Epigenetic Regulation of Skin Cells in Natural Aging and Premature Aging Diseases



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Key Regulators of Epidermal Homeostasis
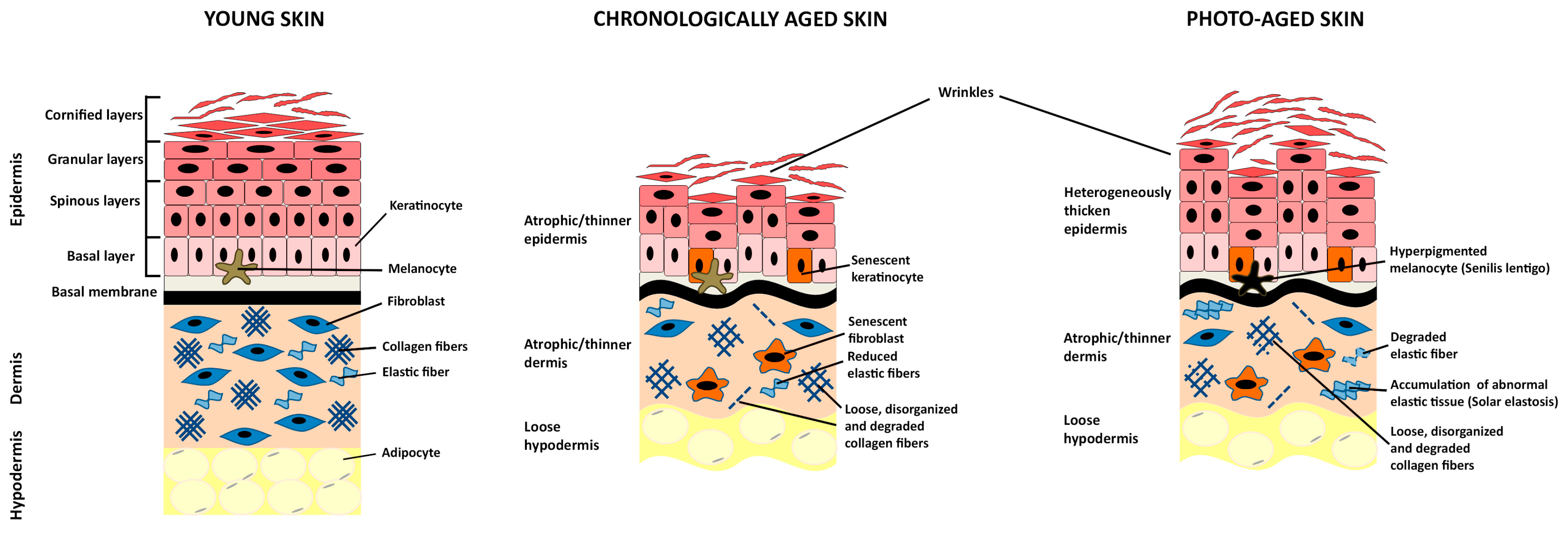
3. Skin Aging
3.1. Chronological Skin Aging
3.2. Photo-Aging
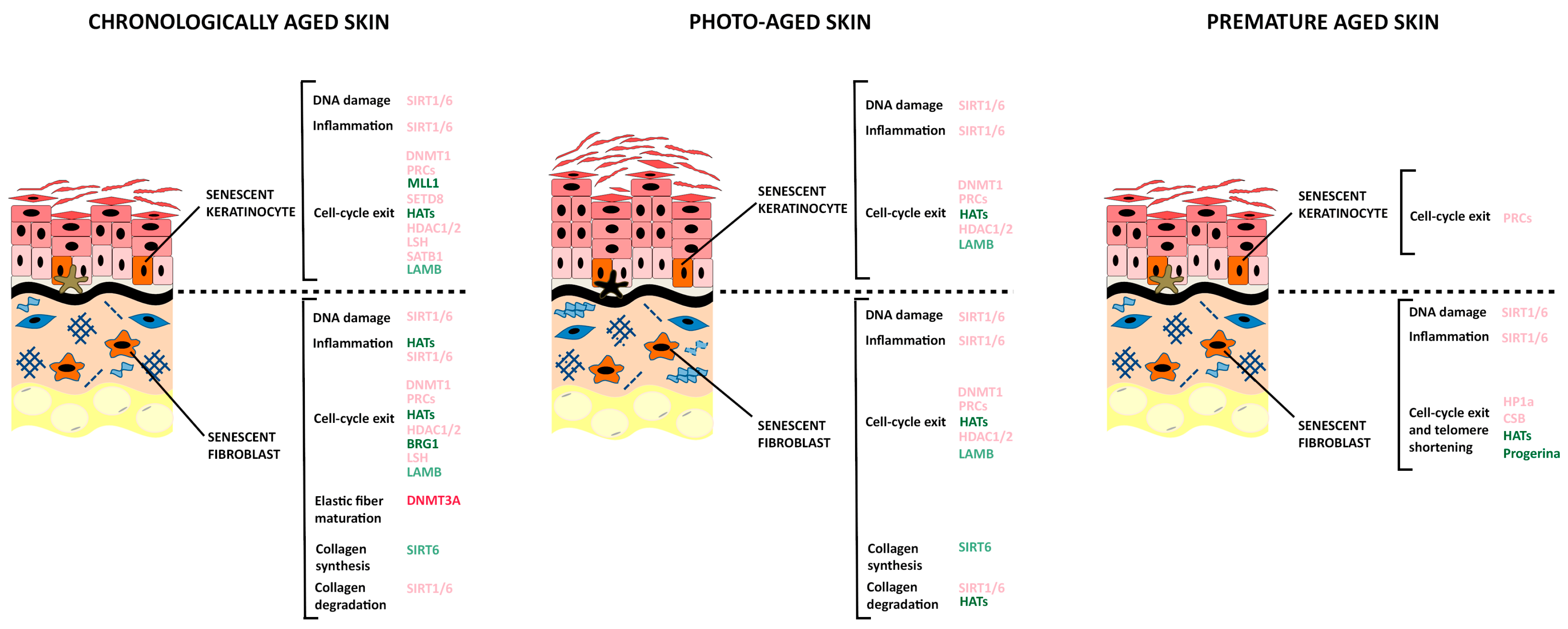
4. Epigenetic Changes in Skin Aging
4.1. DNA Methylation
4.2. Histone Methylation
4.3. Histone Acetylation
4.4. Higher-Order Chromatin Remodeling and Three-Dimensional Genome or Ganization
5. Epigenetic Regulation of Skin Premature Aging Diseases
5.1. Premature Aging Diseases
5.1.1. Progeroid Syndromes Resulting from DNA Repair Alterations
5.1.2. Progeroid Syndromes Associated to Alterations of the Nuclear Architecture
5.1.3. PSs Associated to Alterations of Telomere Metabolism
5.2. Cellular Senescence in Premature Aging Diseases
5.3. Epigenetic Changes in Premature Aging Diseases
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Murphree, R.W. Impairments in Skin Integrity. Nurs. Clin. N. Am. 2017, 52, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.; Li, L.; Fuchs, E. Emerging interactions between skin stem cells and their niches. Nat. Med. 2014, 20, 847–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrandon, Y.; Grasset, N.; Zaffalon, A.; Gorostidi, F.; Claudinot, S.; Droz-Georget, S.L.; Nanba, D.; Rochat, A. Capturing epidermal stemness for regenerative medicine. Semin. Cell Dev. Biol. 2012, 23, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, T.; Rothoeft, T.; Teig, N.; Bauer, J.W.; Pellegrini, G.; De Rosa, L.; Scaglione, D.; Reichelt, J.; Klausegger, A.; Kneisz, D.; et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature 2017, 551, 327–332. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, L.; De Luca, M. Cell biology: Dormant and restless skin stem cells. Nature 2012, 489, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Arseni, L.; Lombardi, A.; Orioli, D. From Structure to Phenotype: Impact of Collagen Alterations on Human Health. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Sriram, G.; Bigliardi, P.L.; Bigliardi-Qi, M. Fibroblast heterogeneity and its implications for engineering organotypic skin models in vitro. Eur. J. Cell Biol. 2015, 94, 483–512. [Google Scholar] [CrossRef] [PubMed]
- Driskell, R.R.; Lichtenberger, B.M.; Hoste, E.; Kretzschmar, K.; Simons, B.D.; Charalambous, M.; Ferron, S.R.; Herault, Y.; Pavlovic, G.; Ferguson-Smith, A.C.; et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 2013, 504, 277–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, R.L.; Adhikary, G.; Rorke, E.A.; Chew, Y.C.; Balasubramanian, S. Polycomb Group Proteins Are Key Regulators of Keratinocyte Function. J. Investig. Dermatol. 2011, 131, 295–301. [Google Scholar] [CrossRef] [Green Version]
- Maurelli, R.; Zambruno, G.; Guerra, L.; Abbruzzese, C.; Dimri, G.; Gellini, M.; Bondanza, S.; Dellambra, E. Inactivation of p16 INK4a ( inhibitor of cyclin-dependent kinase 4A ) immortalizes primary human keratinocytes by maintaining cells in the stem cell compartment. FASEB J. 2006, 20, 1516–1518. [Google Scholar] [CrossRef]
- Cordisco, S.; Maurelli, R.; Bondanza, S.; Stefanini, M.; Zambruno, G.; Guerra, L.; Dellambra, E. Bmi-1 reduction plays a key role in physiological and premature aging of primary human keratinocytes. J. Investig. Dermatol. 2010, 130, 1048–1062. [Google Scholar] [CrossRef] [PubMed]
- Rivetti di Val Cervo, P.; Lena, A.M.; Nicoloso, M.; Rossi, S.; Mancini, M.; Zhou, H.; Saintigny, G.; Dellambra, E.; Odorisio, T.; Mahé, C.; et al. p63-microRNA feedback in keratinocyte senescence. Proc. Natl. Acad. Sci. USA 2012, 109, 1133–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huschtscha, L.I. p16INK4a and the control of cellular proliferative life span. Carcinogenesis 1999, 20, 921–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpless, N.; DePinho, R. Telomeres, stem cells, senescence, and cancer. J. Clin. Investig. 2004, 113. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Bartolome, R.; Callejas-Valera, J.L.; Gutkind, J.S. Control of the epithelial stem cell epigenome: The shaping of epithelial stem cell identity. Curr. Opin. Cell Biol. 2013, 25, 162–169. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, D.; Tinaburri, L.; Dellambra, E. The Role of p16INK4a Pathway in Human Epidermal Stem Cell Self-Renewal, Aging and Cancer. Int. J. Mol. Sci. 2017, 18, 1591. [Google Scholar] [CrossRef] [PubMed]
- Mills, A.A.; Zheng, B.; Wang, X.J.; Vogel, H.; Roop, D.R.; Bradley, A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 1999, 398, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Schweitzer, R.; Sun, D.; Kaghad, M.; Walker, N.; Bronson, R.T.; Tabin, C.; Sharpe, A.; Caput, D.; Crum, C.; et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999, 398, 714–718. [Google Scholar] [CrossRef]
- Missero, C.; Antonini, D. Crosstalk among p53 family members in cutaneous carcinoma. Exp. Dermatol. 2014, 23, 143–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrini, G.; Dellambra, E.; Golisano, O.; Martinelli, E.; Fantozzi, I.; Bondanza, S.; Ponzin, D.; McKeon, F.; De Luca, M. p63 identifies keratinocyte stem cells. Proc. Natl. Acad. Sci. USA 2001, 98, 3156–3161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candi, E.; Amelio, I.; Agostini, M.; Melino, G. MicroRNAs and p63 in epithelial stemness. Cell Death Differ. 2015, 22, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Ellisen, L.W.; Ramsayer, K.D.; Johannessen, C.M.; Yang, A.; Beppu, H.; Minda, K.; Oliner, J.D.; McKeon, F.; Haber, D.A. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol. Cell 2002, 10, 995–1005. [Google Scholar] [CrossRef]
- Truong, A.B.; Kretz, M.; Ridky, T.W.; Kimmel, R.; Khavari, P.A. p63 regulates proliferation and differentiation of developmentally mature keratinocytes. Genes Dev. 2006, 20, 3185–3197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botchkarev, V.A.; Gdula, M.R.; Mardaryev, A.N.; Sharov, A.A.; Fessing, M.Y. Epigenetic Regulation of Gene Expression in Keratinocytes. J. Investig. Dermatol. 2012, 132, 2505–2521. [Google Scholar] [CrossRef] [PubMed]
- Frye, M.; Benitah, S.A. Chromatin regulators in mammalian epidermis. Semin. Cell Dev. Biol. 2012, 23, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Perdigoto, C.N.; Valdes, V.J.; Bardot, E.S.; Ezhkova, E. Epigenetic Regulation of Epidermal Differentiation. Cold Spring Harb. Perspect. Med. 2014, 4, a015263. [Google Scholar] [CrossRef]
- Adam, R.C.; Fuchs, E. The Yin and Yang of Chromatin Dynamics In Stem Cell Fate Selection. Trends Genet. 2016. [CrossRef] [PubMed]
- Avgustinova, A.; Benitah, S.A. Epigenetic control of adult stem cell function. Nat. Rev. Mol. Cell Biol. 2016, 17, 643–658. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investig. 2004, 114, 1299–1307. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Keyes, W.M.; Wu, Y.; Vogel, H.; Guo, X.; Lowe, S.W.; Mills, A.A. p63 deficiency activates a program of cellular senescence and leads to accelerated aging. Genes Dev. 2005, 19, 1986–1999. [Google Scholar] [CrossRef] [Green Version]
- Fisher, G.J.; Kang, S.; Varani, J.; Bata-Csorgo, Z.; Wan, Y.; Datta, S.; Voorhees, J.J. Mechanisms of photoaging and chronological skin aging. Arch. Dermatol. 2002, 138, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Park, H.J. Molecular Mechanisms of Skin Aging and Rejuvenation. In Molecular Mechanisms of the Aging Process and Rejuvenation; InTech: Manila, Philippines, 2016. [Google Scholar] [Green Version]
- Kang, S.; Fisher, G.J.; Voorhees, J.J. Photoaging: Pathogenesis, prevention, and treatment. Clin. Geriatr. Med. 2001, 17, 643–659. [Google Scholar] [CrossRef]
- Fuchs, E. Epithelial Skin Biology. In Current Topics in Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2016; pp. 357–374. [Google Scholar]
- Sharpless, N.E.; DePinho, R.A. How stem cells age and why this makes us grow old. Nat. Rev. Mol. Cell Biol. 2007, 8, 703–713. [Google Scholar] [CrossRef] [PubMed]
- d’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef]
- Ressler, S.; Bartkova, J.; Niederegger, H.; Bartek, J.; Scharffetter-Kochanek, K.; Jansen-Dürr, P.; Wlaschek, M. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell 2006, 5, 379–389. [Google Scholar] [CrossRef]
- Tigges, J.; Krutmann, J.; Fritsche, E.; Haendeler, J.; Schaal, H.; Fischer, J.W.; Kalfalah, F.; Reinke, H.; Reifenberger, G.; Stühler, K.; et al. The hallmarks of fibroblast ageing. Mech. Ageing Dev. 2014, 138, 26–44. [Google Scholar] [CrossRef]
- Krutmann, J.; Bouloc, A.; Sore, G.; Bernard, B.A.; Passeron, T. The skin aging exposome. J. Dermatol. Sci. 2017, 85, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Nũnez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef]
- Herbig, U.; Sedivy, J.M. Regulation of growth arrest in senescence: Telomere damage is not the end of the story. Mech. Ageing Dev. 2006, 127, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, T.; Qin, Z.; Voorhees, J.J.; Fisher, G.J. Cysteine-rich protein 61 (CCN1) mediates replicative senescence-associated aberrant collagen homeostasis in human skin fibroblasts. J. Cell. Biochem. 2012, 113, 3011–3018. [Google Scholar] [CrossRef] [PubMed]
- Yaar, M.; Gilchrest, B.A. Photoageing: Mechanism, prevention and therapy. Br. J. Dermatol. 2007, 157, 874–877. [Google Scholar] [CrossRef] [PubMed]
- Imokawa, G.; Ishida, K. Biological mechanisms underlying the ultraviolet radiation-induced formation of skin wrinkling and sagging I: Reduced skin elasticity, highly associated with enhanced dermal elastase activity, triggers wrinkling and sagging. Int. J. Mol. Sci. 2015, 16, 7753–7775. [Google Scholar] [CrossRef]
- Bernerd, F.; Vioux, C.; Asselineau, D. Evaluation of the Protective Effect of Sunscreens on In Vitro Reconstructed Human Skin Exposed to UVB or UVA Irradiation. Photochem. Photobiol. 2000, 71, 314. [Google Scholar] [CrossRef]
- Bernerd, F.; Asselineau, D. An organotypic model of skin to study photodamage and photoprotection in vitro. J. Am. Acad. Dermatol. 2008, 58, 8–12. [Google Scholar] [CrossRef]
- Duval, C.; Cohen, C.; Chagnoleau, C.; Flouret, V.; Bourreau, E.; Bernerd, F. Key Regulatory Role of Dermal Fibroblasts in Pigmentation as Demonstrated Using a Reconstructed Skin Model: Impact of Photo-Aging. PLoS ONE 2014, 9, e114182. [Google Scholar] [CrossRef]
- Fagot, D.; Asselineau, D.; Bernerd, F. Matrix metalloproteinase-1 production observed after solar-simulated radiation exposure is assumed by dermal fibroblasts but involves a paracrine activation through epidermal keratinocytes. Photochem. Photobiol. 2004, 79, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Bernerd, F.; Marionnet, C.; Duval, C. Solar ultraviolet radiation induces biological alterations in human skin in vitro: Relevance of a well-balanced UVA/UVB protection. Indian J. Dermatol. Venereol. Leprol. 2012, 78, 152. [Google Scholar] [CrossRef] [PubMed]
- D’Errico, M.; Lemma, T.; Calcagnile, A.; Proietti De Santis, L.; Dogliotti, E. Cell type and DNA damage specific response of human skin cells to environmental agents. Mutat. Res. 2007, 614, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Sidler, C.; Kovalchuk, O.; Kovalchuk, I. Epigenetic Regulation of Cellular Senescence and Aging. Front. Genet. 2017, 8, 138. [Google Scholar] [CrossRef] [PubMed]
- Rajarajacholan, U.K.; Riabowol, K. Aging with ING: A comparative study of different forms of stress induced premature senescence. Oncotarget 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, D.; Truman, A.W.; Kron, S.J.; Cote, J. Epigenetic Modifications in Double-Strand Break DNA Damage Signaling and Repair. Clin. Cancer Res. 2010, 16, 4543–4552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contrepois, K.; Coudereau, C.; Benayoun, B.A.; Schuler, N.; Roux, P.-F.; Bischof, O.; Courbeyrette, R.; Carvalho, C.; Thuret, J.-Y.; Ma, Z.; et al. Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression. Nat. Commun. 2017, 8, 14995. [Google Scholar] [CrossRef] [Green Version]
- Rishi, V.; Bhattacharya, P.; Chatterjee, R.; Rozenberg, J.; Zhao, J.; Glass, K.; Fitzgerald, P.; Vinson, C. CpG methylation of half-CRE sequences creates C/EBP binding sites that activate some tissue-specific genes. Proc. Natl. Acad. Sci. USA 2010, 107, 20311–20316. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, L.; Datta, D.; Serrat, J.; Morey, L.; Solanas, G.; Avgustinova, A.; Blanco, E.; Pons, J.I.; Matallanas, D.; Von Kriegsheim, A.; et al. Dnmt3a and Dnmt3b Associate with Enhancers to Regulate Human Epidermal Stem Cell Homeostasis. Cell Stem Cell 2016, 19, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Zampieri, M.; Ciccarone, F.; Calabrese, R.; Franceschi, C.; Bürkle, A.; Caiafa, P. Reconfiguration of DNA methylation in aging. Mech. Ageing Dev. 2015, 151, 60–70. [Google Scholar] [CrossRef]
- Feng, S.; Jacobsen, S.E.; Reik, W. Epigenetic Reprogramming in Plant and Animal Development. Science 2010, 330, 622–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, G.L.; Reuter, J.A.; Webster, D.E.; Zhu, L.; Khavari, P.A. DNMT1 maintains progenitor function in self-renewing somatic tissue. Nature 2010, 463, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, C.; Beerman, I.; Lien, W.H.; Smith, Z.D.; Gu, H.; Boyle, P.; Gnirke, A.; Fuchs, E.; Rossi, D.J.; Meissner, A. DNA Methylation Dynamics during In Vivo Differentiation of Blood and Skin Stem Cells. Mol. Cell 2012, 47, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Mulder, K.W.; Wang, X.; Escriu, C.; Ito, Y.; Schwarz, R.F.; Gillis, J.; Sirokmány, G.; Donati, G.; Uribe-Lewis, S.; Pavlidis, P.; et al. Diverse epigenetic strategies interact to control epidermal differentiation. Nat. Cell Biol. 2012, 14, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; West, J.; Beck, S. Age-associated epigenetic drift: Implications, and a case of epigenetic thrift? Hum. Mol. Genet. 2013, 22, R7–R15. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suñer, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, P.; Esteller, M.; Vaag, A.; Fraga, M.F. The epigenetic basis of twin discordance in age-related diseases. Pediatr. Res. 2007, 61, 38R–42R. [Google Scholar] [CrossRef] [PubMed]
- Grönniger, E.; Weber, B.; Heil, O.; Peters, N.; Stäb, F.; Wenck, H.; Korn, B.; Winnefeld, M.; Lyko, F. Aging and chronic sun exposure cause distinct epigenetic changes in human skin. PLoS Genet. 2010, 6, 6. [Google Scholar] [CrossRef]
- Raddatz, G.; Hagemann, S.; Aran, D.; Söhle, J.; Kulkarni, P.P.; Kaderali, L.; Hellman, A.; Winnefeld, M.; Lyko, F. Aging is associated with highly defined epigenetic changes in the human epidermis. Epigenetics Chromatin 2013, 6, 1–12. [Google Scholar] [CrossRef]
- Bormann, F.; Rodríguez-Paredes, M.; Hagemann, S.; Manchanda, H.; Kristof, B.; Gutekunst, J.; Raddatz, G.; Haas, R.; Terstegen, L.; Wenck, H.; et al. Reduced DNA methylation patterning and transcriptional connectivity define human skin aging. Aging Cell 2016, 15, 563–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, C.M.; Suschek, C.V.; Lin, Q.; Bork, S.; Goergens, M.; Joussen, S.; Pallua, N.; Ho, A.D.; Zenke, M.; Wagner, W. Specific Age-Associated DNA Methylation Changes in Human Dermal Fibroblasts. PLoS ONE 2011, 6, e16679. [Google Scholar] [CrossRef] [PubMed]
- Landan, G.; Cohen, N.M.; Mukamel, Z.; Bar, A.; Molchadsky, A.; Brosh, R.; Horn-Saban, S.; Zalcenstein, D.A.; Goldfinger, N.; Zundelevich, A.; et al. Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat. Genet. 2012, 44, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Lowe, R.; Overhoff, M.G.; Ramagopalan, S.V.; Garbe, J.C.; Koh, J.; Stampfer, M.R.; Beach, D.H.; Rakyan, V.K.; Bishop, C.L. The senescent methylome and its relationship with cancer, ageing and germline genetic variation in humans. Genome Biol. 2015, 16, 194. [Google Scholar] [CrossRef] [PubMed]
- Pérez, R.F.; Tejedor, J.R.; Bayón, G.F.; Fernández, A.F.; Fraga, M.F. Distinct chromatin signatures of DNA hypomethylation in aging and cancer. Aging Cell 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Vandiver, A.R.; Irizarry, R.A.; Hansen, K.D.; Garza, L.A.; Runarsson, A.; Li, X.; Chien, A.L.; Wang, T.S.; Leung, S.G.; Kang, S.; et al. Age and sun exposure-related widespread genomic blocks of hypomethylation in nonmalignant skin. Genome Biol. 2015, 16, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Pastor, W.A.; Shen, Y.; Tahiliani, M.; Liu, D.R.; Rao, A. The Behaviour of 5-Hydroxymethylcytosine in Bisulfite Sequencing. PLoS ONE 2010, 5, e8888. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Jiao, Y.; de Jong, S.; Ophoff, R.A.; Beck, S.; Teschendorff, A.E. An integrative multi-scale analysis of the dynamic DNA methylation landscape in aging. PLoS Genet. 2015, 11, e1004996. [Google Scholar] [CrossRef]
- Li, J.; Jiang, T.-X.; Hughes, M.W.; Wu, P.; Widelitz, R.B.; Fan, G.; Chuong, C. Progressive Alopecia Reveals Decreasing Stem Cell Activation Probability during Aging of Mice with Epidermal Deletion of DNA Methyltransferase 1. J. Investig. Dermatol. 2012, 132, 2681–2690. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.F.; Liu, Y.Z.; Du, R.; Wang, B.; Chen, M.T.; Zhang, Y.Y.; Deng, H.L.; Li, J. Mir-377 induces senescence in human skin fibroblasts by targeting DNA methyltransferase 1. Cell Death Dis. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Zheng, Q.H.; Ma, L.W.; Zhu, W.G.; Zhang, Z.Y.; Tong, T.J. p21Waf1/Cip1 plays a critical role in modulating senescence through changes of DNA methylation. J. Cell. Biochem. 2006, 98, 1230–1248. [Google Scholar] [CrossRef]
- Jung, H.J.; Byun, H.O.; Jee, B.A.; Min, S.; Jeoun, U.W.; Lee, Y.K.; Seo, Y.; Woo, H.G.; Yoon, G. The ubiquitin-like with PHD and ring finger domains 1 (UHRF1)/DNA methyltransferase 1 (DNMT1) axis is a primary regulator of cell senescence. J. Biol. Chem. 2017, 292, 3729–3739. [Google Scholar] [CrossRef] [PubMed]
- Moulin, L.; Cenizo, V.; Antu, A.N.; André, V.; Pain, S.; Sommer, P.; Debret, R. Methylation of LOXL1 Promoter by DNMT3A in Aged Human Skin Fibroblasts. Rejuvenation Res. 2017, 20, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Xie, H.; Xiao, X.; Wang, B.; Du, R.; Liu, Y.; Li, Z.; Wang, J.; Sun, L.; Deng, Z.; et al. Ultraviolet a irradiation induces senescence in human dermal fibroblasts by down-regulating DNMT1 via ZEB1. Aging (Albany NY) 2018, 10, 212–228. [Google Scholar] [CrossRef] [PubMed]
- Lahtz, C.; Kim, S.-I.; Bates, S.E.; Li, A.X.; Wu, X.; Pfeifer, G.P. UVB irradiation does not directly induce detectable changes of DNA methylation in human keratinocytes. F1000Research 2013, 2, 45. [Google Scholar] [CrossRef]
- Wang, D.; Huang, J.H.; Zeng, Q.H.; Gu, C.; Ding, S.; Lu, J.Y.; Chen, J.; Yang, S.B. Increased 5-hydroxymethylcytosine and ten-eleven translocation protein expression in ultraviolet B-irradiated HaCaT cells. Chin. Med. J. 2017, 130, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bardot, E.; Ezhkova, E. Epigenetic regulation of skin: Focus on the Polycomb complex. Cell. Mol. Life Sci. 2012, 69, 2161–2172. [Google Scholar] [CrossRef]
- Gil, J.; O’Loghlen, A. PRC1 complex diversity: Where is it taking us? Trends Cell Biol. 2014, 24, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, K.; O’Neil, K.; Capell, B.C. Histone modifiers: Dynamic regulators of the cutaneous transcriptome. J. Dermatol. Sci. 2017, 89, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Blackledge, N.P.; Farcas, A.M.; Kondo, T.; King, H.W.; McGouran, J.F.; Hanssen, L.L.P.; Ito, S.; Cooper, S.; Kondo, K.; Koseki, Y.; et al. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 2014, 157, 1445–1459. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.; Szabo, V.; Pirity, M.K. Absence of Rybp Compromises Neural Differentiation of Embryonic Stem Cells. Stem Cells Int. 2016, 2016, 4034620. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Tong, H.; Huang, Y.; Yan, Y.; Teng, H.; Xia, Y.; Jiang, Q.; Qin, J. Essential Role for Polycomb Group Protein Pcgf6 in Embryonic Stem Cell Maintenance and a Noncanonical Polycomb Repressive Complex 1 (PRC1) Integrity. J. Biol. Chem. 2017, 292, 2773–2784. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Liu, M.; Ji, H.; Zhu, Y.; Wang, C.; Huang, Y.; Ma, X.; Xing, G.; Xia, Y.; Jiang, Q.; et al. The polycomb group protein Yaf2 regulates the pluripotency of embryonic stem cells in a phosphorylation-dependent manner. J. Biol. Chem. 2018, 293, 12793–12804. [Google Scholar] [CrossRef] [PubMed]
- Sen, G.L.; Webster, D.E.; Barragan, D.I.; Chang, H.Y.; Khavari, P.A. Control of differentiation in a self-renewing mammalian tissue by the histone demethylase JMJD3. Genes Dev. 2008, 22, 1865–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimri, M.; Carroll, J.D.; Cho, J.H.; Dimri, G.P. MicroRNA-141 regulates BMI1 expression and induces senescence in human diploid fibroblasts. Cell Cycle 2013, 12, 3537–3546. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.J.; Kieboom, K.; Marino, S.; DePinho, R.A.; van Lohuizen, M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999, 397, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Tschen, S.-I.; Dhawan, S.; Gurlo, T.; Bhushan, A. Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes 2009, 58, 1312–1320. [Google Scholar] [CrossRef]
- Schuettengruber, B.; Martinez, A.-M.; Iovino, N.; Cavalli, G. Trithorax group proteins: Switching genes on and keeping them active. Nat. Rev. Mol. Cell Biol. 2011, 12, 799–814. [Google Scholar] [CrossRef]
- Hopkin, A.S.; Gordon, W.; Klein, R.H.; Espitia, F.; Daily, K.; Zeller, M.; Baldi, P.; Andersen, B. GRHL3/GET1 and trithorax group members collaborate to activate the epidermal progenitor differentiation program. PLoS Genet. 2012, 8. [Google Scholar] [CrossRef]
- Kotake, Y.; Zeng, Y.; Xiong, Y. DDB1-CUL4 and MLL1 mediate oncogene-induced p16INK4a activation. Cancer Res. 2009, 69, 1809–1814. [Google Scholar] [CrossRef]
- Wu, Z.; Zhang, L. Polycomb group proteins: Novel molecules associated with ultraviolet A-induced photoaging of human skin. Exp. Ther. Med. 2017, 14, 2554–2562. [Google Scholar] [CrossRef] [Green Version]
- Qin, H.; Zhang, G.; Zhang, L. GSK126 (EZH2 inhibitor) interferes with ultraviolet A radiation-induced photoaging of human skin fibroblast cells. Exp. Ther. Med. 2018, 15, 3439–3448. [Google Scholar] [CrossRef] [PubMed]
- Greussing, R.; Hackl, M.; Charoentong, P.; Pauck, A.; Monteforte, R.; Cavinato, M.; Hofer, E.; Scheideler, M.; Neuhaus, M.; Micutkova, L.; et al. Identification of microRNA-mRNA functional interactions in UVB-induced senescence of human diploid fibroblasts. BMC Genom. 2013, 14, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Gesumaria, L.; Matsui, M.S.; Kluz, T.; Costa, M. Solar-simulated ultraviolet radiation induces histone 3 methylation changes in the gene promoters of matrix metalloproteinases 1 and 3 in primary human dermal fibroblasts. Exp. Dermatol. 2015, 24, 384–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driskell, I.; Oda, H.; Blanco, S.; Nascimento, E.; Humphreys, P.; Frye, M. The histone methyltransferase Setd8 acts in concert with c-Myc and is required to maintain skin. EMBO J. 2012, 31, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Takebayashi, S.-I.; Sakamoto, A.; Igata, T.; Nakatsu, Y.; Saitoh, N.; Hino, S.; Nakao, M. The SETD8/PR-Set7 Methyltransferase Functions as a Barrier to Prevent Senescence-Associated Metabolic Remodeling. Cell Rep. 2017, 18, 2148–2161. [Google Scholar] [CrossRef] [PubMed]
- Restelli, M.; Molinari, E.; Marinari, B.; Conte, D.; Gnesutta, N.; Costanzo, A.; Merlo, G.R.; Guerrini, L. FGF8, c-Abl and p300 participate in a pathway that controls stability and function of the ΔNp63 α protein. Hum. Mol. Genet. 2015, 24, 4185–4197. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.-P.; Pickard, A.; McCance, D.J. p300 Alters Keratinocyte Cell Growth and Differentiation through Regulation of p21Waf1/CIP1. PLoS ONE 2010, 5, e8369. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Poi, M.J.; Tsai, M. Regulatory mechanisms of tumor suppressor P16(INK4A) and their relevance to cancer. Biochemistry 2011, 50, 5566–5582. [Google Scholar] [CrossRef]
- Wang, W.; Pan, K.; Chen, Y.; Huang, C.; Zhang, X. The acetylation of transcription factor HBP1 by p300/CBP enhances p16INK4A expression. Nucleic Acids Res. 2012, 40, 981–995. [Google Scholar] [CrossRef]
- Bin, B.-H.; Lee, S.-H.; Bhin, J.; Irié, T.; Kim, S.; Seo, J.; Mishima, K.; Lee, T.R.; Hwang, D.; Fukada, T.; et al. The epithelial zinc transporter ZIP10 epigenetically regulates human epidermal homeostasis by modulating histone acetyltransferase activity. Br. J. Dermatol. 2018. [Google Scholar] [CrossRef]
- Cheng, H.-H.; Wang, K.-H.; Chu, L.; Chang, T.-C.; Kuo, C.-C.; Wu, K.K. Quiescent and proliferative fibroblasts exhibit differential p300 HAT activation through control of 5-methoxytryptophan production. PLoS ONE 2014, 9, e88507. [Google Scholar] [CrossRef] [PubMed]
- LeBoeuf, M.; Terrell, A.; Trivedi, S.; Sinha, S.; Epstein, J.A.; Olson, E.N.; Morrisey, E.E.; Millar, S.E. Hdac1 and Hdac2 act redundantly to control p63 and p53 functions in epidermal progenitor cells. Dev. Cell 2010, 19, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Peterson, L.M.; Wilking-Busch, M.J.; Ndiaye, M.A.; Philippe, C.G.A.C.G.A.; Setaluri, V.; Ahmad, N. Sirtuins in Skin and Skin Cancers. Skin Pharmacol. Physiol. 2017, 30, 216–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohguchi, K.; Itoh, T.; Akao, Y.; Inoue, H.; Nozawa, Y.; Ito, M. SIRT1 modulates expression of matrix metalloproteinases in human dermal fibroblasts. Br. J. Dermatol. 2010, 163, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Baohua, Y.; Li, L. Effects of SIRT6 silencing on collagen metabolism in human dermal fibroblasts. Cell Biol. Int. 2012, 36, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Goyarts, E.C.; Dong, K.; Pelle, E.; Pernodet, N. Effect of SIRT6 knockdown on NF-κB induction and on residual DNA damage in cultured human skin fibroblasts. J. Cosmet. Sci. 2017, 68, 25–33. [Google Scholar] [PubMed]
- Joo, D.; An, S.; Choi, B.G.; Kim, K.; Choi, Y.M.; Ahn, K.J.; An, I.-S.; Cha, H.J. MicroRNA-378b regulates α-1-type 1 collagen expression via sirtuin 6 interference. Mol. Med. Rep. 2017, 16, 8520–8524. [Google Scholar] [CrossRef]
- Vieyra, D.; Loewith, R.; Scott, M.; Bonnefin, P.; Boisvert, F.-M.; Cheema, P.; Pastyryeva, S.; Meijer, M.; Johnston, R.N.; Bazett-Jones, D.P.; et al. Human ING1 proteins differentially regulate histone acetylation. J. Biol. Chem. 2002, 277, 29832–29839. [Google Scholar] [CrossRef]
- Soliman, M.A.; Berardi, P.; Pastyryeva, S.; Bonnefin, P.; Feng, X.; Colina, A.; Young, D.; Riabowol, K. ING1a expression increases during replicative senescence and induces a senescent phenotype. Aging Cell 2008, 7, 783–794. [Google Scholar] [CrossRef] [Green Version]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic mechanisms regulating longevity and aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef]
- Herranz, D.; Muñoz-Martin, M.; Cañamero, M.; Mulero, F.; Martinez-Pastor, B.; Fernandez-Capetillo, O.; Serrano, M. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat. Commun. 2010, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Mostoslavsky, R.; Chua, K.F.; Lombard, D.B.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Mostoslavsky, G.; Franco, S.; Murphy, M.M.; et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Massudi, H.; Grant, R.; Braidy, N.; Guest, J.; Farnsworth, B.; Guillemin, G.J. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS ONE 2012, 7, 1–9. [Google Scholar] [CrossRef]
- Ido, Y.; Duranton, A.; Lan, F.; Weikel, K.A.; Breton, L.; Ruderman, N.B. Resveratrol prevents oxidative stress-induced senescence and proliferative dysfunction by activating the AMPK-FOXO3 cascade in cultured primary human keratinocytes. PLoS ONE 2015, 10, 1–18. [Google Scholar] [CrossRef]
- Carlomosti, F.; D’Agostino, M.; Beji, S.; Torcinaro, A.; Rizzi, R.; Zaccagnini, G.; Maimone, B.; Di Stefano, V.; De Santa, F.; Cordisco, S.; et al. Oxidative Stress-Induced miR-200c Disrupts the Regulatory Loop Among SIRT1, FOXO1, and eNOS. Antioxid. Redox Signal. 2017, 27, 328–344. [Google Scholar] [CrossRef]
- Kim, K.S.; Park, H.-K.; Lee, J.-W.; Kim, Y.I.; Shin, M.K. Investigate correlation between mechanical property and aging biomarker in passaged human dermal fibroblasts. Microsc. Res. Tech. 2015, 78, 277–282. [Google Scholar] [CrossRef] [PubMed]
- de Cabo, R.; Liu, L.; Ali, A.; Price, N.; Zhang, J.; Wang, M.; Lakatta, E.; Irusta, P.M. Serum from calorie-restricted animals delays senescence and extends the lifespan of normal human fibroblasts in vitro. Aging (Albany NY) 2015, 7, 152–166. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Gan, Q.; Han, L.; Li, J.; Zhang, H.; Sun, Y.; Zhang, Z.; Tong, T. SIRT1 overexpression antagonizes cellular senescence with activated ERK/S6k1 signaling in human diploid fibroblasts. PLoS ONE 2008, 3. [Google Scholar] [CrossRef]
- Xu, Z.; Zhang, L.; Zhang, W.; Meng, D.; Zhang, H.; Jiang, Y.; Xu, X.; Meter, M.V.; Seluanov, A.; Gorbunova, V.; et al. Van SIRT6 rescues the age related decline in base excision repair in a PARP1-dependent manner SIRT6 rescues the age related decline in base excision repair in a PARP1-dependent manner. Cell Cycle 2015, 4101, 1551–4005. [Google Scholar] [CrossRef]
- Tinaburri, L.; D’Errico, M.; Sileno, S.; Maurelli, R.; Degan, P.; Magenta, A.; Dellambra, E. miR-200a Modulates the Expression of the DNA Repair Protein OGG1 Playing a Role in Aging of Primary Human Keratinocytes. Oxid. Med. Cell. Longev. 2018, 2018, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Chen, J.; Zeng, Q.; Lu, J.; Tan, L.; Guo, A.; Kang, J.; Yang, S.; Xiang, Y.; Zuo, C.; et al. Chronic sun exposure is associated with distinct histone acetylation changes in human skin. Br. J. Dermatol. 2018, 179, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Pollack, B.P.; Sapkota, B.; Boss, J.M. Ultraviolet Radiation-induced Transcription is Associated with Gene-specific Histone Acetylation. Photochem. Photobiol. 2009, 85, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Shin, J.M.; Eun, H.C.; Chung, J.H. The role of p300 histone acetyltransferase in UV-induced histone modifications and MMP-1 gene transcription. PLoS ONE 2009, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Luo, J. SIRT1 Regulates UV-Induced DNA Repair through Deacetylating XPA. Mol. Cell 2010, 39, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Liu, W.; Niu, T.; Dai, C.; Li, X.; Cui, C.; Zhao, X.; E, Y.; Lu, H. The injury and cumulative effects on human skin by UV exposure from artificial fluorescence emission. Photochem. Photobiol. 2014, 90, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Lu, S.; Kivlin, R.; Wallin, B.; Card, E.; Bagdasarian, A.; Tamakloe, T.; Wang, W.; Song, X.; Chu, W.; et al. SIRT1 confers protection against UVB- and H2O2-induced cell death via modulation of p53 and JNK in cultured skin keratinocytes. J. Cell. Mol. Med. 2009, 13, 3632–3643. [Google Scholar] [CrossRef] [PubMed]
- Wahedi, H.M.; Lee, T.H.; Moon, E.-Y.; Kim, S.Y. Juglone up-regulates sirt1 in skin cells under normal and UVB irradiated conditions. J. Dermatol. Sci. 2016, 81, 210–212. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.W.; Choi, Y.J.; Park, M.H.; Jang, E.J.; Kim, D.H.; Park, B.H.; Yu, B.P.; Chung, H.Y. Molecular insights into SIRT1 protection against UVB-induced skin fibroblast senescence by suppression of oxidative stress and p53 acetylation. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 959–968. [Google Scholar] [CrossRef]
- Lee, J.-S.; Park, K.-Y.; Min, H.-G.; Lee, S.J.; Kim, J.-J.; Choi, J.-S.; Kim, W.-S.; Cha, H.-J. Negative regulation of stress-induced matrix metalloproteinase-9 by Sirt1 in skin tissue. Exp. Dermatol. 2010, 19, 1060–1066. [Google Scholar] [CrossRef]
- Ming, M.; Soltani, K.; Shea, C.R.; Li, X.; He, Y.Y. Dual role of SIRT1 in UVB-induced skin tumorigenesis. Oncogene 2014, 34, 1–7. [Google Scholar] [CrossRef]
- Benavente, C.A.; Schnell, S.A.; Jacobson, E.L. Effects of niacin restriction on sirtuin and PARP responses to photodamage in human skin. PLoS ONE 2012, 7, e42276. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.; Grether-Beck, S.; Singh, M.; Kuck, F.; Jakob, S.; Kefalas, A.; Altinoluk-Hambüchen, S.; Graffmann, N.; Schneider, M.; Lindecke, A.; et al. MicroRNA-15b regulates mitochondrial ROS production and the senescence-associated secretory phenotype through sirtuin 4/SIRT4. Aging (Albany NY) 2016, 8, 484–505. [Google Scholar] [CrossRef] [PubMed]
- Botchkarev, V.A. The Molecular Revolution in Cutaneous Biology: Chromosomal Territories, Higher-Order Chromatin Remodeling, and the Control of Gene Expression in Keratinocytes. J. Investig. Dermatol. 2017, 137, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Fessing, M.Y.; Mardaryev, A.N.; Gdula, M.R.; Sharov, A.A.; Sharova, T.Y.; Rapisarda, V.; Gordon, K.B.; Smorodchenko, A.D.; Poterlowicz, K.; Ferone, G.; et al. p63 regulates Satb1 to control tissue-specific chromatin remodeling during development of the epidermis. J. Cell Biol. 2011, 194, 825–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keyes, W.M.; Pecoraro, M.; Aranda, V.; Vernersson-Lindahl, E.; Li, W.; Vogel, H.; Guo, X.; Garcia, E.L.; Michurina, T.V.; Enikolopov, G.; et al. A ΔNp63α is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell 2011, 8, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Mardaryev, A.N.; Gdula, M.R.; Yarker, J.L.; Emelianov, V.U.; Emelianov, V.N.; Poterlowicz, K.; Sharov, A.A.; Sharova, T.Y.; Scarpa, J.A.; Joffe, B.; et al. p63 and Brg1 control developmentally regulated higher-order chromatin remodelling at the epidermal differentiation complex locus in epidermal progenitor cells. Development 2014, 141, 101–111. [Google Scholar] [CrossRef]
- Rapisarda, V.; Malashchuk, I.; Asamaowei, I.E.; Poterlowicz, K.; Fessing, M.Y.; Sharov, A.A.; Karakesisoglou, I.; Botchkarev, V.A.; Mardaryev, A. p63 Transcription Factor Regulates Nuclear Shape and Expression of Nuclear Envelope-Associated Genes in Epidermal Keratinocytes. J. Investig. Dermatol. 2017, 137, 2157–2167. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, L.; Benitah, S.A. Epigenetic regulation of adult stem cell function. FEBS J. 2015, 282, 1589–1604. [Google Scholar] [CrossRef]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef]
- Indra, A.K.; Dupé, V.; Bornert, J.-M.; Messaddeq, N.; Yaniv, M.; Mark, M.; Chambon, P.; Metzger, D. Temporally controlled targeted somatic mutagenesis in embryonic surface ectoderm and fetal epidermal keratinocytes unveils two distinct developmental functions of BRG1 in limb morphogenesis and skin barrier formation. Development 2005, 132, 4533–4544. [Google Scholar] [CrossRef] [Green Version]
- Jaubert, J.; Cheng, J.; Segre, J.A. Ectopic expression of kruppel like factor 4 (Klf4) accelerates formation of the epidermal permeability barrier. Development 2003, 130, 2767–2777. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Tang, J.; Lopez-Pajares, V.; Tao, S.; Qu, K.; Crabtree, G.R.; Khavari, P.A. ACTL6a Enforces the Epidermal Progenitor State by Suppressing SWI/SNF-Dependent Induction of KLF4. Cell Stem Cell 2013, 12, 193–203. [Google Scholar] [CrossRef]
- Panatta, E.; Lena, A.M.; Mancini, M.; Affinati, M.; Smirnov, A.; Annicchiarico-Petruzzelli, M.; Piro, M.C.; Campione, E.; Bianchi, L.; Mazzanti, C.; et al. Kruppel-like factor 4 regulates keratinocyte senescence. Biochem. Biophys. Res. Commun. 2018, 499, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Zhuang, X.; Yao, Y.-G.; Zhang, R. BRG1 is required for formation of senescence-associated heterochromatin foci induced by oncogenic RAS or BRCA1 loss. Mol. Cell. Biol. 2013, 33, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Park, E.-J.; Lee, H.-S.; Kim, S.J.; Hur, S.-K.; Imbalzano, A.N.; Kwon, J. Mammalian SWI/SNF complexes facilitate DNA double-strand break repair by promoting gamma-H2AX induction. EMBO J. 2006, 25, 3986–3997. [Google Scholar] [CrossRef]
- Muegge, K. Lsh: A guardian of heterochromatin at repeat elements. Biochem. Cell Biol. 2005, 83, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Xi, S.; Zhu, H.; Xu, H.; Schmidtmann, A.; Geiman, T.M.; Muegge, K. Lsh controls Hox gene silencing during development. Proc. Natl. Acad. Sci. USA 2007, 104, 14366–14371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myant, K.; Stancheva, I. LSH cooperates with DNA methyltransferases to repress transcription. Mol. Cell. Biol. 2008, 28, 215–226. [Google Scholar] [CrossRef]
- Zhou, R.; Han, L.; Li, G.; Tong, T. Senescence delay and repression of p16INK4a by Lsh via recruitment of histone deacetylases in human diploid fibroblasts. Nucleic Acids Res. 2009, 37, 5183–5196. [Google Scholar] [CrossRef]
- Niu, J.; Chen, T.; Han, L.; Wang, P.; Li, N.; Tong, T. Transcriptional activation of the senescence regulator Lsh by E2F1. Mech. Ageing Dev. 2011, 132, 180–186. [Google Scholar] [CrossRef]
- Ohlsson, R.; Bartkuhn, M.; Renkawitz, R. CTCF shapes chromatin by multiple mechanisms: The impact of 20 years of CTCF research on understanding the workings of chromatin. Chromosoma 2010, 119, 351–560. [Google Scholar] [CrossRef] [PubMed]
- Witcher, M.; Emerson, B.M. Epigenetic silencing of the p16(INK4a) tumor suppressor is associated with loss of CTCF binding and a chromatin boundary. Mol. Cell 2009, 34, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Rosa-Garrido, M.; Ceballos, L.; Alonso-Lecue, P.; Abraira, C.; Delgado, M.D.; Gandarillas, A. A cell cycle role for the epigenetic factor CTCF-L./BORIS. PLoS ONE 2012, 7, e39371. [Google Scholar] [CrossRef] [PubMed]
- Han, H.-J.; Russo, J.; Kohwi, Y.; Kohwi-Shigematsu, T. SATB1 reprogrammes gene expression to promote breast tumour growth and metastasis. Nature 2008, 452, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Lena, A.M.; Mancini, M.; Rivetti di Val Cervo, P.; Saintigny, G.; Mahé, C.; Melino, G.; Candi, E. MicroRNA-191 triggers keratinocytes senescence by SATB1 and CDK6 downregulation. Biochem. Biophys. Res. Commun. 2012, 423, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Agrelo, R.; Kishimoto, H.; Novatchkova, M.; Peraza, V.; Paolino, M.; Souabni, A.; Wutz, A. SATB1 collaborates with loss of p16 in cellular transformation. Oncogene 2013, 32, 5492–5500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dechat, T.; Pfleghaar, K.; Sengupta, K.; Shimi, T.; Shumaker, D.K.; Solimando, L.; Goldman, R.D. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008, 22, 832–853. [Google Scholar] [CrossRef]
- Shimi, T.; Pfleghaar, K.; Kojima, S.; Pack, C.-G.; Solovei, I.; Goldman, A.E.; Adam, S.A.; Shumaker, D.K.; Kinjo, M.; Cremer, T.; et al. The A- and B-type nuclear lamin networks: Microdomains involved in chromatin organization and transcription. Genes Dev. 2008, 22, 3409–3421. [Google Scholar] [CrossRef]
- Jung, H.-J.; Tatar, A.; Tu, Y.; Nobumori, C.; Yang, S.H.; Goulbourne, C.N.; Herrmann, H.; Fong, L.G.; Young, S.G. An absence of nuclear lamins in keratinocytes leads to ichthyosis, defective epidermal barrier function, and intrusion of nuclear membranes and endoplasmic reticulum into the nuclear chromatin. Mol. Cell. Biol. 2014, 34, 4534–4544. [Google Scholar] [CrossRef]
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; Hamanaka, R.B.; Goldman, A.E.; Lucas, C.A.; Shumaker, D.K.; Kosak, S.T.; Chandel, N.S.; Goldman, R.D. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev. 2011, 25, 2579–2593. [Google Scholar] [CrossRef] [Green Version]
- Dreesen, O.; Chojnowski, A.; Ong, P.F.; Zhao, T.Y.; Common, J.E.; Lunny, D.; Lane, E.B.; Lee, S.J.; Vardy, L.A.; Stewart, C.L.; Colman, A. Lamin B1 fluctuations have differential effects on cellular proliferation and senescence. J. Cell Biol. 2013, 200, 605–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClintock, D.; Ratner, D.; Lokuge, M.; Owens, D.M.; Gordon, L.B.; Collins, F.S.; Djabali, K. The mutant form of Lamin A that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PLoS ONE 2007, 2. [Google Scholar] [CrossRef] [PubMed]
- Kudlow, B.A.; Stanfel, M.N.; Burtner, C.R.; Johnston, E.D.; Kennedy, B.K. Suppression of proliferative defects associated with processing-defective lamin A mutants by hTERT or inactivation of p53. Mol. Biol. Cell 2008, 19, 5238–5248. [Google Scholar] [CrossRef] [PubMed]
- Dreesen, O.; Ong, P.F.; Chojnowski, A.; Colman, A. The contrasting roles of lamin B1 in cellular aging and human disease. Nucleus 2013, 4, 283–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.S.; Dreesen, O. Biomarkers of cellular senescence and skin aging. Front. Genet. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Carrero, D.; Soria-Valles, C.; López-Otín, C. Hallmarks of progeroid syndromes: Lessons from mice and reprogrammed cells. Dis. Model. Mech. 2016, 9, 719–735. [Google Scholar] [CrossRef]
- Navarro, C.L.; Cau, P.; Lévy, N. Molecular bases of progeroid syndromes. Hum. Mol. Genet. 2006, 15, 151–161. [Google Scholar] [CrossRef]
- Capell, B.C.; Tlougan, B.E.; Orlow, S.J. From the rarest to the most common: Insights from progeroid syndromes into skin cancer and aging. J. Investig. Dermatol. 2009, 129, 2340–2350. [Google Scholar] [CrossRef]
- Croteau, D.L.; Popuri, V.; Opresko, P.L.; Bohr, V.A. Human RecQ Helicases in DNA Repair, Recombination, and Replication. Annu. Rev. Biochem. 2014, 83, 519–552. [Google Scholar] [CrossRef] [Green Version]
- Stefanini, M.; Botta, E.; Lanzafame, M.; Orioli, D. Trichothiodystrophy: From basic mechanisms to clinical implications. DNA Repair 2010, 9, 2–10. [Google Scholar] [CrossRef]
- Warrick, E.; Garcia, M.; Chagnoleau, C.; Chevallier, O.; Bergoglio, V.; Sartori, D.; Mavilio, F.; Angulo, J.F.; Avril, M.-F.; Sarasin, A.; et al. Preclinical Corrective Gene Transfer in Xeroderma Pigmentosum Human Skin Stem Cells. Mol. Ther. 2012, 20, 798–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanzafame, M.; Vaz, B.; Nardo, T.; Botta, E.; Orioli, D.; Stefanini, M. From laboratory tests to functional characterisation of Cockayne syndrome. Mech. Ageing Dev. 2013, 134, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Bagby, G. Recent advances in understanding hematopoiesis in Fanconi Anemia. F1000Research 2018, 7, 105. [Google Scholar] [CrossRef] [Green Version]
- Alderton, G.K.; Joenje, H.; Varon, R.; Børglum, A.D.; Jeggo, P.A.; O’Driscoll, M. Seckel syndrome exhibits cellular features demonstrating defects in the ATR-signalling pathway. Hum. Mol. Genet. 2004, 13, 3127–3138. [Google Scholar] [CrossRef] [Green Version]
- Marechal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Taylor, A.M.R.A.M.R.; Groom, A.; Byrd, P.J. Ataxia-telangiectasia-like disorder (ATLD)-Its clinical presentation and molecular basis. DNA Repair 2004, 3, 1219–1225. [Google Scholar] [CrossRef]
- McKinnon, P.J. ATM and the Molecular Pathogenesis of Ataxia Telangiectasia. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 303–321. [Google Scholar] [CrossRef]
- Lin, F.; Worman, H.J. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 1993, 268, 16321–16326. [Google Scholar] [CrossRef]
- Ramírez, C.L.; Cadiñanos, J.; Varela, I.; Freije, J.M.P.J.M.P.; López-Otín, C. Human progeroid syndromes, aging and cancer: New genetic and epigenetic insights into old questions. Cell. Mol. Life Sci. 2007, 64, 155–170. [Google Scholar] [CrossRef]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef] [PubMed]
- Jamin, A.; Wiebe, M.S. Barrier to Autointegration Factor (BANF1): Interwoven roles in nuclear structure, genome integrity, innate immunity, stress responses and progeria. Curr. Opin. Cell Biol. 2015, 34, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Rankin, J.; Ellard, S. The laminopathies: A clinical review. Clin. Genet. 2006, 70, 261–274. [Google Scholar] [CrossRef]
- Shawi, M.; Autexier, C. Telomerase, senescence and ageing. Mech. Ageing Dev. 2008, 129, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Glousker, G.; Touzot, F.; Revy, P.; Tzfati, Y.; Savage, S.A. Unraveling the pathogenesis of Hoyeraal-Hreidarsson syndrome, a complex telomere biology disorder. Br. J. Haematol. 2015, 170, 457–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallis, C.V.; Sheerin, A.N.; Green, M.H.L.M.H.L.; Jones, C.J.; Kipling, D.; Faragher, R.G.A. Fibroblast clones from patients with Hutchinson-Gilford progeria can senesce despite the presence of telomerase. Exp. Gerontol. 2004, 39, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Pascucci, B.; Fragale, A.; Marabitti, V.; Leuzzi, G.; Salvatore Calcagnile, A.; Parlanti, E.; Franchitto, A.; Dogliotti, E.; D’Errico, M. CSA and CSB play a role in the response to DNA breaks. Oncotarget 2018, 9, 11581–11591. [Google Scholar] [CrossRef]
- Cordisco, S.; Tinaburri, L.; Teson, M.; Orioli, D.; Cardin, R.; Degan, P.; Stefanini, M.; Zambruno, G.; Guerra, L.; Dellambra, E. Cockayne Syndrome Type A Protein Protects Primary Human Keratinocytes from Senescence. J. Investig. Dermatol. 2018, 1–13. [Google Scholar] [CrossRef]
- Pascucci, B.; Lemma, T.; Iorio, E.; Giovannini, S.; Vaz, B.; Iavarone, I.; Calcagnile, A.; Narciso, L.; Degan, P.; Podo, F.; et al. An altered redox balance mediates the hypersensitivity of Cockayne syndrome primary fibroblasts to oxidative stress. Aging Cell 2012, 11, 520–529. [Google Scholar] [CrossRef] [Green Version]
- Scheibye-Knudsen, M.; Croteau, D.L.; Bohr, V.A. Mitochondrial deficiency in Cockayne syndrome. Mech. Ageing Dev. 2013, 134, 275–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheibye-Knudsen, M.; Mitchell, S.J.; Fang, E.F.; Iyama, T.; Ward, T.; Wang, J.; Dunn, C.A.; Singh, N.; Veith, S.; Hasan-Olive, M.M.; et al. A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 2014, 20, 840–855. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; Bailey, A.D.; Weiner, A.M. Cockayne syndrome group B protein (CSB) plays a general role in chromatin maintenance and remodeling. Proc. Natl. Acad. Sci. USA 2006, 103, 9613–9618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arseni, L.; Lanzafame, M.; Compe, E.; Fortugno, P.; Afonso-Barroso, A.; Peverali, F.A.; Lehmann, A.R.; Zambruno, G.; Egly, J.-M.; Stefanini, M.; et al. TFIIH-dependent MMP-1 overexpression in trichothiodystrophy leads to extracellular matrix alterations in patient skin. Proc. Natl. Acad. Sci. USA 2015, 112, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Varela, I.; Cadiñanos, J.; Pendás, A.M.; Gutiérrez-Fernández, A.; Folgueras, A.R.; Sánchez, L.M.; Zhou, Z.; Rodríguez, F.J.; Stewart, C.L.; Vega, J.A.; et al. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature 2005, 437, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Chojnowski, A.; Ong, P.F.; Wong, E.S.M.E.S.M.; Lim, J.S.Y.J.S.Y.; Mutalif, R.A.; Navasankari, R.; Dutta, B.; Yang, H.; Liow, Y.Y.; Sze, S.K.; et al. Progerin reduces LAP2α-telomere association in Hutchinson-Gilford progeria. Elife 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Risques, R.A.; Martin, G.M.; Rabinovitch, P.S.; Oshima, J. Accelerated telomere shortening and replicative senescence in human fibroblasts overexpressing mutant and wild-type lamin A. Exp. Cell Res. 2008, 314, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Scaffidi, P.; Misteli, T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat. Cell Biol. 2008, 10, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Espada, J.; Varela, I.; Flores, I.; Ugalde, A.P.; Cadiñanos, J.; Pendás, A.M.; Stewart, C.L.; Tryggvason, K.; Blasco, M.A.; Freije, J.M.P.J.M.P.; et al. Nuclear envelope defects cause stem cell dysfunction in premature-aging mice. J. Cell Biol. 2008, 181, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Loi, M.; Cenni, V.; Duchi, S.; Squarzoni, S.; Otin, C.L.; Foisner, R.; Lattanzi, G.; Capanni, C. Barrier-to-Autointegration Factor (BAF) involvement in prelamin A-related chromatin organization changes. Oncotarget 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 2005, 11, 440–445. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Li, J.; Suzuki, K.; Qu, J.; Wang, P.; Zhou, J.; Liu, X.; Ren, R.; Xu, X.; Ocampo, A.; et al. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 2015, 348, 1160–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shumaker, D.K.; Dechat, T.; Kohlmaier, A.; Adam, S.A.; Bozovsky, M.R.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Khuon, S.; Collins, F.S.; et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. USA 2006, 103, 8703–8708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarg, B.; Koutzamani, E.; Helliger, W.; Rundquist, I.; Lindner, H.H. Postsynthetic trimethylation of histone H4 at lysine 20 in mammalian tissues is associated with aging. J. Biol. Chem. 2002, 277, 39195–39201. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.M.; Jaber-Hijazi, F.; Cole, J.J.; Robertson, N.A.; Pawlikowski, J.S.; Norris, K.T.; Criscione, S.W.; Pchelintsev, N.A.; Piscitello, D.; Stong, N.; et al. Mapping H4K20me3 onto the chromatin landscape of senescent cells indicates a function in control of cell senescence and tumor suppression through preservation of genetic and epigenetic stability. Genome Biol. 2016, 17, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Mulia, J.C.; Desprat, R.; Trevilla-Garcia, C.; Cornacchia, D.; Schwerer, H.; Sasaki, T.; Sima, J.; Fells, T.; Studer, L.; Lemaitre, J.-M.; et al. DNA replication timing alterations identify common markers between distinct progeroid diseases. Proc. Natl. Acad. Sci. USA 2017, 2017, 11613. [Google Scholar] [CrossRef] [PubMed]
- Le May, N.; Mota-Fernandes, D.; Vélez-Cruz, R.; Iltis, I.; Biard, D.; Egly, J.M. NER Factors Are Recruited to Active Promoters and Facilitate Chromatin Modification for Transcription in the Absence of Exogenous Genotoxic Attack. Mol. Cell 2010, 38, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.M.; Schmitt, N.; Hoffmann-Rohrer, U.; Schäfer, A.; Grummt, I.; Mayer, C. TAF12 Recruits Gadd45a and the Nucleotide Excision Repair Complex to the Promoter of rRNA Genes Leading to Active DNA Demethylation. Mol. Cell 2009, 33, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Le May, N.; Fradin, D.; Iltis, I.; Bougnères, P.; Egly, J.-M. XPG and XPF Endonucleases Trigger Chromatin Looping and DNA Demethylation for Accurate Expression of Activated Genes. Mol. Cell 2012, 47, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, T.L.A.T.L.A.; Rapicavoli, N.A.; Wu, A.R.; Qu, K.; Quake, S.R.; Chang, H.Y. Dynamic chromatin localization of sirt6 shapes stress- and aging-related transcriptional networks. PLoS Genet. 2011, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Edelman, J.R. Unstable Heterochromatin in Fanconi ’s Anemia Cells. Cytologia (Tokyo) 2001, 66, 403–407. [Google Scholar] [CrossRef]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orioli, D.; Dellambra, E. Epigenetic Regulation of Skin Cells in Natural Aging and Premature Aging Diseases. Cells 2018, 7, 268. https://doi.org/10.3390/cells7120268
Orioli D, Dellambra E. Epigenetic Regulation of Skin Cells in Natural Aging and Premature Aging Diseases. Cells. 2018; 7(12):268. https://doi.org/10.3390/cells7120268
Chicago/Turabian StyleOrioli, Donata, and Elena Dellambra. 2018. "Epigenetic Regulation of Skin Cells in Natural Aging and Premature Aging Diseases" Cells 7, no. 12: 268. https://doi.org/10.3390/cells7120268
APA StyleOrioli, D., & Dellambra, E. (2018). Epigenetic Regulation of Skin Cells in Natural Aging and Premature Aging Diseases. Cells, 7(12), 268. https://doi.org/10.3390/cells7120268