Epigenetic Erosion in Adult Stem Cells: Drivers and Passengers of Aging




Abstract
:
1. Introduction
2. Spermatogonial Stem Cells
2.1. DNA Methylation Changes during Spermatogenesis
2.2. Dynamic Regulation of Histone Modifications in SSC Development
3. Hematopoietic Stem Cells
3.1. Impact of DNA Damage and Inflammation on the HSC Epigenome
3.2. Epigenetic Erosion of Hematopoietic Stem Cells and Clonal Aberration with Age
3.3. Link between Aging-Induced Epimutations and Hematologic Cancer Formation
3.4. Therapeutic Implications of HSC Aging
4. Muscle Stem Cells
4.1. Impact of H3K4me3 and H3K27me3 Marks on MuSC Activation
4.2. Aberrant Regulation of H3K4me3 and H3K27me3 in MuSC Aging
4.3. H4K20 Methylation Marks Play a Pivotal Role in Establishing MuSC Quiescence
4.4. Link between Epigenetic Regulation, Metabolism, and Muscle Aging
5. Conclusions
Funding
Conflicts of Interest
References
- Ramalho-Santos, M.; Willenbring, H. On the origin of the term “stem cell”. Cell Stem Cell 2007, 1, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Ermolaeva, M.; Neri, F.; Ori, A.; Rudolph, K.L. Cellular and epigenetic drivers of stem cell ageing. Nat. Rev. Mol. Cell Biol. 2018, 19, 594–610. [Google Scholar] [CrossRef] [PubMed]
- Cheung, T.H.; Rando, T.A. Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol. 2013, 14, 329–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rooij, D.G. The nature and dynamics of spermatogonial stem cells. Development 2017, 144, 3022–3030. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, D.G.; Griswold, M.D. Questions about spermatogonia posed and answered since 2000. J. Androl. 2012, 33, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Burton, A.; Torres-Padilla, M.E. Chromatin dynamics in the regulation of cell fate allocation during early embryogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.Q.; Dean, J. Reprogramming the genome to totipotency in mouse embryos. Trends Cell Biol. 2015, 25, 82–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frans, E.M.; McGrath, J.J.; Sandin, S.; Lichtenstein, P.; Reichenberg, A.; Langstrom, N.; Hultman, C.M. Advanced paternal and grandpaternal age and schizophrenia: A three-generation perspective. Schizophr. Res. 2011, 133, 120–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janecka, M.; Mill, J.; Basson, M.A.; Goriely, A.; Spiers, H.; Reichenberg, A.; Schalkwyk, L.; Fernandes, C. Advanced paternal age effects in neurodevelopmental disorders-review of potential underlying mechanisms. Transl. Psychiatry 2017, 7, e1019. [Google Scholar] [CrossRef] [PubMed]
- Sales, V.M.; Ferguson-Smith, A.C.; Patti, M.E. Epigenetic mechanisms of transmission of metabolic disease across generations. Cell Metab. 2017, 25, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Grow, E.J.; Yi, C.; Mlcochova, H.; Maher, G.J.; Lindskog, C.; Murphy, P.J.; Wike, C.L.; Carrell, D.T.; Goriely, A.; et al. Chromatin and single-cell RNA-seq profiling reveal dynamic signaling and metabolic transitions during human spermatogonial stem cell development. Cell Stem Cell 2017, 21, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Hammoud, S.S.; Low, D.H.; Yi, C.; Carrell, D.T.; Guccione, E.; Cairns, B.R. Chromatin and transcription transitions of mammalian adult germline stem cells and spermatogenesis. Cell Stem Cell 2014, 15, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Hammoud, S.S.; Low, D.H.; Yi, C.; Lee, C.L.; Oatley, J.M.; Payne, C.J.; Carrell, D.T.; Guccione, E.; Cairns, B.R. Transcription and imprinting dynamics in developing postnatal male germline stem cells. Genes Dev. 2015, 29, 2312–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oakes, C.C.; La Salle, S.; Smiraglia, D.J.; Robaire, B.; Trasler, J.M. Developmental acquisition of genome-wide DNA methylation occurs prior to meiosis in male germ cells. Dev. Biol. 2007, 307, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Kubo, N.; Toh, H.; Shirane, K.; Shirakawa, T.; Kobayashi, H.; Sato, T.; Sone, H.; Sato, Y.; Tomizawa, S.; Tsurusaki, Y.; et al. DNA methylation and gene expression dynamics during spermatogonial stem cell differentiation in the early postnatal mouse testis. BMC Genom. 2015, 16, 624. [Google Scholar] [CrossRef] [PubMed]
- Bourc’his, D.; Bestor, T.H. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 2004, 431, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Dion, V.; Lin, Y.; Hubert, L., Jr.; Waterland, R.A.; Wilson, J.H. Dnmt1 deficiency promotes CAG repeat expansion in the mouse germline. Hum. Mol. Genet. 2008, 17, 1306–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneda, M.; Okano, M.; Hata, K.; Sado, T.; Tsujimoto, N.; Li, E.; Sasaki, H. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 2004, 429, 900–903. [Google Scholar] [CrossRef] [PubMed]
- Webster, K.E.; O’Bryan, M.K.; Fletcher, S.; Crewther, P.E.; Aapola, U.; Craig, J.; Harrison, D.K.; Aung, H.; Phutikanit, N.; Lyle, R.; et al. Meiotic and epigenetic defects in Dnmt3L-knockout mouse spermatogenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 4068–4073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aston, K.I.; Uren, P.J.; Jenkins, T.G.; Horsager, A.; Cairns, B.R.; Smith, A.D.; Carrell, D.T. Aberrant sperm DNA methylation predicts male fertility status and embryo quality. Fertil. Steril. 2015, 104, 1388–1397. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.S.; Moley, K.H.; Wangler, M.; Feinberg, A.P.; Debaun, M.R. Association between Beckwith-Wiedemann syndrome and assisted reproductive technology: A case series of 19 patients. Fertil. Steril. 2005, 83, 349–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chopra, M.; Amor, D.J.; Sutton, L.; Algar, E.; Mowat, D. Russell-Silver syndrome due to paternal H19/IGF2 hypomethylation in a patient conceived using intracytoplasmic sperm injection. Reprod. Biomed. Online 2010, 20, 843–847. [Google Scholar] [CrossRef] [PubMed]
- DeBaun, M.R.; Niemitz, E.L.; Feinberg, A.P. Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am. J. Hum. Genet. 2003, 72, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Marees, T.; Dommering, C.J.; Imhof, S.M.; Kors, W.A.; Ringens, P.J.; van Leeuwen, F.E.; Moll, A.C. Incidence of retinoblastoma in Dutch children conceived by IVF: An expanded study. Hum. Reprod. 2009, 24, 3220–3224. [Google Scholar] [CrossRef] [PubMed]
- Schieve, L.A.; Meikle, S.F.; Ferre, C.; Peterson, H.B.; Jeng, G.; Wilcox, L.S. Low and very low birth weight in infants conceived with use of assisted reproductive technology. N. Engl. J. Med. 2002, 346, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, S.; Bashiri, R.; Ghadiri-Anari, A.; Nadjarzadeh, A. Antioxidant supplements and semen parameters: An evidence based review. Int. J. Reprod. Biomed. (Yazd) 2016, 14, 729–736. [Google Scholar] [CrossRef] [Green Version]
- Boxmeer, J.C.; Macklon, N.S.; Lindemans, J.; Beckers, N.G.; Eijkemans, M.J.; Laven, J.S.; Steegers, E.A.; Steegers-Theunissen, R.P. IVF outcomes are associated with biomarkers of the homocysteine pathway in monofollicular fluid. Hum. Reprod. 2009, 24, 1059–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, W.Y.; Merkus, H.M.; Thomas, C.M.; Menkveld, R.; Zielhuis, G.A.; Steegers-Theunissen, R.P. Effects of folic acid and zinc sulfate on male factor subfertility: A double-blind, randomized, placebo-controlled trial. Fertil. Steril. 2002, 77, 491–498. [Google Scholar] [CrossRef]
- Dong, H.; Wang, Y.; Zou, Z.; Chen, L.; Shen, C.; Xu, S.; Zhang, J.; Zhao, F.; Ge, S.; Gao, Q.; et al. Abnormal methylation of imprinted genes and cigarette smoking: assessment of their association with the risk of male infertility. Reprod. Sci. 2017, 24, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Laqqan, M.; Tierling, S.; Alkhaled, Y.; Porto, C.L.; Solomayer, E.F.; Hammadeh, M.E. Aberrant DNA methylation patterns of human spermatozoa in current smoker males. Reprod. Toxicol. 2017, 71, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Ouko, L.A.; Shantikumar, K.; Knezovich, J.; Haycock, P.; Schnugh, D.J.; Ramsay, M. Effect of alcohol consumption on CpG methylation in the differentially methylated regions of H19 and IG-DMR in male gametes: Implications for fetal alcohol spectrum disorders. Alcohol. Clin. Exp. Res. 2009, 33, 1615–1627. [Google Scholar] [CrossRef] [PubMed]
- Stouder, C.; Somm, E.; Paoloni-Giacobino, A. Prenatal exposure to ethanol: A specific effect on the H19 gene in sperm. Reprod. Toxicol. 2011, 31, 507–512. [Google Scholar] [CrossRef] [PubMed]
- De Castro Barbosa, T.; Ingerslev, L.R.; Alm, P.S.; Versteyhe, S.; Massart, J.; Rasmussen, M.; Donkin, I.; Sjogren, R.; Mudry, J.M.; Vetterli, L.; et al. High-fat diet reprograms the epigenome of rat spermatozoa and transgenerationally affects metabolism of the offspring. Mol. Metab. 2016, 5, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Godmann, M.; Auger, V.; Ferraroni-Aguiar, V.; Di Sauro, A.; Sette, C.; Behr, R.; Kimmins, S. Dynamic regulation of histone H3 methylation at lysine 4 in mammalian spermatogenesis. Biol. Reprod. 2007, 77, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Luense, L.J.; Wang, X.; Schon, S.B.; Weller, A.H.; Lin Shiao, E.; Bryant, J.M.; Bartolomei, M.S.; Coutifaris, C.; Garcia, B.A.; Berger, S.L. Comprehensive analysis of histone post-translational modifications in mouse and human male germ cells. Epigenet. Chromatin 2016, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Payne, C.; Braun, R.E. Histone lysine trimethylation exhibits a distinct perinuclear distribution in Plzf-expressing spermatogonia. Dev. Biol. 2006, 293, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Shima, J.E.; McLean, D.J.; McCarrey, J.R.; Griswold, M.D. The murine testicular transcriptome: Characterizing gene expression in the testis during the progression of spermatogenesis. Biol. Reprod. 2004, 71, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Goudarzi, A.; Shiota, H.; Rousseaux, S.; Khochbin, S. Genome-scale acetylation-dependent histone eviction during spermatogenesis. J. Mol. Biol. 2014, 426, 3342–3349. [Google Scholar] [CrossRef] [PubMed]
- Sonnack, V.; Failing, K.; Bergmann, M.; Steger, K. Expression of hyperacetylated histone H4 during normal and impaired human spermatogenesis. Andrologia 2002, 34, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Steilmann, C.; Paradowska, A.; Bartkuhn, M.; Vieweg, M.; Schuppe, H.C.; Bergmann, M.; Kliesch, S.; Weidner, W.; Steger, K. Presence of histone H3 acetylated at lysine 9 in male germ cells and its distribution pattern in the genome of human spermatozoa. Reprod. Fertil. Dev. 2011, 23, 997–1011. [Google Scholar] [CrossRef] [PubMed]
- Kofman, A.E.; Huszar, J.M.; Payne, C.J. Transcriptional analysis of histone deacetylase family members reveal similarities between differentiating and aging spermatogonial stem cells. Stem Cell Rev. Rep. 2013, 9, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Erkek, S.; Hisano, M.; Liang, C.Y.; Gill, M.; Murr, R.; Dieker, J.; Schubeler, D.; van der Vlag, J.; Stadler, M.B.; Peters, A.H. Molecular determinants of nucleosome retention at CpG-rich sequences in mouse spermatozoa. Nat. Struct. Mol. Biol. 2013, 20, 868–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Royo, H.; Stadler, M.B.; Peters, A. Alternative computational analysis shows no evidence for nucleosome enrichment at repetitive sequences in mammalian spermatozoa. Dev. Cell 2016, 37, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Van de Werken, C.; van der Heijden, G.W.; Eleveld, C.; Teeuwssen, M.; Albert, M.; Baarends, W.M.; Laven, J.S.; Peters, A.H.; Baart, E.B. Paternal heterochromatin formation in human embryos is H3K9/HP1 directed and primed by sperm-derived histone modifications. Nat. Commun. 2014, 5, 5868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siklenka, K.; Erkek, S.; Godmann, M.; Lambrot, R.; McGraw, S.; Lafleur, C.; Cohen, T.; Xia, J.; Suderman, M.; Hallett, M.; et al. Disruption of histone methylation in developing sperm impairs offspring health transgenerationally. Science 2015, 350. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, T.G.; Aston, K.I.; Pflueger, C.; Cairns, B.R.; Carrell, D.T. Age-associated sperm DNA methylation alterations: Possible implications in offspring disease susceptibility. PLoS Genet. 2014, 10, e1004458. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Luo, M.; Jeong, M.; Rodriguez, B.; Xia, Z.; Hannah, R.; Wang, H.; Le, T.; Faull, K.F.; Chen, R.; et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell 2014, 14, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Orford, K.W.; Scadden, D.T. Deconstructing stem cell self-renewal: Genetic insights into cell-cycle regulation. Nat. Rev. Genet. 2008, 9, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Hirao, A.; Arai, F.; Matsuoka, S.; Takubo, K.; Hamaguchi, I.; Nomiyama, K.; Hosokawa, K.; Sakurada, K.; Nakagata, N.; et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 2004, 431, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Nijnik, A.; Woodbine, L.; Marchetti, C.; Dawson, S.; Lambe, T.; Liu, C.; Rodrigues, N.P.; Crockford, T.L.; Cabuy, E.; Vindigni, A.; et al. DNA repair is limiting for haematopoietic stem cells during ageing. Nature 2007, 447, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Bryder, D.; Seita, J.; Nussenzweig, A.; Hoeijmakers, J.; Weissman, I.L. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 2007, 447, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Walter, D.; Lier, A.; Geiselhart, A.; Thalheimer, F.B.; Huntscha, S.; Sobotta, M.C.; Moehrle, B.; Brocks, D.; Bayindir, I.; Kaschutnig, P.; et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 2015, 520, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Mohrin, M.; Bourke, E.; Alexander, D.; Warr, M.R.; Barry-Holson, K.; Le Beau, M.M.; Morrison, C.G.; Passegue, E. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell 2010, 7, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Milyavsky, M.; Gan, O.I.; Trottier, M.; Komosa, M.; Tabach, O.; Notta, F.; Lechman, E.; Hermans, K.G.; Eppert, K.; Konovalova, Z.; et al. A distinctive DNA damage response in human hematopoietic stem cells reveals an apoptosis-independent role for p53 in self-renewal. Cell Stem Cell 2010, 7, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Denkinger, M.D.; Leins, H.; Schirmbeck, R.; Florian, M.C.; Geiger, H. HSC Aging and senescent immune remodeling. Trends Immunol. 2015, 36, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Krtolica, A.; Beausejour, C.M.; Parrinello, S.; Hodgson, J.G.; Chin, K.; Desprez, P.Y.; Campisi, J. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS ONE 2010, 5, e9188. [Google Scholar] [CrossRef] [PubMed]
- King, K.Y.; Goodell, M.A. Inflammatory modulation of HSCs: Viewing the HSC as a foundation for the immune response. Nat. Rev. Immunol. 2011, 11, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Quinton, L.J.; Nelson, S.; Boe, D.M.; Zhang, P.; Zhong, Q.; Kolls, J.K.; Bagby, G.J. The granulocyte colony-stimulating factor response after intrapulmonary and systemic bacterial challenges. J. Infect. Dis. 2002, 185, 1476–1482. [Google Scholar] [CrossRef] [PubMed]
- Schuettpelz, L.G.; Borgerding, J.N.; Christopher, M.J.; Gopalan, P.K.; Romine, M.P.; Herman, A.C.; Woloszynek, J.R.; Greenbaum, A.M.; Link, D.C. G-CSF regulates hematopoietic stem cell activity, in part, through activation of Toll-like receptor signaling. Leukemia 2014, 28, 1851–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldridge, M.T.; King, K.Y.; Goodell, M.A. Inflammatory signals regulate hematopoietic stem cells. Trends Immunol. 2011, 32, 57–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagai, Y.; Garrett, K.P.; Ohta, S.; Bahrun, U.; Kouro, T.; Akira, S.; Takatsu, K.; Kincade, P.W. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 2006, 24, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Mitroulis, I.; Ruppova, K.; Wang, B.; Chen, L.S.; Grzybek, M.; Grinenko, T.; Eugster, A.; Troullinaki, M.; Palladini, A.; Kourtzelis, I.; et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell 2018, 172, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.; Oser, G.M.; Jaworski, M.; Blanco-Bose, W.E.; Laurenti, E.; Adolphe, C.; Essers, M.A.; Macdonald, H.R.; Trumpp, A. Dormant and self-renewing hematopoietic stem cells and their niches. Ann. N. Y. Acad. Sci. 2007, 1106, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Florian, M.C.; Dorr, K.; Niebel, A.; Daria, D.; Schrezenmeier, H.; Rojewski, M.; Filippi, M.D.; Hasenberg, A.; Gunzer, M.; Scharffetter-Kochanek, K.; et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell 2012, 10, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Florian, M.C.; Nattamai, K.J.; Dorr, K.; Marka, G.; Uberle, B.; Vas, V.; Eckl, C.; Andra, I.; Schiemann, M.; Oostendorp, R.A.; et al. A canonical to non-canonical Wnt signalling switch in haematopoietic stem-cell ageing. Nature 2013, 503, 392–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, Y.; Li, Y.; Zhang, W.; Yuan, S.; Winkler, R.; Krohnert, U.; Han, J.; Lin, T.; Zhou, Y.; Miao, P.; et al. Sepsis-induced thymic atrophy is associated with defects in early lymphopoiesis. Stem Cells 2016, 34, 2902–2915. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, H.; Fritsch, K.; Kovtonyuk, L.V.; Saito, Y.; Yakkala, C.; Jacobs, K.; Ahuja, A.K.; Lopes, M.; Hausmann, A.; Hardt, W.D.; et al. Pathogen-induced TLR4-TRIF innate immune signaling in hematopoietic stem cells promotes proliferation but reduces competitive fitness. Cell Stem Cell 2017, 21, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Yu, V.W.C.; Yusuf, R.Z.; Oki, T.; Wu, J.; Saez, B.; Wang, X.; Cook, C.; Baryawno, N.; Ziller, M.J.; Lee, E.; et al. Epigenetic memory underlies cell-autonomous heterogeneous behavior of hematopoietic stem cells. Cell 2016, 167, 1310–1322. [Google Scholar] [CrossRef] [PubMed]
- Buschbeck, M.; Hake, S.B. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.; Vallania, F.; Warsinske, H.C.; Donato, M.; Schaffert, S.; Chang, S.E.; Dvorak, M.; Dekker, C.L.; Davis, M.M.; Utz, P.J.; et al. Single-cell chromatin modification profiling reveals increased epigenetic variations with aging. Cell 2018, 173, 1385–1397. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, S.; Zhang, Y.; Langfelder, P.; Kahn, R.S.; Boks, M.P.; van Eijk, K.; van den Berg, L.H.; Ophoff, R.A. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012, 13, R97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jockel, K.H.; Erbel, R.; Muhleisen, T.W.; et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Belden, W.J. Long non-coding RNAs have age-dependent diurnal expression that coincides with age-related changes in genome-wide facultative heterochromatin. BMC Genom. 2018, 19, 777. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Kahler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- McKerrell, T.; Park, N.; Moreno, T.; Grove, C.S.; Ponstingl, H.; Stephens, J.; Understanding Society Scientific Group; Crawley, C.; Craig, J.; Scott, M.A.; et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 2015, 10, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Cuartero, S.; Weiss, F.D.; Dharmalingam, G.; Guo, Y.; Ing-Simmons, E.; Masella, S.; Robles-Rebollo, I.; Xiao, X.; Wang, Y.F.; Barozzi, I.; et al. Control of inducible gene expression links cohesin to hematopoietic progenitor self-renewal and differentiation. Nat. Immunol. 2018, 19, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, S.; Natarajan, P.; Ebert, B.L. Clonal hematopoiesis and atherosclerosis. N. Engl. J. Med. 2017, 377, 1401–1402. [Google Scholar] [CrossRef] [PubMed]
- Buscarlet, M.; Tessier, A.; Provost, S.; Mollica, L.; Busque, L. Human blood cell levels of 5-hydroxymethylcytosine (5hmC) decline with age, partly related to acquired mutations in TET2. Exp. Hematol. 2016, 44, 1072–1084. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thol, F.; Damm, F.; Ludeking, A.; Winschel, C.; Wagner, K.; Morgan, M.; Yun, H.; Gohring, G.; Schlegelberger, B.; Hoelzer, D.; et al. Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J. Clin. Oncol. 2011, 29, 2889–2896. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.J.; Ding, L.; Shen, D.; Shao, J.; Grillot, M.; McLellan, M.; Fulton, R.; Schmidt, H.; Kalicki-Veizer, J.; O’Laughlin, M.; et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia 2011, 25, 1153–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejar, R.; Stevenson, K.E.; Caughey, B.A.; Abdel-Wahab, O.; Steensma, D.P.; Galili, N.; Raza, A.; Kantarjian, H.; Levine, R.L.; Neuberg, D.; et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J. Clin. Oncol. 2012, 30, 3376–3382. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Shen, H.; Xu, T.; Yang, Z.; Qiu, H.; Sun, A.; Chen, S.; Wu, D.; Xu, Y. Persistent DNMT3A mutation burden in DNMT3A mutated adult cytogenetically normal acute myeloid leukemia patients in long-term remission. Leuk. Res. 2016, 49, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Ivey, A.; Hills, R.K.; Simpson, M.A.; Jovanovic, J.V.; Gilkes, A.; Grech, A.; Patel, Y.; Bhudia, N.; Farah, H.; Mason, J.; et al. Assessment of minimal residual disease in standard-risk AML. N. Engl. J. Med. 2016, 374, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Jeziskova, I.; Musilova, M.; Culen, M.; Foltankova, V.; Dvorakova, D.; Mayer, J.; Racil, Z. Distribution of mutations in DNMT3A gene and the suitability of mutations in R882 codon for MRD monitoring in patients with AML. Int. J. Hematol. 2015, 102, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Corces-Zimmerman, M.R.; Hong, W.J.; Weissman, I.L.; Medeiros, B.C.; Majeti, R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc. Natl. Acad. Sci. USA 2014, 111, 2548–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, R.; Wang, P.; Parton, T.; Zhou, Y.; Chrysovergis, K.; Rockowitz, S.; Chen, W.Y.; Abdel-Wahab, O.; Wade, P.A.; Zheng, D.; et al. Epigenetic perturbations by Arg882-mutated DNMT3A potentiate aberrant stem cell gene-expression program and acute leukemia development. Cancer Cell 2016, 30, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Rodriguez, B.; Mayle, A.; Park, H.J.; Lin, X.; Luo, M.; Jeong, M.; Curry, C.V.; Kim, S.B.; Ruau, D.; et al. DNMT3A loss drives enhancer hypomethylation in FLT3-ITD-associated leukemias. Cancer Cell 2016, 30, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Su, J.; Jeong, M.; Ko, M.; Huang, Y.; Park, H.J.; Guzman, A.; Lei, Y.; Huang, Y.H.; Rao, A.; et al. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat. Genet. 2016, 48, 1014–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platzbecker, U.; Santini, V.; Mufti, G.J.; Haferlach, C.; Maciejewski, J.P.; Park, S.; Sole, F.; van de Loosdrecht, A.A.; Haase, D. Update on developments in the diagnosis and prognostic evaluation of patients with myelodysplastic syndromes (MDS): Consensus statements and report from an expert workshop. Leuk. Res. 2012, 36, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Santini, V. Novel therapeutic strategies: Hypomethylating agents and beyond. Hematol. Am. Soc. Hematol. Educ. Program 2012, 2012, 65–73. [Google Scholar] [CrossRef]
- Paul, T.A.; Bies, J.; Small, D.; Wolff, L. Signatures of polycomb repression and reduced H3K4 trimethylation are associated with p15INK4b DNA methylation in AML. Blood 2010, 115, 3098–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Y.; Lennartsson, A.; Gaidzik, V.I.; Deneberg, S.; Karimi, M.; Bengtzen, S.; Hoglund, M.; Bullinger, L.; Dohner, K.; Lehmann, S. Differential methylation in CN-AML preferentially targets non-CGI regions and is dictated by DNMT3A mutational status and associated with predominant hypomethylation of HOX genes. Epigenetics 2014, 9, 1108–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.J.; Xu, J.; Gu, Z.H.; Pan, C.M.; Lu, G.; Shen, Y.; Shi, J.Y.; Zhu, Y.M.; Tang, L.; Zhang, X.W.; et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat. Genet. 2011, 43, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Traina, F.; Visconte, V.; Elson, P.; Tabarroki, A.; Jankowska, A.M.; Hasrouni, E.; Sugimoto, Y.; Szpurka, H.; Makishima, H.; O’Keefe, C.L.; et al. Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia 2014, 28, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Rau, R.E.; Rodriguez, B.A.; Luo, M.; Jeong, M.; Rosen, A.; Rogers, J.H.; Campbell, C.T.; Daigle, S.R.; Deng, L.; Song, Y.; et al. DOT1L as a therapeutic target for the treatment of DNMT3A-mutant acute myeloid leukemia. Blood 2016, 128, 971–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dykstra, B.; Olthof, S.; Schreuder, J.; Ritsema, M.; de Haan, G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J. Exp. Med. 2011, 208, 2691–2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ocampo, A.; Reddy, P.; Martinez-Redondo, P.; Platero-Luengo, A.; Hatanaka, F.; Hishida, T.; Li, M.; Lam, D.; Kurita, M.; Beyret, E.; et al. In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell 2016, 167, 1719–1733. [Google Scholar] [CrossRef] [PubMed]
- Leonard, W.R.; Robertson, M.L.; Snodgrass, J.J.; Kuzawa, C.W. Metabolic correlates of hominid brain evolution. Comp. Biochem. Physiol. 2003, 136, 5–15. [Google Scholar] [CrossRef]
- Mauro, A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef] [PubMed]
- Almada, A.E.; Wagers, A.J. Molecular circuitry of stem cell fate in skeletal muscle regeneration, ageing and disease. Nat. Rev. Mol. Cell Biol. 2016, 17, 267–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marty, E.; Liu, Y.; Samuel, A.; Or, O.; Lane, J. A review of sarcopenia: Enhancing awareness of an increasingly prevalent disease. Bone 2017, 105, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Prat, L.; Munoz-Canoves, P. Aging, metabolism and stem cells: Spotlight on muscle stem cells. Mol. Cell. Endocrinol. 2017, 445, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.H. The altered fate of aging satellite cells is determined by signaling and epigenetic changes. Front. Genet. 2015, 6, 59. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; Wang, Y.X.; Rudnicki, M.A. Building muscle: Molecular regulation of myogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Kawabe, Y.; Wang, Y.X.; McKinnell, I.W.; Bedford, M.T.; Rudnicki, M.A. Carm1 regulates Pax7 transcriptional activity through MLL1/2 recruitment during asymmetric satellite stem cell divisions. Cell Stem Cell 2012, 11, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cheung, T.H.; Charville, G.W.; Hurgo, B.M.; Leavitt, T.; Shih, J.; Brunet, A.; Rando, T.A. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 2013, 4, 189–204. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, R.J.; Kubicek, S.; Schreiber, S.L.; Karlseder, J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat. Struct. Mol. Biol. 2010, 17, 1218–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Han, S.; Cousin, W.; Conboy, I.M. Age-specific functional epigenetic changes in p21 and p16 in injury-activated satellite cells. Stem Cells 2015, 33, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Schworer, S.; Becker, F.; Feller, C.; Baig, A.H.; Kober, U.; Henze, H.; Kraus, J.M.; Xin, B.; Lechel, A.; Lipka, D.B.; et al. Epigenetic stress responses induce muscle stem-cell ageing by Hoxa9 developmental signals. Nature 2016, 540, 428–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boonsanay, V.; Zhang, T.; Georgieva, A.; Kostin, S.; Qi, H.; Yuan, X.; Zhou, Y.; Braun, T. Regulation of skeletal muscle stem cell quiescence by Suv4-20h1-dependent facultative heterochromatin formation. Cell Stem Cell 2016, 18, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Schotta, G.; Sengupta, R.; Kubicek, S.; Malin, S.; Kauer, M.; Callen, E.; Celeste, A.; Pagani, M.; Opravil, S.; De La Rosa-Velazquez, I.A.; et al. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev. 2008, 22, 2048–2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bierhoff, H.; Dammert, M.A.; Brocks, D.; Dambacher, S.; Schotta, G.; Grummt, I. Quiescence-induced LncRNAs trigger H4K20 trimethylation and transcriptional silencing. Mol. Cell 2014, 54, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Shogren-Knaak, M.; Peterson, C.L. Switching on chromatin: Mechanistic role of histone H4-K16 acetylation. Cell Cycle 2006, 5, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, A.; Scher, M.; Lee, D.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol. Cell 2004, 16, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Ryall, J.G.; Dell’Orso, S.; Derfoul, A.; Juan, A.; Zare, H.; Feng, X.; Clermont, D.; Koulnis, M.; Gutierrez-Cruz, G.; Fulco, M.; et al. The NAD(+)-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell 2015, 16, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Hoppel, C.L. Oxidative phosphorylation and aging. Ageing Res. Rev. 2006, 5, 402–433. [Google Scholar] [CrossRef] [PubMed]
- Cerletti, M.; Jang, Y.C.; Finley, L.W.; Haigis, M.C.; Wagers, A.J. Short-term calorie restriction enhances skeletal muscle stem cell function. Cell Stem Cell 2012, 10, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Francaux, M.; Deldicque, L. Exercise and the control of muscle mass in human. Pflugers Arch. Eur. J. Physiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sincennes, M.C.; Brun, C.E.; Rudnicki, M.A. Concise Review: Epigenetic Regulation of Myogenesis in Health and Disease. Stem Cells Transl. Med. 2016, 5, 282–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartorelli, V.; Puri, P.L.; Hamamori, Y.; Ogryzko, V.; Chung, G.; Nakatani, Y.; Wang, J.Y.; Kedes, L. Acetylation of MyoD directed by PCAF is necessary for the execution of the muscle program. Mol. Cell 1999, 4, 725–734. [Google Scholar] [CrossRef]
- Minetti, G.C.; Colussi, C.; Adami, R.; Serra, C.; Mozzetta, C.; Parente, V.; Fortuni, S.; Straino, S.; Sampaolesi, M.; Di Padova, M.; et al. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat. Med. 2006, 12, 1147–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, M.E.; Bhattacharya, A.; Sataranatarajan, K.; Qaisar, R.; Sloane, L.; Rahman, M.M.; Kinter, M.; Van Remmen, H. The histone deacetylase inhibitor butyrate improves metabolism and reduces muscle atrophy during aging. Aging Cell 2015, 14, 957–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]




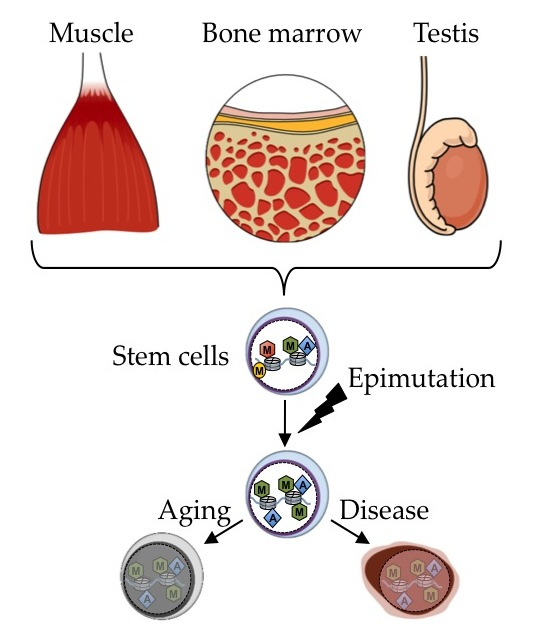{kind=link}
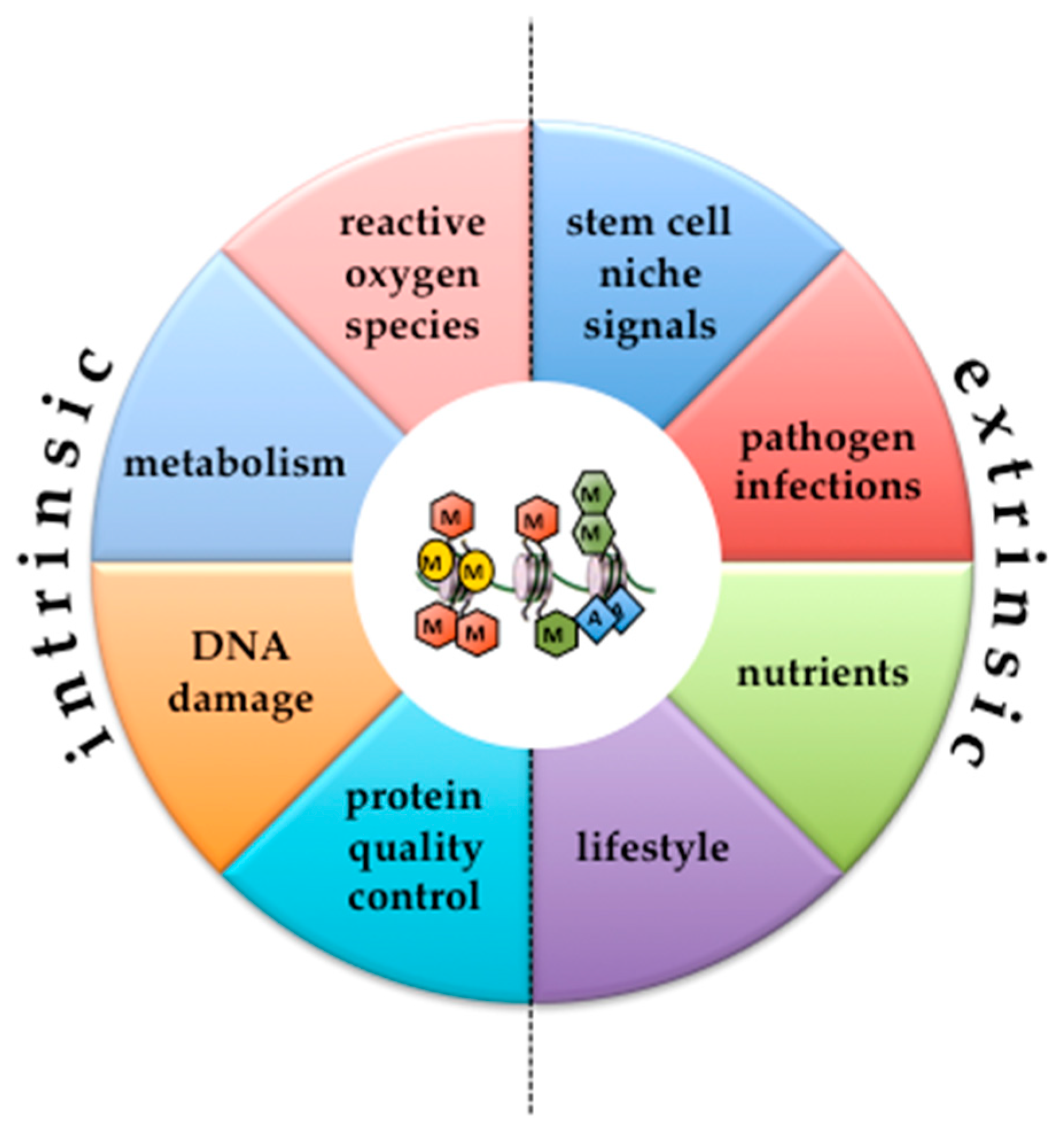{kind=link}
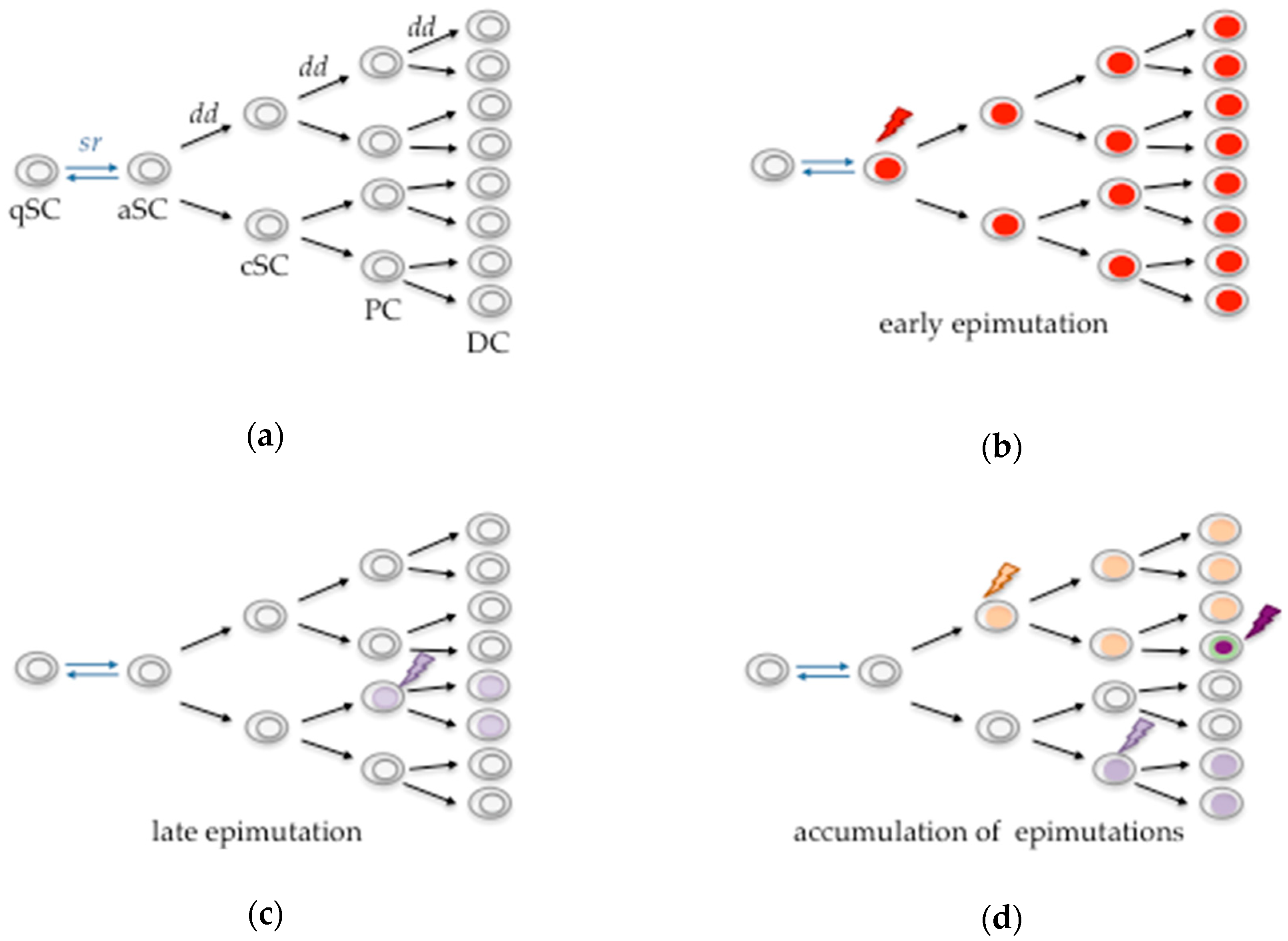{kind=link}
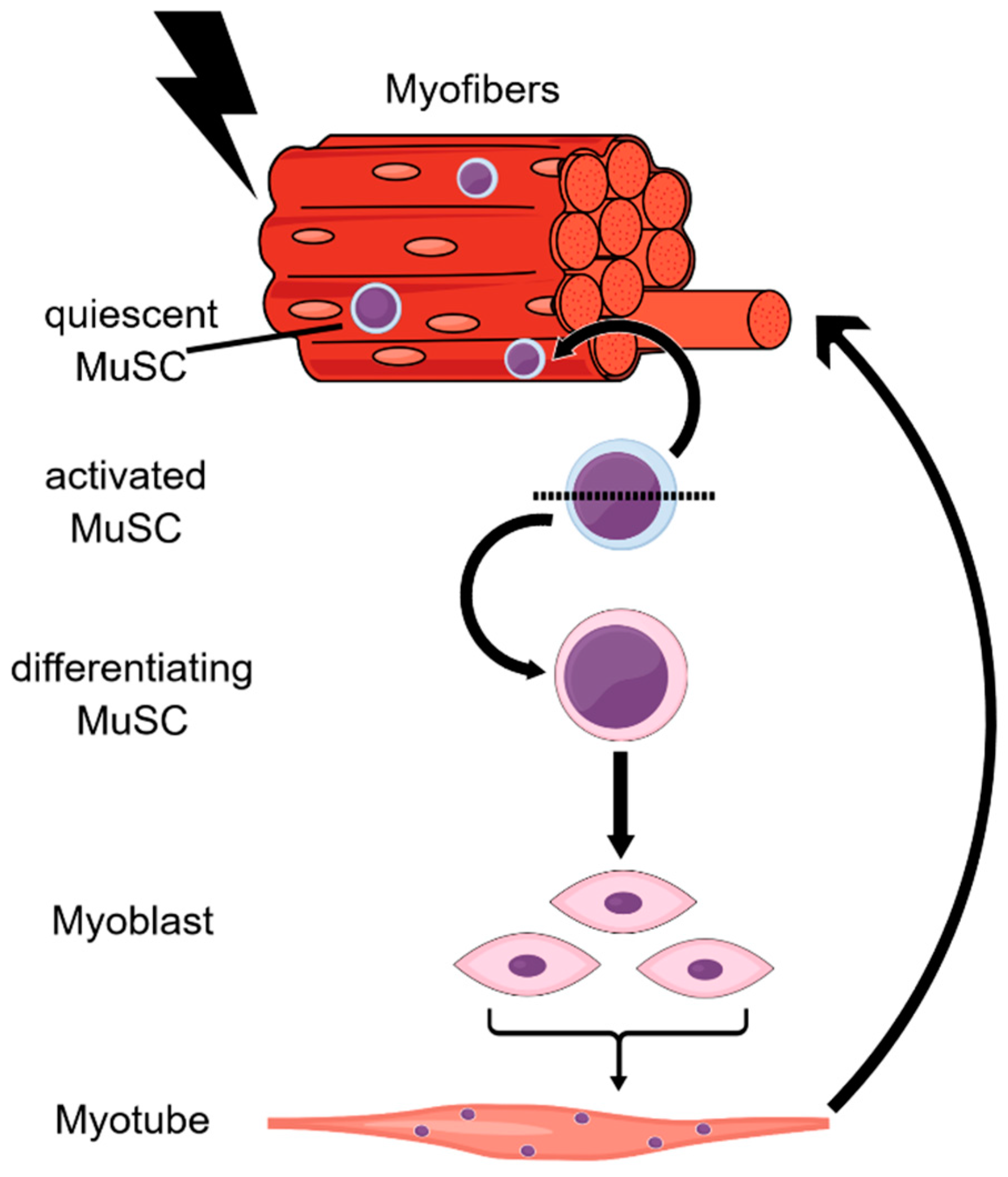{kind=link}
| Young MuSCs | Old MuSCs | |
|---|---|---|
| Quiescent MuSCs | H3K4me3 ● H3K27me3 ● bivalency ● H4K20me3 ↑ H4K16ac ↓ | H3K4me3 ↓ H3K27me3 ↑ (at histone genes & globally) H4K16ac ↑ |
| Activated MuSCs | H3K4me3 ● H3K27me3 ↑ bivalency ↑ H4K20me3 ↓ H4K16ac ↑ | H3K4me3 ↑↑ (at Hoxa9 locus) H3K27me 3● H4K16ac ↑↑ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kosan, C.; Heidel, F.H.; Godmann, M.; Bierhoff, H. Epigenetic Erosion in Adult Stem Cells: Drivers and Passengers of Aging. Cells 2018, 7, 237. https://doi.org/10.3390/cells7120237
Kosan C, Heidel FH, Godmann M, Bierhoff H. Epigenetic Erosion in Adult Stem Cells: Drivers and Passengers of Aging. Cells. 2018; 7(12):237. https://doi.org/10.3390/cells7120237
Chicago/Turabian StyleKosan, Christian, Florian H. Heidel, Maren Godmann, and Holger Bierhoff. 2018. "Epigenetic Erosion in Adult Stem Cells: Drivers and Passengers of Aging" Cells 7, no. 12: 237. https://doi.org/10.3390/cells7120237
APA StyleKosan, C., Heidel, F. H., Godmann, M., & Bierhoff, H. (2018). Epigenetic Erosion in Adult Stem Cells: Drivers and Passengers of Aging. Cells, 7(12), 237. https://doi.org/10.3390/cells7120237