C.G.; Desaubry, L. Prohibitin ligands in cell death and survival: Mode of action and therapeutic potential. Chem. Biol. 2013, 20, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Perez-Perarnau, A.; Preciado, S.; Palmeri, C.M.; Moncunill-Massaguer, C.; Iglesias-Serret, D.; Gonzalez-Girones, D.M.; Miguel, M.; Karasawa, S.; Sakamoto, S.; Cosialls, A.M.; et al. A trifluorinated thiazoline scaffold leading to pro-apoptotic agents targeting prohibitins. Angew. Chem. Int. Ed. Engl. 2014, 53, 10150–10154. [Google Scholar] [CrossRef] [PubMed]
- Nijtmans, L.G.; Artal, S.M.; Grivell, L.A.; Coates, P.J. The mitochondrial PHB complex: Roles in mitochondrial respiratory complex assembly, ageing and degenerative disease. Cell Mol. Life Sci. 2002, 59, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Nyomba, B.G. Prohibitin–At the crossroads of obesity-linked diabetes and cancer. Exp. Biol. Med. (Maywood) 2017, 242, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Merkwirth, C.; Martinelli, P.; Korwitz, A.; Morbin, M.; Bronneke, H.S.; Jordan, S.D.; Rugarli, E.I.; Langer, T. Loss of prohibitin membrane scaffolds impairs mitochondrial architecture and leads to tau hyperphosphorylation and neurodegeneration. PLoS Genet. 2012, 8, e1003021. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.X.Z.; Ande, S.R.; Mishra, S. Prohibitin: A new player in immunometabolism and in linking obesity and inflammation with cancer. Cancer Lett. 2018, 415, 208–216. [Google Scholar] [CrossRef]
- Piper, P.W.; Jones, G.W.; Bringloe, D.; Harris, N.; MacLean, M.; Mollapour, M. The shortened replicative life span of prohibitin mutants of yeast appears to be due to defective mitochondrial segregation in old mother cells. Aging Cell 2002, 1, 149–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schleicher, M.; Shepherd, B.R.; Suarez, Y.; Fernandez-Hernando, C.; Yu, J.; Pan, Y.; Acevedo, L.M.; Shadel, G.S.; Sessa, W.C. Prohibitin-1 maintains the angiogenic capacity of endothelial cells by regulating mitochondrial function and senescence. J. Cell Biol. 2008, 180, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasashima, K.; Ohta, E.; Kagawa, Y.; Endo, H. Mitochondrial functions and estrogen receptor-dependent nuclear translocation of pleiotropic human prohibitin 2. J. Biol. Chem. 2006, 281, 36401–36410. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, D.; Kumar, D.; Bhadra, U.; Devi, T.A.; Bhadra, M.P. Prohibitin confers cytoprotection against ISO-induced hypertrophy in H9c2 cells via attenuation of oxidative stress and modulation of Akt/Gsk-3beta signaling. Mol. Cell Biochem. 2017, 425, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef] [PubMed]
- Schleit, J.; Johnson, S.C.; Bennett, C.F.; Simko, M.; Trongtham, N.; Castanza, A.; Hsieh, E.J.; Moller, R.M.; Wasko, B.M.; Delaney, J.R.; et al. Molecular mechanisms underlying genotype-dependent responses to dietary restriction. Aging Cell 2013, 12, 1050–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bausewein, T.; Mills, D.J.; Langer, J.D.; Nitschke, B.; Nussberger, S.; Kuhlbrandt, W. Cryo-EM Structure of the TOM Core Complex from Neurospora crassa. Cell 2017, 170, 693–700. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernando-Rodríguez, B.; Artal-Sanz, M. Mitochondrial Quality Control Mechanisms and the PHB (Prohibitin) Complex. Cells 2018, 7, 238. https://doi.org/10.3390/cells7120238
Hernando-Rodríguez B, Artal-Sanz M. Mitochondrial Quality Control Mechanisms and the PHB (Prohibitin) Complex. Cells. 2018; 7(12):238. https://doi.org/10.3390/cells7120238
Chicago/Turabian StyleHernando-Rodríguez, Blanca, and Marta Artal-Sanz. 2018. "Mitochondrial Quality Control Mechanisms and the PHB (Prohibitin) Complex" Cells 7, no. 12: 238. https://doi.org/10.3390/cells7120238
APA StyleHernando-Rodríguez, B., & Artal-Sanz, M. (2018). Mitochondrial Quality Control Mechanisms and the PHB (Prohibitin) Complex. Cells, 7(12), 238. https://doi.org/10.3390/cells7120238